
Molecular Studies for the Early Detection of Philadelphia-Negative Myeloproliferative Neoplasms

 , ,
, ,
Abstract
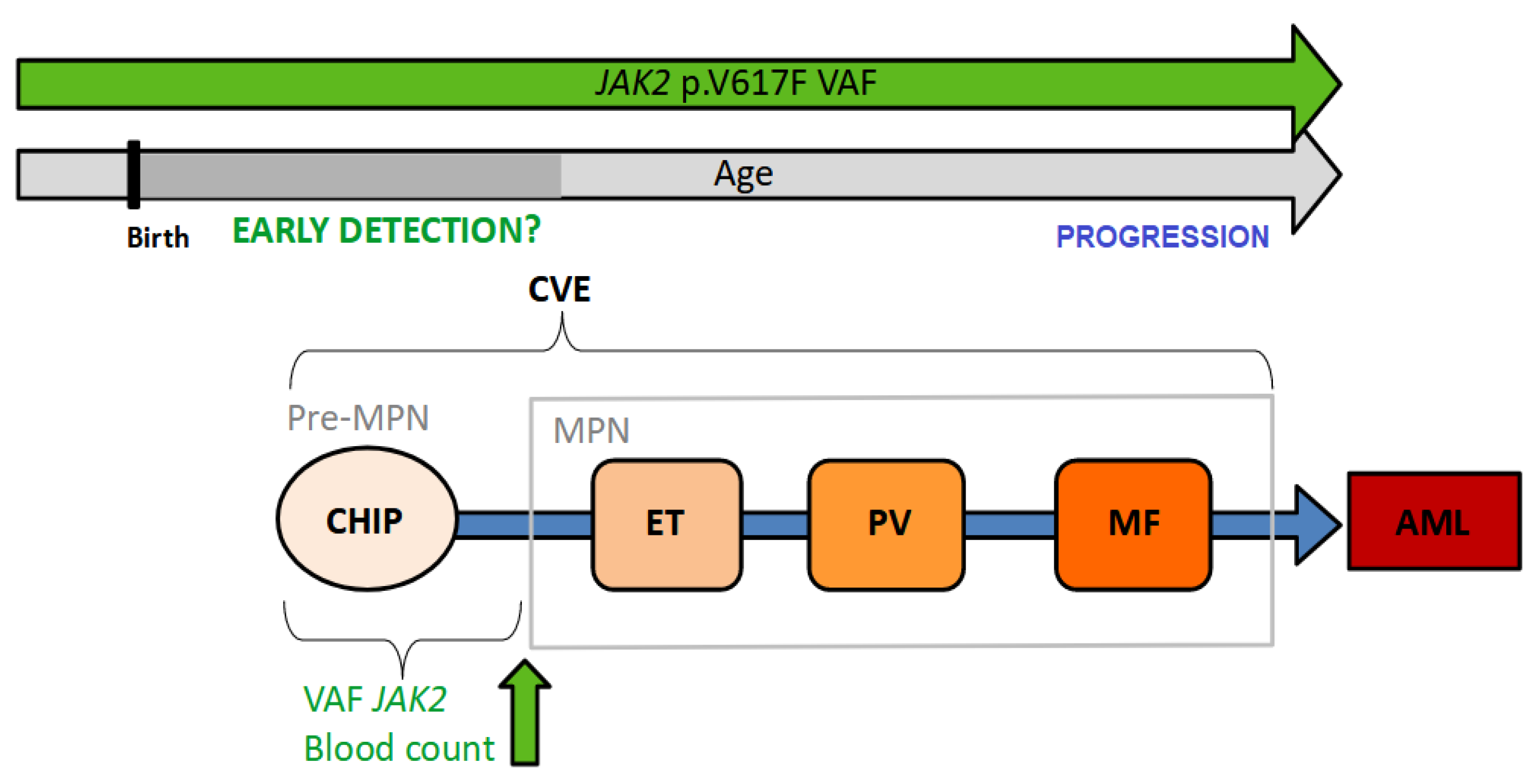
:1. Introduction
2. Clonal Hematopoiesis of Indeterminate Potential (CHIP)
3. Variant Allele Frequency
4. Latency
5. Clonal Expansion
5.1. Other Mutations
5.2. Germline Predisposition
5.3. Inflammation
6. Can Early-Phase MPN Be Detected in the Clinic?
6.1. Screening Feasibility
6.2. Possible Intervention
7. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Khoury, J.D.; Solary, E.; Abla, O.; Akkari, Y.; Alaggio, R.; Apperley, J.F.; Bejar, R.; Berti, E.; Busque, L.; Chan, J.K.C.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia 2022, 36, 1703–1719. [Google Scholar] [CrossRef]
- How, J.; Garcia, J.S.; Mullally, A. Biology and therapeutic targeting of molecular mechanisms in MPN. Blood 2023, 141, 1922–1933. [Google Scholar] [CrossRef] [PubMed]
- Langabeer, S.E.; Andrikovics, H.; Asp, J.; Bellosillo, B.; Carillo, S.; Haslam, K.; Kjaer, L.; Lippert, E.; Mansier, O.; Oppliger Leibundgut, E.; et al. Molecular diagnostics of myeloproliferative neoplasms. Eur. J. Haematol. 2015, 95, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Campbell, P.J.; Scott, L.M.; Buck, G.; Wheatley, K.; East, C.L.; Marsden, J.T.; Duffy, A.; Boyd, E.M.; Bench, A.J.; Scott, M.A.; et al. Definition of subtypes of essential thrombocythaemia and relation to polycythaemia vera based on JAK2 V617F mutation status: A prospective study. Lancet 2005, 366, 1945–1953. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pietra, D.; Della Porta, M.G.; Boveri, E.; Pascutto, C.; Vanelli, L.; Arcaini, L.; Burcheri, S.; Malcovati, L.; et al. Relation between JAK2 (V617F) mutation status, granulocyte activation, and constitutive mobilization of CD34 cells into peripheral blood in myeloproliferative disorders. Blood 2006, 107, 3676–3682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alshemmari, S.H.; Rajaan, R.; Ameen, R.; Al-Drees, M.A.; Almosailleakh, M.R. JAK2V617F allele burden in patients with myeloproliferative neoplasms. Ann. Hematol. 2014, 93, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Kralovics, R.; Passamonti, F.; Buser, A.S.; Teo, S.S.; Tiedt, R.; Passweg, J.R.; Tichelli, A.; Cazzola, M.; Skoda, R.C. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N. Engl. J. Med. 2005, 352, 1779–1790. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Wadleigh, M.; Cools, J.; Ebert, B.L.; Wernig, G.; Huntly, B.J.; Boggon, T.J.; Wlodarska, I.; Clark, J.J.; Moore, S.; et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell 2005, 7, 387–397. [Google Scholar] [CrossRef] [Green Version]
- Kralovics, R.; Guan, Y.; Prchal, J.T. Acquired uniparental disomy of chromosome 9p is a frequent stem cell defect in polycythemia vera. Exp. Hematol. 2002, 30, 229–236. [Google Scholar] [CrossRef]
- Jamieson, C.H.; Gotlib, J.; Durocher, J.A.; Chao, M.P.; Mariappan, M.R.; Lay, M.; Jones, C.; Zehnder, J.L.; Lilleberg, S.L.; Weissman, I.L. The JAK2 V617F mutation occurs in hematopoietic stem cells in polycythemia vera and predisposes toward erythroid differentiation. Proc. Natl. Acad. Sci. USA 2006, 103, 6224–6229. [Google Scholar] [CrossRef]
- Swerdlow, S.H. (Ed.) WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2017. [Google Scholar]
- Enblom, A.; Lindskog, E.; Hasselbalch, H.; Hersby, D.; Bak, M.; Tetu, J.; Girodon, F.; Andréasson, B. High rate of abnormal blood values and vascular complications before diagnosis of myeloproliferative neoplasms. Eur. J. Intern. Med. 2015, 26, 344–347. [Google Scholar] [CrossRef]
- Hultcrantz, M.; Björkholm, M.; Dickman, P.W.; Landgren, O.; Derolf, Å.R.; Kristinsson, S.Y.; Andersson, T.M.L. Risk for arterial and venous thrombosis in patients with myeloproliferative neoplasms: A population-based cohort study. Ann Inter. Med. 2018, 168, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Tam, C.S.; Nussenzveig, R.M.; Popat, U.; Bueso-Ramos, C.E.; Thomas, D.A.; Cortes, J.A.; Champlin, R.E.; Ciurea, S.E.; Manshouri, T.; Pierce, S.M.; et al. The Natural History and Treatment Outcome of Blast Phase BCR-ABL—Myeloproliferative Neoplasms. Blood 2008, 112, 1628–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Guglielmelli, P.; Larson, D.R.; Finke, C.; Wassie, E.A.; Pieri, L.; Gangat, N.; Fjerza, R.; Belachew, A.A.; Lasho, T.L.; et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood 2014, 124, 2507–2615. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Screening and Early Detection Cancer. Cancer, WHO Fact Sheet. May 2010. Available online: https://www.who.int/europe/news-room/fact-sheets/item/cancer-screening-and-early-detection-of-cancer (accessed on 9 January 2023).
- Srivastava, S.; Ghosh, S.; Kagan, J.; Mazurchuk, R. The PreCancer Atlas (PCA). Trends Cancer 2018, 4, 513–514. [Google Scholar] [CrossRef]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Kefala, M.; Gjerdrum, L.M.R.; Hasselbalch, H.C.; Ellervik, C. Early detection of myeloproliferative neoplasms in a Danish general population study. Leukemia 2021, 35, 2706–2709. [Google Scholar] [CrossRef]
- Jelinek, J.; Oki, Y.; Gharibyan, V.; Bueso-Ramos, C.; Prchal, J.T.; Verstovsek, S.; Beran, M.; Estey, E.; Kantarjian, H.M.; Issa, J.P. JAK2 mutation 1849G > T is rare in acute leukemias but can be found in CMML, Philadelphia chromosome-negative CML, and megakaryocytic leukemia. Blood 2005, 106, 3370–3373. [Google Scholar] [CrossRef] [Green Version]
- Levine, R.L.; Loriaux, M.; Huntly, B.J.; Loh, M.L.; Beran, M.; Stoffregen, E.; Berger, R.; Clark, J.J.; Willis, S.G.; Nguyen, K.T.; et al. The JAK2V617F activating mutation occurs in chronic myelomonocytic leukemia and acute myeloid leukemia, but not in acute lymphoblastic leukemia or chronic lymphocytic leukemia. Blood 2005, 106, 3377–3379. [Google Scholar] [CrossRef] [Green Version]
- Sidon, P.; El Housni, H.; Dessars, B.; Heimann, P. The JAK2V617F mutation is detectable at very low level in peripheral blood of healthy donors. Leukemia 2006, 20, 1622. [Google Scholar] [CrossRef]
- Lynch, M. Evolution of the mutation rate. Trends Genet. 2010, 26, 345–352. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.M. Natural selection and the concept of a protein space. Nature 1970, 225, 563–564. [Google Scholar] [CrossRef]
- Bowling, S.; Lawlor, K.; Rodríguez, T.A. Cell competition: The winners and losers of fitness selection. Development 2019, 146, dev167486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martincorena, I.; Campbell, P.J. Somatic mutation in cancer and normal cells. Science 2015, 349, 1483–1489. [Google Scholar] [CrossRef]
- Osorio, F.G.; Rosendahl Huber, A.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee-Six, H.; Øbro, N.F.; Shepherd, M.S.; Grossmann, S.; Dawson, K.; Belmonte, M.; Osborne, R.J.; Huntly, B.J.P.; Martincorena, I.; Anderson, E.; et al. Population dynamics of normal human blood inferred from somatic mutations. Nature 2018, 561, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Ebert, B.L. Clonal hematopoiesis in human aging and disease. Science 2019, 366, eaan4673. [Google Scholar] [CrossRef]
- Mead, A.J.; Mullally, A. Myeloproliferative neoplasm stem cells. Blood 2017, 129, 1607–1616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kralovics, R. Genetic complexity of myeloproliferative neoplasms. Leukemia 2008, 22, 1841–1848. [Google Scholar] [CrossRef] [Green Version]
- Loh, P.R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8342 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef]
- Niroula, A.; Sekar, A.; Murakami, M.A.; Trinder, M.; Agrawal, M.; Wong, W.J.; Bick, A.G.; Uddin, M.M.; Gibson, C.J.; Griffin, G.K.; et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat. Med. 2021, 27, 1921–1927. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Mooney, L.; Goodyear, C.S.; Chandra, T.; Kirschner, K.; Copland, M.; Petrie, M.C.; Lang, N.N. Clonal haematopoiesis of indeterminate potential: Intersections between inflammation, vascular disease and heart failure. Clin. Sci. 2021, 135, 991–1007. [Google Scholar] [CrossRef]
- Segura-Díaz, A.; Stuckey, R.; Florido, Y.; González-Martín, J.M.; López-Rodríguez, J.F.; Sánchez-Sosa, S.; González-Pérez, E.; Sáez Perdomo, M.N.; Perera, M.D.M.; de la Iglesia, S.; et al. Thrombotic Risk Detection in Patients with Polycythemia Vera: The Predictive Role of DNMT3A/TET2/ASXL1 Mutations. Cancers 2020, 12, 934. [Google Scholar] [CrossRef] [Green Version]
- Barbui, T.; Finazzi, G.; Carobbio, A.; Thiele, J.; Passamonti, F.; Rumi, E.; Ruggeri, M.; Rodeghiero, F.; Randi, M.L.; Bertozzi, I.; et al. Development and validation of an International Prognostic Score of thrombosis in World Health Organization-essential thrombocythemia (IPSET-thrombosis). Blood 2012, 120, 5128–5133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Zhang, Q.; Luo, J.; Xing, S.; Li, Q.; Krantz, S.B.; Fu, X.; Zhao, Z.J. JAK2(V617F): Prevalence in a large Chinese hospital population. Blood 2007, 109, 339–342. [Google Scholar] [CrossRef] [Green Version]
- Cordua, S.; Kjaer, L.; Skov, V.; Pallisgaard, N.; Hasselbalch, H.C.; Ellervik, C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood 2019, 134, 469–479. [Google Scholar] [CrossRef]
- Wernig, G.; Mercher, T.; Okabe, R.; Levine, R.L.; Lee, B.H.; Gilliland, D.G. Expression of Jak2V617F causes a polycythemia vera-like disease with associated myelofibrosis in a murine bone marrow transplant model. Blood 2006, 107, 4274–4281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacout, C.; Pisani, D.F.; Tulliez, M.; Gachelin, F.M.; Vainchenker, W.; Villeval, J.L. JAK2V617F expression in murine hematopoietic cells leads to MPD mimicking human PV with secondary myelofibrosis. Blood 2006, 108, 1652–1660. [Google Scholar] [CrossRef] [Green Version]
- Lundberg, P.; Takizawa, H.; Kubovcakova, L.; Guo, G.; Hao-Shen, H.; Dirnhofer, S.; Orkin, S.H.; Manz, M.G.; Skoda, R.C. Myeloproliferative neoplasms can be initiated from a single hematopoietic stem cell expressing JAK2-V617F. J. Exp. Med. 2014, 211, 2213–2230. [Google Scholar] [CrossRef]
- Jovanovic, J.V.; Ivey, A.; Vannucchi, A.M.; Lippert, E.; Oppliger Leibundgut, E.; Cassinat, B.; Pallisgaard, N.; Maroc, N.; Hermouet, S.; Nickless, G.; et al. Establishing optimal quantitative-polymerase chain reaction assays for routine diagnosis and tracking of minimal residual disease in JAK2-V617F-associated myeloproliferative neoplasms: A joint European LeukemiaNet/MPN&MPNr-EuroNet study. Leukemia 2013, 27, 2032–2039. [Google Scholar] [PubMed] [Green Version]
- Lippert, E.; Girodon, F.; Hammond, E.; Jelinek, J.; Reading, N.S.; Fehse, B.; Hanlon, K.; Hermans, M.; Richard, C.; Swierczek, S.; et al. Concordance of assays designed for the quantification of JAK2V617F: A multicenter study. Haematologica 2009, 94, 38–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippert, E.; Mansier, O.; Migeon, M.; Denys, B.; Nilsson, A.; Rosmond, C.; Lodé, L.; Ugo, V.; Lascaux, A.; Bellosillo, B.; et al. Clinical and biological characterization of patients with low (0.1–2%) JAK2V617F allele burden at diagnosis. Haematologica 2014, 99, e98–e101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perricone, M.; Polverelli, N.; Martinelli, G.; Catani, L.; Ottaviani, E.; Zuffa, E.; Franchini, E.; Dizdari, A.; Forte, D.; Sabattini, E.; et al. The relevance of a low JAK2V617F allele burden in clinical practice: A monocentric study. Oncotarget 2017, 8, 37239–37249. [Google Scholar] [CrossRef] [Green Version]
- Link-Lenczowska, D.; Pallisgaard, N.; Cordua, S.; Zawada, M.; Czekalska, S.; Krochmalczyk, D.; Kanduła, Z.; Sacha, T. A comparison of qPCR and ddPCR used for quantification of the JAK2 V617F allele burden in Ph negative MPNs. Ann. Hematol. 2018, 97, 2299–2308. [Google Scholar] [CrossRef] [Green Version]
- Passamonti, F.; Rumi, E. Clinical relevance of JAK2 (V617F) mutant allele burden. Haematologica 2009, 94, 7–10. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Rambaldi, A.; Barosi, G.; Marchioli, R.; Marfisi, R.M.; Finazzi, G.; Guerini, V.; Fabris, F.; et al. Clinical profile of homozygous JAK2 617V>F mutation in patients with polycythemia vera or essential thrombocythemia. Blood 2007, 110, 840–846. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Longo, G.; Pancrazzi, A.; Ponziani, V.; Bogani, C.; Ferrini, P.R.; Rambaldi, A.; Guerini, V.; et al. Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007, 21, 1952–1959. [Google Scholar] [CrossRef] [Green Version]
- Kirschner, M.M.; Schemionek, M.; Schubert, C.; Chatain, N.; Sontag, S.; Isfort, S.; Ortiz-Brüchle, N.; Schmitt, K.; Krüger, L.; Zerres, K.; et al. Dissecting Genomic Aberrations in Myeloproliferative Neoplasms by Multiplex-PCR and Next Generation Sequencing. PLoS ONE 2015, 10, e0123476. [Google Scholar] [CrossRef] [Green Version]
- Vannucchi, A.M.; Antonioli, E.; Guglielmelli, P.; Pardanani, A.; Tefferi, A. Clinical correlates of JAK2V617F presence or allele burden in myeloproliferative neoplasms: A critical reappraisal. Leukemia 2008, 22, 1299–1307. [Google Scholar] [CrossRef]
- Vannucchi, A.M.; Barosi, G.; Rambaldi, A.; Marchioli, R.; Barbui, T. Clinical Significance of JAK2V617F Homozygosity in the Chronic Myeloproliferative Disorders. A Study on 1306 Patients. Blood 2006, 108, 664. [Google Scholar] [CrossRef]
- Passamonti, F.; Rumi, E.; Pietra, D.; Elena, C.; Boveri, E.; Arcaini, L.; Roncoroni, E.; Astori, C.; Merli, M.; Boggi, S.; et al. A prospective study of 338 patients with polycythemia vera: The impact of JAK2 (V617F) allele burden and leukocytosis on fibrotic or leukemic disease transformation and vascular complications. Leukemia 2010, 24, 1574–1579. [Google Scholar] [CrossRef] [Green Version]
- Malak, S.; Labopin, M.; Saint-Martin, C.; Bellanne-Chantelot, C.; Najman, A.; French Group of Familial Myeloproliferative Disorders. Long term follow up of 93 families with myeloproliferative neoplasms: Life expectancy and implications of JAK2V617F in the occurrence of complications. Blood Cells Mol. Dis. 2012, 49, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Grinfeld, J.; Nangalia, J.; Baxter, E.J.; Wedge, D.C.; Angelopoulos, N.; Cantrill, R.; Godfrey, A.L.; Papaemmanuil, E.; Gundem, G.; MacLean, C.; et al. Classification and Personalized Prognosis in Myeloproliferative Neoplasms. N. Engl. J. Med. 2018, 379, 1416–1430. [Google Scholar] [CrossRef] [PubMed]
- McKerrell, T.; Park, N.; Chi, J.; Collord, G.; Moreno, T.; Ponstingl, H.; Dias, J.; Gerasimou, P.; Melanthiou, K.; Prokopiou, C.; et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv. 2017, 1, 968–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Egeren, D.; Escabi, J.; Nguyen, M.; Liu, S.; Reilly, C.R.; Patel, S.; Kamaz, B.; Kalyva, M.; DeAngelo, D.J.; Galinsky, I.; et al. Reconstructing the Lineage Histories and Differentiation Trajectories of Individual Cancer Cells in Myeloproliferative Neoplasms. Cell Stem Cell 2021, 28, 514–523.e9. [Google Scholar] [CrossRef]
- Abu-Zeinah, G.; Di Giandomenico, S.; Choi, D.; Cruz, T.; Erdos, K.; Taylor, E.; Ritchie, E.K.; Silver, R.T.; Scandura, J.M. Hematopoietic fitness of JAK2V617F myeloproliferative neoplasms is linked to clinical outcome. Blood Adv. 2022, 6, 5477–5481. [Google Scholar] [CrossRef]
- Nienhold, R.; Ashcroft, P.; Zmajkovic, J.; Rai, S.; Rao, T.N.; Drexler, B.; Meyer, S.C.; Lundberg, P.; Passweg, J.R.; Leković, D.; et al. MPN patients with low mutant JAK2 allele burden show late expansion restricted to erythroid and megakaryocytic lineages. Blood 2020, 136, 2591–2595. [Google Scholar] [CrossRef]
- Nangalia, J.; Mitchell, E.; Green, A.R. Clonal approaches to understanding the impact of mutations on hematologic disease development. Blood 2019, 133, 1436–1445. [Google Scholar] [CrossRef] [Green Version]
- Williams, N.; Lee, J.; Mitchell, E.; Moore, L.; Baxter, E.J.; Hewinson, J.; Dawson, K.J.; Menzies, A.; Godfrey, A.L.; Green, A.R.; et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature 2022, 60, 162–168. [Google Scholar] [CrossRef]
- Weinstock, J.S.; Gopakumar, J.; Burugula, B.B.; Uddin, M.M.; Jahn, N.; Belk, J.A.; Bouzid, H.; Daniel, B.; Miao, Z.; Ly, N.; et al. Aberrant activation of TCL1A promotes stem cell expansion in clonal haematopoiesis. Nature 2023, 616, 755–763. [Google Scholar] [CrossRef]
- Hansen, J.W.; Pedersen, D.A.; Larsen, L.A.; Husby, S.; Clemmensen, S.B.; Hjelmborg, J.; Favero, F.; Weischenfeldt, J.; Christensen, K.; Grønbæk, K. Clonal hematopoiesis in elderly twins: Concordance, discordance, and mortality. Blood 2020, 135, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Sousos, N.; Ní Leathlobhair, M.; Simoglou Karali, C.; Louka, E.; Bienz, N.; Royston, D.; Clark, S.A.; Hamblin, A.; Howard, K.; Mathews, V.; et al. In utero origin of myelofibrosis presenting in adult monozygotic twins. Nat. Med. 2022, 28, 1207–1211. [Google Scholar] [CrossRef] [PubMed]
- Gale, R.E.; Allen, A.J.; Nash, M.J.; Linch, D.C. Long-term serial analysis of X-chromosome inactivation patterns and JAK2 V617F mutant levels in patients with essential thrombocythemia show that minor mutant-positive clones can remain stable for many years. Blood 2007, 109, 1241–1243. [Google Scholar] [CrossRef] [PubMed]
- Fabre, M.A.; de Almeida, J.G.; Fiorillo, E.; Mitchell, E.; Damaskou, A.; Rak, J.; Orrù, V.; Marongiu, M.; Chapman, M.S.; Vijayabaskar, M.S.; et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature 2022, 606, 335–342. [Google Scholar] [CrossRef]
- Ortmann, C.A.; Kent, D.G.; Nangalia, J.; Silber, Y.; Wedge, D.C.; Grinfeld, J.; Baxter, E.J.; Massie, C.E.; Papaemmanuil, E.; Menon, S.; et al. Effect of mutation order on myeloproliferative neoplasms. N. Engl. J. Med. 2015, 7, 372. [Google Scholar] [CrossRef] [Green Version]
- Nangalia, J.; Nice, F.L.; Wedge, D.C.; Godfrey, A.L.; Grinfeld, J.; Thakker, C.; Massie, C.E.; Baxter, J.; Sewell, D.; Silber, Y.; et al. DNMT3A mutations occur early or late in patients with myeloproliferative neoplasms and mutation order influences phenotype. Haematologica 2015, 100, e438–e442. [Google Scholar] [CrossRef] [Green Version]
- Landgren, O.; Goldin, L.R.; Kristinsson, S.Y.; Helgadottir, E.A.; Samuelsson, J.; Björkholm, M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood 2008, 112, 2199–2204. [Google Scholar] [CrossRef]
- Bick, A.G.; Weinstock, J.S.; Nandakumar, S.K.; Fulco, C.P.; Bao, E.L.; Zekavat, S.M.; Szeto, M.D.; Liao, X.; Leventhal, M.J.; Nasser, J.; et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature 2020, 586, 763–768. [Google Scholar] [CrossRef]
- Jones, A.V.; Chase, A.; Silver, R.T.; Oscier, D.; Zoi, K.; Wang, Y.L.; Cario, H.; Pahl, H.L.; Collins, A.; Reiter, A.; et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat. Genet. 2009, 41, 446–449. [Google Scholar] [CrossRef] [Green Version]
- Kilpivaara, O.; Mukherjee, S.; Schram, A.M.; Wadleigh, M.; Mullally, A.; Ebert, B.L.; Bass, A.; Marubayashi, S.; Heguy, A.; Garcia-Manero, G.; et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2V617F-positive myeloproliferative neoplasms. Nat. Genet. 2009, 41, 455–459. [Google Scholar] [CrossRef] [Green Version]
- Olcaydu, D.; Harutyunyan, A.; Jäger, R.; Berg, T.; Gisslinger, B.; Pabinger, I.; Gisslinger, H.; Kralovics, R. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat. Genet. 2009, 41, 450–454. [Google Scholar] [CrossRef]
- Hinds, D.A.; Barnholt, K.E.; Mesa, R.A.; Kiefer, A.K.; Do, C.B.; Eriksson, N.; Mountain, J.L.; Francke, U.; Tung, J.Y.; Nguyen, H.M.; et al. Germ line variants predispose to both JAK2 V617F clonal hematopoiesis and myeloproliferative neoplasms. Blood 2016, 128, 1121–1128. [Google Scholar] [CrossRef] [Green Version]
- Kar, S.P.; Quiros, P.M.; Gu, M.; Jiang, T.; Mitchell, J.; Langdon, R.; Iyer, V.; Barcena, C.; Vijayabaskar, M.S.; Fabre, M.A.; et al. Genome-wide analyses of 200,453 individuals yield new insights into the causes and consequences of clonal hematopoiesis. Nat. Genet. 2022, 54, 1155–1166. [Google Scholar] [CrossRef]
- Zink, F.; Stacey, S.N.; Norddahl, G.L.; Frigge, M.L.; Magnusson, O.T.; Jonsdottir, I.; Thorgeirsson, T.E.; Sigurdsson, A.; Gudjonsson, S.A.; Gudmundsson, J.; et al. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood 2017, 130, 742–752. [Google Scholar] [CrossRef] [Green Version]
- Trifa, A.P.; Bănescu, C.; Tevet, M.; Bojan, A.; Dima, D.; Urian, L.; Török-Vistai, T.; Popov, V.M.; Zdrenghea, M.; Petrov, L.; et al. TERT rs2736100 A>C SNP and JAK2 46/1 haplotype significantly contribute to the occurrence of JAK2 V617F and CALR mutated myeloproliferative neoplasms—A multicentric study on 529 patients. Br. J. Haematol. 2016, 174, 218–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, E.L.; Nandakumar, S.K.; Liao, X.; Bick, A.G.; Karjalainen, J.; Tabaka, M.; Gan, O.I.; Havulinna, A.S.; Kiiskinen, T.T.J.; Lareau, C.A.; et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature 2020, 586, 769–775. [Google Scholar] [CrossRef]
- Sanders, M.A.; Chew, E.; Flensburg, C.; Zeilemaker, A.; Miller, S.E.; Al Hinai, A.S.; Bajel, A.; Luiken, B.; Rijken, M.; Mclennan, T.; et al. MBD4 guards against methylation damage and germ line deficiency predisposes to clonal hematopoiesis and early-onset AML. Blood 2018, 132, 1526–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.J.; Genovese, G.; Halvardson, J.; Ulirsch, J.C.; Wright, D.J.; Terao, C.; Davidsson, O.B.; Day, F.R.; Sulem, P.; Jiang, Y.; et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature 2019, 575, 652–657. [Google Scholar] [CrossRef]
- Silver, A.J.; Bick, A.G.; Savona, M.R. Germline risk of clonal haematopoiesis. Nat. Rev. Genet. 2021, 22, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Fleischman, A.G.; Aichberger, K.J.; Luty, S.B.; Bumm, T.G.; Petersen, C.L.; Doratotaj, S.; Vasudevan, K.B.; LaTocha, D.H.; Yang, F.; Press, R.D.; et al. TNFα facilitates clonal expansion of JAK2V617F positive cells in myeloproliferative neoplasms. Blood 2011, 118, 6392–6398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: Is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood 2012, 119, 3219–3224. [Google Scholar] [CrossRef] [Green Version]
- Kristinsson, S.Y.; Landgren, O.; Samuelsson, J.; Björkholm, M.; Goldin, L.R. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica 2010, 95, 1216–1220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marty, C.; Lacout, C.; Droin, N.; Le Couédic, J.P.; Ribrag, V.; Solary, E.; Vainchenker, W.; Villeval, J.L.; Plo, I. A role for reactive oxygen species in JAK2 V617F myeloproliferative neoplasm progression. Leukemia 2013, 27, 2187–2195. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.R.C.; Nix, D.; Gregory, M.; Ciorba, M.A.; Ostrander, E.L.; Newberry, R.D.; Spencer, D.H.; Challen, G.A. Inflammatory cytokines promote clonal hematopoiesis with specific mutations in ulcerative colitis patients. Exp. Hematol. 2019, 80, 36–41.e33. [Google Scholar] [CrossRef] [PubMed]
- Lai, H.Y.; Brooks, S.A.; Craver, B.M.; Morse, S.J.; Nguyen, T.K.; Haghighi, N.; Garbati, M.R.; Fleischman, A.G. Defective negative regulation of Toll-like receptor signaling leads to excessive TNF-alpha in myeloproliferative neoplasm. Blood Adv. 2019, 3, 122–131. [Google Scholar] [CrossRef] [Green Version]
- Masselli, E.; Carubbi, C.; Cambò, B.; Pozzi, G.; Gobbi, G.; Mirandola, P.; Follini, E.; Pagliaro, L.; Di Marcantonio, D.; Bonatti, F.; et al. The −2518 A/G polymorphism of the monocyte chemoattractant protein-1 as a candidate genetic predisposition factor for secondary myelofibrosis and biomarker of disease severity. Leukemia 2018, 32, 2266–2270. [Google Scholar] [CrossRef] [Green Version]
- Bick, A.G.; Pirruccello, J.P.; Griffin, G.K.; Gupta, N.; Gabriel, S.; Saleheen, D.; Libby, P.; Kathiresan, S.; Natarajan, P. Genetic Interleukin 6 Signaling Deficiency Attenuates Cardiovascular Risk in Clonal Hematopoiesis. Circulation 2020, 141, 124–131. [Google Scholar] [CrossRef]
- Biagi, E.; Candela, M.; Franceschi, C.; Brigidi, P. The aging gut microbiota: New perspectives. Ageing Res. Rev. 2011, 10, 428–429. [Google Scholar] [CrossRef]
- Meisel, M.; Hinterleitner, R.; Pacis, A.; Chen, L.; Earley, Z.M.; Mayassi, T.; Pierre, J.F.; Ernest, J.D.; Galipeau, H.J.; Thuille, N.; et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature 2018, 557, 580–584. [Google Scholar] [CrossRef]
- Oliver, A.; El Alaoui, K.; Haunschild, C.; Avelar-Barragan, J.; Mendez Luque, L.F.; Whiteson, K.; Fleischman, A.G. Fecal Microbial Community Composition in Myeloproliferative Neoplasm Patients Is Associated with an Inflammatory State. Microbiol. Spectr. 2022, 10, e0003222. [Google Scholar] [CrossRef]
- Mendez Luque, L.F.; Avelar-Barragan, J.; Nguyen, H.; Nguyen, J.; Soyfer, E.M.; Liu, J.; Chen, J.H.; Mehrotra, N.; Kosiorek, H.E.; Dueck, A.; et al. The Nutrient Trial (NUTRitional Intervention among myEloproliferative Neoplasms): Feasibility Phase. medRxiv 2023. [Google Scholar] [CrossRef]
- Latchney, S.E.; Calvi, L.M. The aging hematopoietic stem cell niche: Phenotypic and functional changes and mechanisms that contribute to hematopoietic aging. Semin. Hematol. 2017, 54, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, W.W.; Schrier, S.L.; Weissman, I.L. Age-associated changes in human hematopoietic stem cells. Semin. Hematol 2017, 54, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.A.; Hillary, R.F.; McCartney, D.L.; Terradas-Terradas, M.; Higham, J.; Sproul, D.; Deary, I.J.; Kirschner, K.; Marioni, R.E.; Chandra, T. Age-related clonal haemopoiesis is associated with increased epigenetic age. Curr. Biol. 2019, 29, R786–R787. [Google Scholar] [CrossRef] [Green Version]
- Nachun, D.; Lu, A.T.; Bick, A.G.; Natarajan, P.; Weinstock, J.; Szeto, M.D.; Kathiresan, S.; Abecasis, G.; Taylor, K.D.; Guo, X.; et al. Clonal hematopoiesis associated with epigenetic aging and clinical outcomes. Aging Cell 2021, 20, e13366. [Google Scholar] [CrossRef] [PubMed]
- Genovese, G.; Kähler, A.K.; Handsaker, R.E.; Lindberg, J.; Rose, S.A.; Bakhoum, S.F.; Chambert, K.; Mick, E.; Neale, B.M.; Fromer, M.; et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014, 371, 2477–2487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marusyk, A.; DeGregori, J. Declining cellular fitness with age promotes cancer initiation by selecting for adaptive oncogenic mutations. Biochim. Biophys. Acta. 2008, 1785, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Piris-Villaespesa, M.; Álvarez-Larrán, A.; Saez-Marín, A.; Nuñez-Torrón, C.; Muñoz-Martin, G.; Sánchez, R.; Del Castillo, F.J.; Villarrubia, J.; Lopez-Jimenez, J.; Martinez-Lopez, J.; et al. Development and validation of a sequential two-step algorithm for the screening of individuals with potential polycythaemia vera. Sci. Rep. 2021, 11, 209. [Google Scholar] [CrossRef]
- Arber, D.A.; Orazi, A.; Hasserjian, R.; Thiele, J.; Borowitz, M.J.; Le Beau, M.M.; Bloomfield, C.D.; Cazzola, M.; Vardiman, J.W. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016, 127, 2391–2405. [Google Scholar] [CrossRef] [PubMed]
- Osman, A.E.W.G. When are idiopathic and clonal cytopenias of unknown significance (ICUS or CCUS)? Hematol. Am. Soc. Hematol. Educ. Program 2021, 2021, 399–404. [Google Scholar] [CrossRef]
- Nielsen, C.; Bojesen, S.E.; Nordestgaard, B.G.; Kofoed, K.F.; Birgens, H.S. JAK2V617F somatic mutation in the general population: Myeloproliferative neoplasm development and progression rate. Haematologica 2014, 99, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Wolfe, R.; Woods, R.L.; Tonkin, A.M.; Donnan, G.A.; Nelson, M.R.; Reid, C.M.; Lockery, J.E.; Kirpach, B.; Storey, E.; et al. Effect of Aspirin on Cardiovascular Events and Bleeding in the Healthy Elderly. New Engl. J. Med. 2018, 379, 1509–1518. [Google Scholar] [CrossRef] [PubMed]
- Craver, B.M.; Ramanathan, G.; Hoang, S.; Chang, X.; Mendez Luque, L.F.; Brooks, S.; Lai, H.Y.; Fleischman, A.G. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020, 4, 312–321. [Google Scholar] [CrossRef]
- Scherber, R.M.; Langlais, B.T.; Geyer, H.; Dueck, A.; Kosoriek, H.; Johnston, C.; Padrnos, L.; Palmer, J.; Fleischman, A.G.; Mesa, R.A. Nutrition and Supplement Use Characteristics in the Myeloproliferative Neoplasms: Results from the Nutrient Survey. Blood 2017, 130 (Suppl. S1), 2193. [Google Scholar]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef] [Green Version]
- Agathocleous, M.; Meacham, C.E.; Burgess, R.J.; Piskounova, E.; Zhao, Z.; Crane, G.M.; Cowin, B.L.; Bruner, E.; Murphy, M.M.; Chen, W.; et al. Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 2017, 549, 476–481. [Google Scholar] [CrossRef] [Green Version]
- Cimmino, L.; Dolgalev, I.; Wang, Y.; Yoshimi, A.; Martin, G.H.; Wang, J.; Ng, V.; Xia, B.; Witkowski, M.T.; Mitchell-Flack, M.; et al. Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 2017, 170, 1079–1095.e20. [Google Scholar] [CrossRef] [Green Version]
- Magrì, A.; Germano, G.; Lorenzato, A.; Lamba, S.; Chilà, R.; Montone, M.; Amodio, V.; Ceruti, T.; Sassi, F.; Arena, S.; et al. High-dose vitamin C enhances cancer immunotherapy. Sci. Transl. Med. 2020, 12, eaay8707. [Google Scholar] [CrossRef] [PubMed]
- Kiladjian, J.J.; Cassinat, B.; Chevret, S.; Turlure, P.; Cambier, N.; Roussel, M.; Bellucci, S.; Grandchamp, B.; Chomienne, C.; Fenaux, P. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. Blood 2008, 112, 3065–3072. [Google Scholar] [CrossRef]
- Rao, T.N.; Hansen, N.; Stetka, J.; Luque Paz, D.; Kalmer, M.; Hilfiker, J.; Endele, M.; Ahmed, N.; Kubovcakova, L.; Rybarikova, M.; et al. JAK2-V617F and interferon-α induce megakaryocyte-biased stem cells characterized by decreased long-term functionality. Blood 2021, 137, 2139–2151. [Google Scholar] [CrossRef]
- Kiladjian, J.J. Long-term treatment with interferon alfa for myeloproliferative neoplasms. Lancet Haematol. 2017, 4, e150–e151. [Google Scholar] [CrossRef]
- Guarda, G.; Braun, M.; Staehli, F.; Tardivel, A.; Mattmann, C.; Förster, I.; Farlik, M.; Decker, T.; Du Pasquier, R.A.; Romero, P.; et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011, 34, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Svensson, E.C.; Madar, A.; Campbell, C.D.; He, Y.; Sultan, M.; Healey, M.L.; Xu, H.; D’Aco, K.; Fernandez, A.; Wache-Mainier, C.; et al. TET2-Driven Clonal Hematopoiesis Predicts Enhanced Response to Canakinumab in the CANTOS Trial: An Exploratory Analysis. Circulation 2018, 138, A15111. [Google Scholar]
- Rai, S.; Hansen, N.; Hao-Shen, H.; Dirnhofer, S.; Tata, N.R.; Skoda, R.C. IL-1β secreted from mutant cells carrying JAK2-V617F favors early clonal expansion and promotes MPN disease initiation and progression. Blood 2019, 134 (Suppl. S1), 307. [Google Scholar] [CrossRef]
- McMullin, M.F. Diagnostic workflow for hereditary erythrocytosis and thrombocytosis. Hematol. Am. Soc. Hematol. Educ. Program 2019, 2019, 391–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hasselbalch, H.C. Smoking as a contributing factor for development of polycythemia vera and related neoplasms. Leuk. Res. 2015, 39, 1137–1145. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, K.M.; Bak, M.; Sørensen, A.L.; Zwisler, A.D.; Ellervik, C.; Larsen, M.K.; Hasselbalch, H.C.; Tolstrup, J.S. Smoking is associated with increased risk of myeloproliferative neoplasms: A general population-based cohort study. Cancer Med. 2018, 7, 5796–5802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tefferi, A.; Vannucchi, A.M. Genetic Risk Assessment in Myeloproliferative Neoplasms. Mayo Clin. Proc. 2017, 92, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Hermange, G.; Rakotonirainy, A.; Bentriou, M.; Tisserand, A.; El-Khoury, M.; Girodon, F.; Marzac, C.; Vainchenker, W.; Plo, I.; Cournède, P.H. Inferring the initiation and development of myeloproliferative neoplasms. Proc. Natl. Acad. Sci. USA 2022, 119, e2120374119. [Google Scholar] [CrossRef] [PubMed]
- Al Assaf, C.; Van Obbergh, F.; Billiet, J.; Lierman, E.; Devos, T.; Graux, C.; Hervent, A.S.; Emmerechts, J.; Tousseyn, T.; De Paepe, P.; et al. Analysis of phenotype and outcome in essential thrombocythemia with CALR or JAK2 mutations. Haematologica 2015, 100, 893–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pérez Encinas, M.M.; Sobas, M.; Gómez-Casares, M.T.; Abuin Blanco, A.; Noya Pereira, M.S.; Raya, J.M.; Andrade-Campos, M.M.; Álvarez Larrán, A.; Lewandowski, K.; Łukasz, S.; et al. The risk of thrombosis in essential thrombocythemia is associated with the type of CALR mutation: A multicentre collaborative study. Eur. J. Haematol. 2021, 106, 371–379. [Google Scholar] [CrossRef]
- Nangalia, J.; Green, A.R. Myeloproliferative neoplasms: From origins to outcomes. Blood 2017, 130, 2475–2483. [Google Scholar] [CrossRef] [PubMed]
- Steensma, D.P.; Bolton, K.L. What to tell your patient with clonal hematopoiesis and why: Insights from 2 specialized clinics. Blood 2020, 136, 1623–1631. [Google Scholar] [CrossRef]
- Sirinukunwattana, K.; Aberdeen, A.; Theissen, H.; Sousos, N.; Psaila, B.; Mead, A.J.; Turner, G.D.H.; Rees, G.; Rittscher, J.; Royston, D. Artificial intelligence-based morphological fingerprinting of megakaryocytes: A new tool for assessing disease in MPN patients. Blood Adv. 2020, 4, 3284–3294. [Google Scholar] [CrossRef] [PubMed]
- Mosquera-Orgueira, A.; Pérez-Encinas, M.; Hernández-Sánchez, A.; González-Martínez, T.; Arellano-Rodrigo, E.; Martínez-Elicegui, J.; Villaverde-Ramiro, Á.; Raya, J.M.; Ayala, R.; Ferrer-Marín, F.; et al. Machine Learning Improves Risk Stratification in Myelofibrosis: An Analysis of the Spanish Registry of Myelofibrosis. HemaSphere 2022, 7, e818. [Google Scholar] [CrossRef]

{kind=link}
| Gene | Function | dbSNP | Effect | Reference |
|---|---|---|---|---|
| JAK2 46/1 haplotype | Key driver mutation in MPN. | rs1327494 | Higher risk of acquiring JAK2 p.V617F-mutated CHIP or MPN. | [71,72,73,74] |
| TET2 | Role in myelopoiesis. Loss of this gene has been associated with MPN and CHIP. | rs144418061 | Variant specific to African ancestry in an intergenic region near TET2. Carriers of the A allele have a 2.4-fold increased risk of CHIP. | [71] |
| rs1548483 | SNP upstream of TET2 associated with JAK2-mutated CHIP and MPN. | [75] | ||
| rs79901204 | Disrupts a TET2 distal enhancer resulting in decreased TET2 expression, increased HSC self-renewal, and increased risk of acquiring any CHIP driver mutation. | [71] | ||
| TERT | Telomere enzyme reverse transcriptase. Key role in telomere length maintenance. | rs7705526 | Frequent variant in the 5th intron of TERT associated with CHIP, and specifically with JAK2-mutated CHIP and MPN. | [71,75,78] |
| rs34002450 | The A allele confers a 1.3-fold increased risk of developing CHIP. | [71,79] | ||
| rs2853677 | Identified in European populations. | [78] | ||
| rs13156167 E2D;rs2086132 | Associated specifically with DNMT3A-mutated CHIP. | [76] | ||
| rs2736100 | Associated with TET2-mutated CHIP and MPN. | [76] | ||
| CHEK2 | DNA damage repair. | rs555607708 E2D; | Higher risk of being a JAK2 p.V617F carrier and developing multiple mCAs. | [31,75] |
| rs62237617 | The T allele conferred a large increase in the risk of DNMT3A-mutated CHIP. | [78] | ||
| ATM | DNA damage repair. | rs1800056 | Higher risk of being a JAK2 p.V617F carrier. | [75] |
| rs11212666 | Associated specifically with DNMT3A-mutated CHIP in a European population. | [78] | ||
| SH2B3 | Mutations identified in MPN and result in aberrant JAK-STAT signaling. | rs7310615 | Associated with JAK2-mutated CHIP and MPN. | [75] |
| GFI1B | Transcriptional repressor with key role in hematopoiesis. | rs524137 | SNP in the enhancer region resulted in 2.7-fold increase in the expansion of HSC. | [80] |
| rs621940 | Higher risk of being a JAK2 p.V617F carrier. | [75] | ||
| SMC4 | Encodes condensin subunit with key role in chromosome segregation. | rs12632224 | Identified in European populations. | [78] |
| PARP1 | DNA damage repair. | rs138994074 | Associated specifically with DNMT3A-mutated CHIP in a European population. | [78] |
| CD164 | HSC migration/homing. | rs35452836 | Identified in European populations. | [78] |
| ENPP6 | Enzyme with a role in choline metabolism. | rs13130545 | Identified in European populations. | [78] |
| SETBP1 | Myeloid oncogenesis. | rs8088824 | Associated specifically with DNMT3A-mutated CHIP in a European population. | [78] |
| MBD4 | Role in DNA mismatch repair. | rs79901204 | Individuals with the A allele have a 2.4-fold increased risk for CHIP due to the disruption of a TET2 enhancer resulting in decreased TET2 expression. Particularly prone to DNMT3A-mutated CHIP. | [81] |
| TCL1A | Factor that enhances cell proliferation. | rs2887399 | Carriers of the T allele have 1.2-fold increased risk of CHIP, particularly DNMT3A-mutated CHIP in the European population. The variant is also associated with mosaic loss of chromosome Y. | [71,78,82] |
| rs10131341 | Associated specifically with TET2-mutated CHIP in European population. | [78] | ||
| KPNA4-TRIM59 locus | Role in nuclear protein import. | rs1210060191 | Carriers of this common variant (1-base pair deletion) have a 1.2-fold increased risk of CHIP, including JAK2-mutated CHIP. | [71] |
| PINT | No known role in HSC biology. | rs58270997 | Higher risk of being a JAK2 p.V617F carrier. | [75] |
| TMEM209 | Integral protein of nuclear envelope. | rs79633204 | Associated specifically with TET2-mutated CHIP in a European population. | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stuckey, R.; Bilbao-Sieyro, C.; Segura-Díaz, A.; Gómez-Casares, M.T. Molecular Studies for the Early Detection of Philadelphia-Negative Myeloproliferative Neoplasms. Int. J. Mol. Sci. 2023, 24, 12700. https://doi.org/10.3390/ijms241612700
Stuckey R, Bilbao-Sieyro C, Segura-Díaz A, Gómez-Casares MT. Molecular Studies for the Early Detection of Philadelphia-Negative Myeloproliferative Neoplasms. International Journal of Molecular Sciences. 2023; 24(16):12700. https://doi.org/10.3390/ijms241612700
Chicago/Turabian StyleStuckey, Ruth, Cristina Bilbao-Sieyro, Adrián Segura-Díaz, and María Teresa Gómez-Casares. 2023. "Molecular Studies for the Early Detection of Philadelphia-Negative Myeloproliferative Neoplasms" International Journal of Molecular Sciences 24, no. 16: 12700. https://doi.org/10.3390/ijms241612700
APA StyleStuckey, R., Bilbao-Sieyro, C., Segura-Díaz, A., & Gómez-Casares, M. T. (2023). Molecular Studies for the Early Detection of Philadelphia-Negative Myeloproliferative Neoplasms. International Journal of Molecular Sciences, 24(16), 12700. https://doi.org/10.3390/ijms241612700