
Unraveling the Molecular Puzzle: Exploring Gene Networks across Diverse EMT Status of Cell Lines

Abstract
:1. Introduction
2. Investigating System Changes in Epithelial–Mesenchymal Transition through Personalized Gene Network Analysis
2.1. Computational Strategies
2.1.1. NetworkProfiler
2.1.2. Robust NetworkProfiler
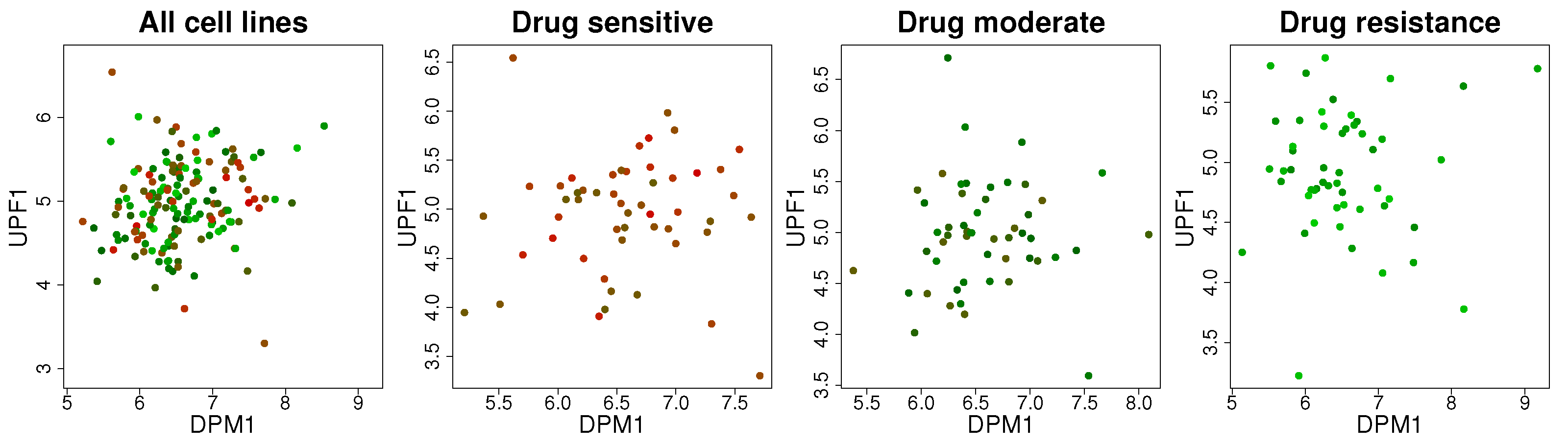
2.2. Uncovering Changes in Gene Regulatory Networks in the Epithelial–Mesenchymal Transition
- The expression of miR-141 had a strong positive effect on the expression of E-cadherin in epithelial-like cells, whereas this effect decreased as the transition from epithelial- to mesenchymal-like cell lines occurred.
- The expression of ZEB1 had a weak negative effect on the expression of E-cadherin in epithelial-like cells, whereas this effect increased as the transition from epithelial- to mesenchymal-like cell lines occurred.
- miR-141 and ZEB1 had a strong negative effect on each other only in epithelial-like cells.
- As the transition from epithelial-like cells to mesenchymal-like cells occurred, the expression levels of miR-141 and E-cadherin decreased, whereas the expression level of ZEB1 increased.
2.3. Limitations of Current Personalized Gene Network Analysis
3. Explainable Artificial Intelligence (XAI) for Comprehensive Gene Network Analysis
3.1. Method: Tensor Reconstruction-Based Interpretable Prediction (TRIP)
3.2. Comprehensive Interpretation of the Massive Multiple Gene Networks across Varying EMT Statuses
4. Discussion
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Daoud, M.; Mayo, M. A survey of neural network-based cancer prediction models from microarray data. Artif. Intell. Med. 2019, 97, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Kovacs, I.; Barabasi, A. Network-based prediction of drug combinations. Nat. Commun. 2019, 10, 1197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fout, A.; Byrd, J.; Shariat, B.; Ben-Hur, A. Protein interface prediction using graph convolutional networks. In Proceedings of the NIPS’17: Proceedings of the 31st International Conference on Neural Information Processing Systems, Long Beach, CA, USA, 4–9 December 2017; pp. 6533–6542.
- Shimamura, T.; Imoto, S.; Shimada, Y.; Hosono, Y.; Niida, A.; Nagasaki, M.; Yamaguchi, R.; Takahashi, T.; Miyano, S. A novel network profiling analysis reveals system changes in epithelial-mesenchymal transition. PLoS ONE 2011, 6, e20804. [Google Scholar] [CrossRef] [PubMed]
- Emmert-Streib, F.; Dehmer, M.; Haibe-Kains, B. Gene regulatory networks and their applications: Understanding biological and medical problems in terms of networks. Front. Cell Dev. Biol. 2014, 2, 38. [Google Scholar]
- Badia-I-Mompel, P.; Wessels, L.; Müller-Dott, S.; Trimbour, R.; Ramirez Flores, R.O.; Argelaguet, R.; Saez-Rodriguez, J. Gene regulatory network inference in the era of single-cell multi-omics. Nat. Rev. Genet. 2023, 1–16. [Google Scholar]
- Zhao, M.; He, W.; Tang, J.; Zou, Q.; Guo, F. A comprehensive overview and critical evaluation of gene regulatory network inference technologies. Brief. Bioinform. 2021, 22, bbab009. [Google Scholar] [CrossRef]
- Lavin, D.P.; Tiwari, V.K. Unresolved Complexity in the Gene Regulatory Network Underlying EMT. Front. Oncol. 2020, 10, 554. [Google Scholar] [CrossRef]
- Fazilaty, H.; Rago, L.; Kass Youssef, K.; Ocana, O.H.; Garcia-Asencio, F.; Arcas, A.; Galceran, J.; Nieto, M.A. A gene regulatory network to control EMT programs in development and disease. Nat. Commun. 2019, 10, 5115. [Google Scholar]
- Maruhashi, K.; Park, H.; Yamaguchi, R.; Miyano, S. Linear Tensor Projection Revealing Nonlinearity. arXiv 2020, arXiv:2007.03912. [Google Scholar]
- Park, H.; Maruhashi, K.; Yamaguchi, R.; Imoto, S.; Miyano, S. Global gene network exploration based on explainable artificial intelligence approach. PLoS ONE 2020, 15, e0241508. [Google Scholar]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B 2005, 67, 301–320. [Google Scholar] [CrossRef] [Green Version]
- Hoerl, A.E.; Kennard, R.W. Ridge regression: Biased estimation for nonorthogonal problems. Techonometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Hastie, T.; Tibshirani, R. Varying-Coefficient Models. J. R. Stat. Soc. Ser. B 1993, 4, 757–796. [Google Scholar] [CrossRef]
- Park, H.; Shimamura, T.; Miyano, S.; Imoto, S. Robust prediction of anti-cancer drug sensitivity and sensitivity-specific biomarker. PLoS ONE 2014, 9, e108990. [Google Scholar] [CrossRef]
- Khan, J.A.; Van Aelst, S.; Zamar, R.H. Robust linear model selection based on least angle regression. J. Am. Stat. Assoc. 2007, 102, 1289–1299. [Google Scholar] [CrossRef] [Green Version]
- Niida, A.; Smith, A.D.; Imoto, S.; Aburatani, H.; Zhang, M.Q.; Akiyama, T. Gene set-based module discovery in the breast cancer transcriptome. BMC Bioinf. 2009, 10, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef]
- Comijn, J.; Berx, G.; Vermassen, P.; Verschueren, K.; van Grunsven, L.; Bruyneel, E.; Mareel, M.; Huylebroeck, D.; van Roy, F. The two-handed E box binding zinc finger protein SIP1 downregulates Ecadherin and induces invasion. Mol. Cell 2001, 7, 1267–1278. [Google Scholar] [CrossRef] [Green Version]
- Yori, J.L.; Johnson, E.; Zhou, G.; Jain, M.K.; Keri, R.A. Kruppel-like factor 4 inhibits epithelial-tomesenchymal transition through regulation of E-cadherin gene expression. J. Biol. Chem. 2010, 285, 16854–16863. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Washington, M.K.; Crawford, H.C. Loss of FOXA1/2 is essential for the epithelialto-mesenchymal transition in pancreatic cancer. Cancer Res. 2010, 70, 2115–2125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sobrado, V.R.; Moreno-Bueno, G.; Cubillo, E.; Holt, L.J.; Nieto, M.A.; Portillo, F.; Cano, A. The class I bHLH factors E2-2A and E2-2B regulate EMT. J. Cell Sci. 2009, 122, 1014–1024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, M.; Zhang, J.; Wang, M.; Lanting, L.; Yuan, H.; Rossi, J.J.; Natarajan, R. MicroRNA-192 in diabetic kidney glomeruli and its function in TGF-beta-induced collagen expression via inhibition of E-box repressors. Proc. Natl. Acad. Sci. USA 2007, 104, 3432–3437. [Google Scholar] [CrossRef] [PubMed]
- Eger, A.; Aigner, K.; Sonderegger, S.; Dampier, B.; Oehler, S.; Schreiber, M.; Berx, G.; Cano, A.; Beug, H.; Foisner, R. DeltaEF1 is a transcriptional repressor of E-cadherin and regulates epithelial plasticity in breast cancer cells. Oncogene 2005, 4, 2375–2385. [Google Scholar] [CrossRef] [Green Version]
- Hajra, K.M.; Chen, D.Y.; Fearon, E.R. The SLUG zinc-finger protein represses E-cadherin in breast cancer. Cancer Res. 2002, 62, 1613–1618. [Google Scholar]
- Bracken, C.P.; Gregory, P.A.; Kolesnikoff, N.; Bert, A.G.; Wang, J.; Shannon, M.F.; Goodall, G.J. A double-negative feedback loop between ZEB1-SIP1 and the microRNA-200 family regulates epithelial-mesenchymal transition. Cancer Res. 2008, 68, 7846–7854. [Google Scholar] [CrossRef] [Green Version]
- Daniel, J.G.; Panizzi, J.R. Spatiotemporal expression profile of embryonic and adult ankyrin repeat and EF-hand domain containing protein 1-encoding genes ankef1a and ankef1b in zebrafish. Gene Exp. Patterns. 2019, 34, 119069. [Google Scholar] [CrossRef]
- Wang, S.; Yan, S.; Zhu, S.; Zhao, Y.; Yan, J.; Xiao, Z.; Bi, J.; Qiu, J.; Zhang, D.; Hong, Z.; et al. FOXF1 Induces Epithelial-Mesenchymal Transition in Colorectal Cancer Metastasis by Transcriptionally Activating SNAI1. Neoplasia 2018, 20, 996–1007. [Google Scholar] [CrossRef]
- Wei, H.J.; Nickoloff, J.A.; Chen, W.H.; Liu, H.Y.; Lo, W.C.; Chang, Y.T.; Yang, P.C.; Wu, C.W.; Williams, D.F.; Gelovani, J.G.; et al. FOXF1 mediates mesenchymal stem cell fusion-induced reprogramming of lung cancer cells. Oncotarget 2014, 5, 9514–9529. [Google Scholar] [CrossRef] [Green Version]
- Lo, P.K. The controversial role of forkhead box F2 (FOXF2) transcription factor in breast cancer. PRAS Open. 2017, 1, 9. [Google Scholar]
- Cai, J.; Tian, A.X.; Wang, Q.S.; Kong, P.Z.; Du, X.; Li, X.Q.; Feng, Y.M. FOXF2 suppresses the FOXC2-mediated epithelial-mesenchymal transition and multidrug resistance of basal-like breast cancer. Cancer Lett. 2015, 367, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwasaki, H.; Nakano, K.; Shinkai, K.; Kunisawa, Y.; Hirahashi, M.; Oda, Y.; Onishi, H.; Katano, M. Hedgehog Gli3 activator signal augments tumorigenicity of colorectal cancer via upregulation of adherence-related genes. Cancer Sci. 2013, 104, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.F.S.D.; Miguita, L.; De Andrade, N.P.; Heguedusch, D.; Rodini, C.O. GLI3 knockdown decreases stemness, cell proliferation and invasion in oral squamous cell carcinoma. Int. J. Oncol. 2018, 53, 2458–2472. [Google Scholar] [CrossRef] [Green Version]
- Chung, V.Y.; Tan, T.Z.; Tan, M.; Wong, M.K.; Kuay, K.T.; Yang, Z.; Ye, J.; Muller, J.; Koh, C.M.; Guccione, E.; et al. GRHL2-miR-200-ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci. Rep. 2016, 6, 19943. [Google Scholar] [CrossRef] [Green Version]
- Xiang, J.; Fu, X.; Ran, W.; Wang, Z. Grhl2 reduces invasion and migration through inhibition of TGFβ-induced EMT in gastric cancer. Oncogenesis 2017, 6, e284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieply, B.; Farris, J.; Denvir, J.; Ford, H.L.; Frisch, S.M. Epithelial-mesenchymal transition and tumor suppression are controlled by a reciprocal feedback loop between ZEB1 and Grainyhead-like-2. Cancer Res. 2013, 73, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Mooney, S.M.; Talebian, V.; Jolly, M.K.; Jia, D.; Gromala, M.; Levine, H.; McConkey, B.J. The GRHL2/ZEB Feedback Loop-A Key Axis in the Regulation of EMT in Breast Cancer. J. Cell Biochem. 2017, 118, 2559–2570. [Google Scholar] [CrossRef]
- Alimirah, F.; Chen, J.; Davis, F.J.; Choubey, D. IFI16 in human prostate cancer. Mol. Cancer Res. 2007, 5, 251–259. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Zhao, Z.; Ni, Z.; Zhao, Y.; Du, W.; Chen, S. IFI16 restoration in hepatocellular carcinoma induces tumour inhibition via activation of p53 signals and inflammasome. Cell Prolif. 2017, 50, e12392. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Ghosh, A.; Kumar, B.; Chandran, B. IFI16, a nuclear innate immune DNA sensor, mediates epigenetic silencing of herpesvirus genomes by its association with H3K9 methyltransferases SUV39H1 and GLP. eLife 2019, 8, e49500. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.Y.; Xiao, W.L.; Chen, C.M.; Lo, L.J.; Wong, F.H. IRF6 is the mediator of TGFβ3 during regulation of the epithelial mesenchymal transition and palatal fusion. Sci. Rep. 2015, 5, 12791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Cheng, P.; Wang, J.; Qiu, X.; Zhang, X. IRF6 Is Directly Regulated by ZEB1 and ELF3, and Predicts a Favorable Prognosis in Gastric Cancer. Front. Oncol. 2019, 9, 200. [Google Scholar]
- Shimada, H.; Abe, S.; Kohno, T.; Satohisa, S.; Konno, Y. Loss of tricellular tight junction protein LSR promotes cell invasion and migration via upregulation of TEAD1/AREG in human endometrial cancer. Sci. Rep. 2017, 7, 37049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsana, P.; Amend, S.R.; Hernandez, J.; Pienta, K.J.; Battle, A. Identifying global expression patterns and key regulators in epithelial to mesenchymal transition through multi-study integration. BMC Cancer. 2017, 17, 447. [Google Scholar] [CrossRef] [Green Version]
- Reaves, D.K.; Fagan-Solis, K.D.; Dunphy, K.; Oliver, S.D.; Scott, D.W.; Fleming, J.M. The role of lipolysis stimulated lipoprotein receptor in breast cancer and directing breast cancer cell behavior. PLoS ONE 2014, 9, e91747. [Google Scholar] [CrossRef]
- Takano, K.; Kakuki, T.; Obata, K.; Nomura, K.; Miyata, R.; Kondo, A.; Kurose, M.; Kakiuchi, A.; Kaneko, Y.; Kohno, T.; et al. The Behavior and Role of Lipolysis-stimulated Lipoprotein Receptor, a Component of Tricellular Tight Junctions, in Head and Neck Squamous Cell Carcinomas. Anticancer Res. 2016, 36, 5895–5904. [Google Scholar] [CrossRef]
- Liu, J.; Wu, Q.; Wang, Y.; Wei, Y.; Wu, H.; Duan, L.; Zhang, Q.; Wu, Y. Ovol2 induces mesenchymal-epithelial transition via targeting ZEB1 in osteosarcoma. Onco Targets Ther. 2018, 11, 2963–2973. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, G.; Kannius-Janson, M. Forkhead Box F1 promotes breast cancer cell migration by upregulating lysyl oxidase and suppressing Smad2/3 signaling. BMC Cancer 2016, 16, 142. [Google Scholar] [CrossRef] [Green Version]
- Roca, H.; Hernandez, J.; Weidner, S.; McEachin, R.C.; Fuller, D. Transcription factors OVOL1 and OVOL2 induce the mesenchymal to epithelial transition in human cancer. PLoS ONE 2013, 8, e76773. [Google Scholar] [CrossRef]
- Hong, T.; Watanabe, K.; Ta, C.H.; Villarreal-Ponce, A.; Nie, Q.; Dai, X. An Ovol2-Zeb1 Mutual Inhibitory Circuit Governs Bidirectional and Multi-step Transition between Epithelial and Mesenchymal States. PLoS Comput. Biol. 2015, 11, e1004569. [Google Scholar] [CrossRef]
- Zhang, Y.; Liao, Y.; Chen, C.; Sun, W.; Sun, X.; Liu, Y.; Xu, E.; Lai, M.; Zhang, H. p38 regulated FOXC1 stability is required for colorectal cancer metastasis. J. Pathol. 2020, 250, 217–230. [Google Scholar] [CrossRef]
- Chandhoke, A.S.; Karve, K.; Dadakhujaev, S.; Netherton, S.; Deng, L.; Bonni, S. The ubiquitin ligase Smurf2 suppresses TGFβ-induced epithelial-mesenchymal transition in a sumoylation-regulated manner. Cell Death Differ. 2016, 23, 876–888. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Tong, J.; He, F.; Yu, X.; Fan, L.; Hu, J.; Tan, J.; Chen, Z. miR-141 regulates TGF-β1-induced epithelial-mesenchymal transition through repression of HIPK2 expression in renal tubular epithelial cells. Int. J. Mol. Med. 2014, 35, 311–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Heldin, C.H. Mechanisms of TGFβ Induced Epithelial-Mesenchymal Transition. J. Clin. Med. 2016, 5, 63. [Google Scholar] [PubMed] [Green Version]
- Saito, R.A.; Watabe, T.; Horiguchi, K.; Kohyama, T.; Saitoh, M.; Nagase, T.; Miyazono, K. Thyroid transcription factor-1 inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 2009, 69, 2783–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, W.; Chen, X. P63 regulates tubular formation via epithelial-to-mesenchymal transition. Oncogene 2014, 33, 1548–1557. [Google Scholar] [CrossRef] [Green Version]
- Olsen, J.R.; Oyan, A.M.; Rostad, K.; Hellem, M.R.; Liu, J.; Li, L.; Micklem, D.R.; Haugen, H.; Lorens, J.B.; Rotter, V.; et al. p63 attenuates epithelial to mesenchymal potential in an experimental prostate cell model. PLoS ONE 2013, 8, e62547. [Google Scholar]
- Lindsay, J.; McDade, S.S.; Pickard, A.; McCloskey, K.D.; McCance, D.J. Role of DeltaNp63gamma in epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 3915–3924. [Google Scholar] [CrossRef] [Green Version]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
| Gene | Components | References | In NetworkProfiler |
|---|---|---|---|
| AFF1 | 3 | - | - |
| ANKRD5 | 1 | [28] | - |
| FOXF1 | 2 | [29,30] | - |
| FOXF2 | 1 | [31,32] | - |
| GLI3 | 2 | [33,34] | - |
| GRHL2 | 1 | [35,36,37,38] | ◯ |
| IFI16 | 2, 3 | [39,40,41,42] | - |
| IRF6 | 3 | [43,44] | ◯ |
| KANK2 | 3 | - | - |
| LSR | 2 | [45,46,47,48] | ◯ |
| MAFB | 3 | - | - |
| OVOL2 | 2 | [49,50,51,52] | ◯ |
| PCBD1 | 2 | - | - |
| SOX13 | 2 | [53] | - |
| TGFB1I1 | 2 | [54,55,56,57] | - |
| TP63 | 2, 3 | [58,59,60] | - |
| ZNF91 | 1 | - | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H. Unraveling the Molecular Puzzle: Exploring Gene Networks across Diverse EMT Status of Cell Lines. Int. J. Mol. Sci. 2023, 24, 12784. https://doi.org/10.3390/ijms241612784
Park H. Unraveling the Molecular Puzzle: Exploring Gene Networks across Diverse EMT Status of Cell Lines. International Journal of Molecular Sciences. 2023; 24(16):12784. https://doi.org/10.3390/ijms241612784
Chicago/Turabian StylePark, Heewon. 2023. "Unraveling the Molecular Puzzle: Exploring Gene Networks across Diverse EMT Status of Cell Lines" International Journal of Molecular Sciences 24, no. 16: 12784. https://doi.org/10.3390/ijms241612784