
Impact of Drug Administration Routes on the In Vivo Efficacy of the Natural Product Sorangicin A Using a Staphylococcus aureus Infection Model in Zebrafish Embryos
 , , , and
, , , and
Abstract
:1. Introduction
2. Results
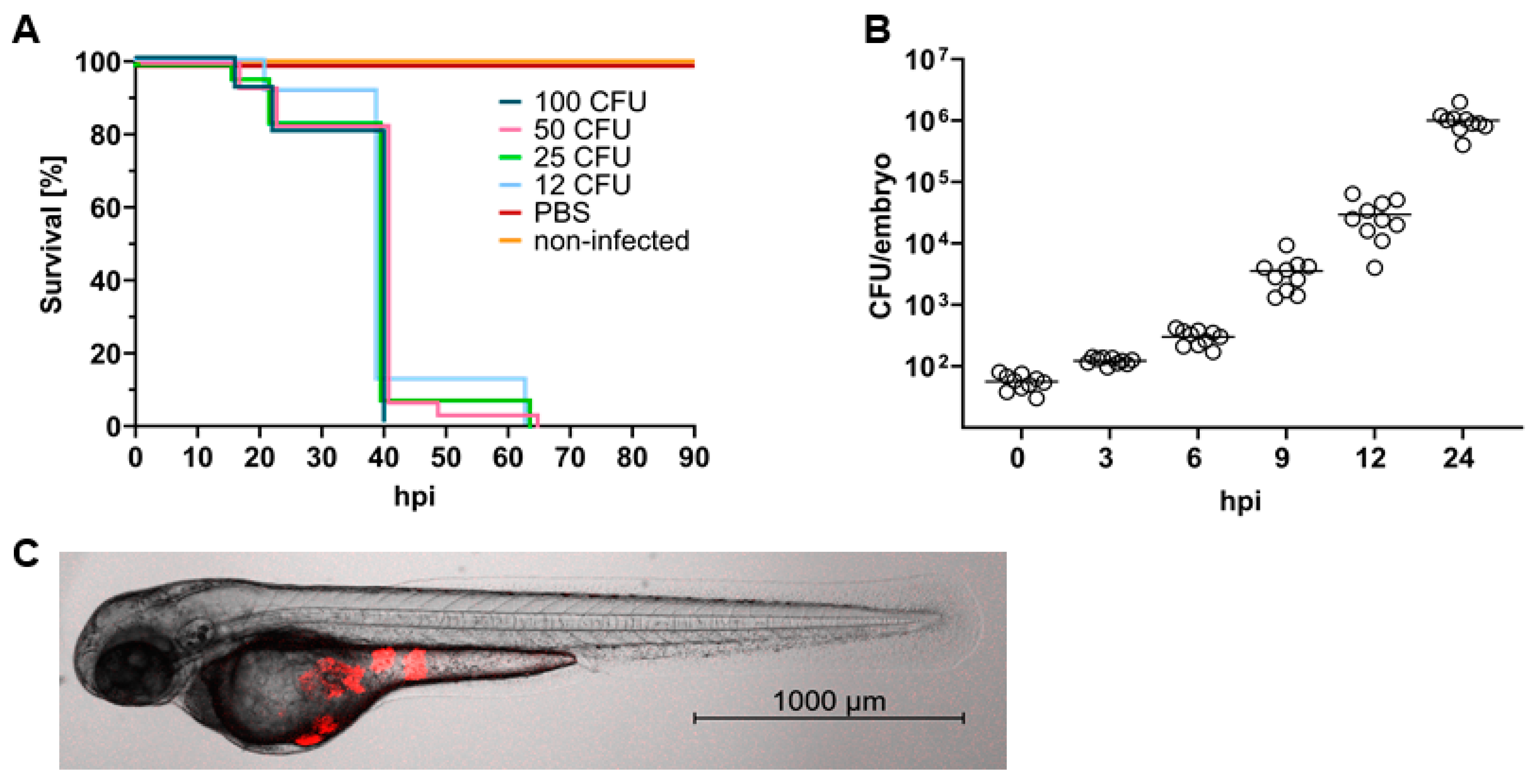
2.1. S. aureus Causes a Lethal Infection in Zebrafish Embryos
2.2. Experimental Design to Assess In Vivo Efficacy of Drugs against S. aureus in Zebrafish Embryos
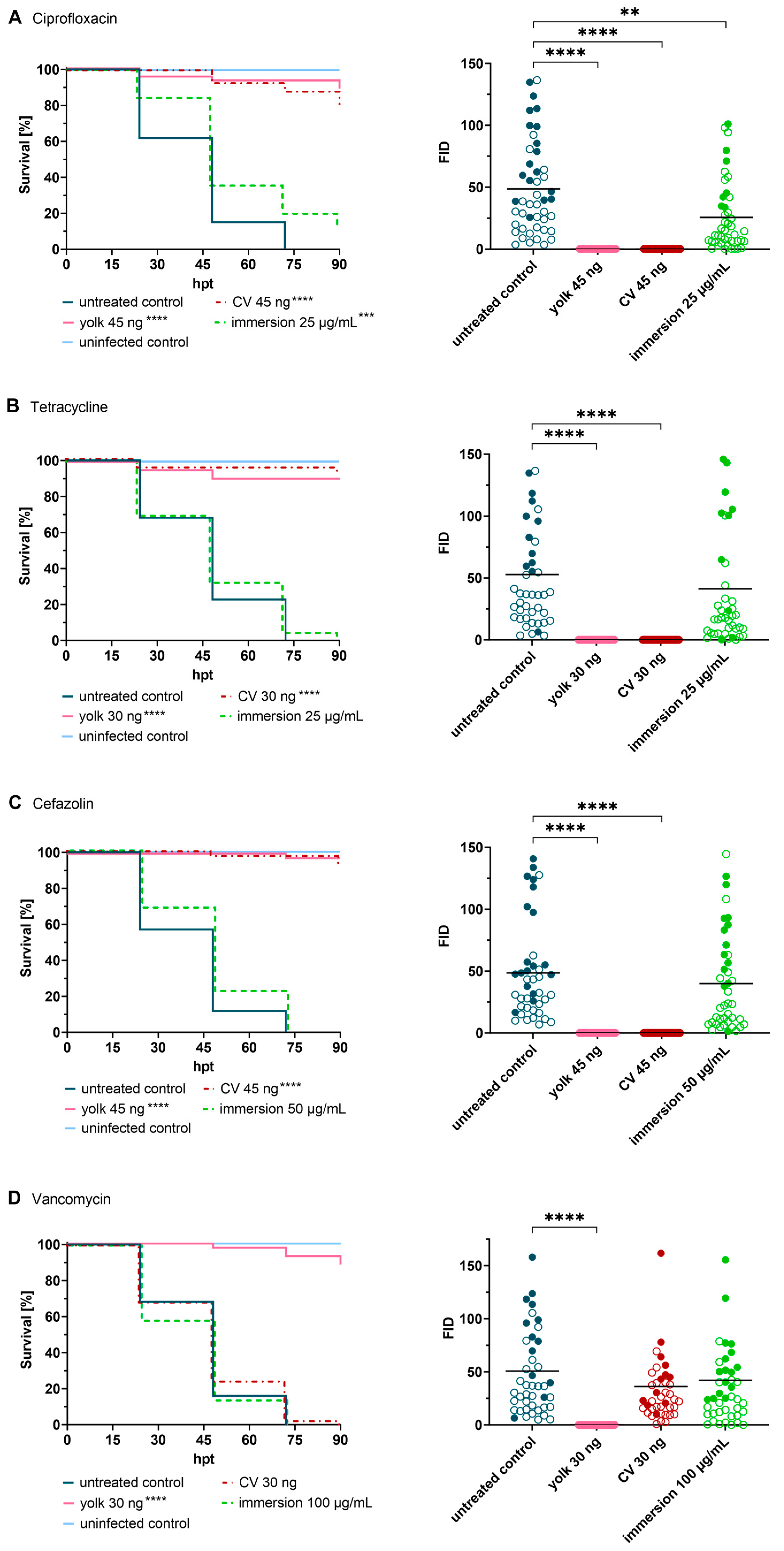
2.3. Impact of the Route of Administration on Drug Activity
2.4. In Vivo Evaluation of the Natural Product Sorangicin A
3. Discussion
4. Materials and Methods
4.1. Zebrafish Lines and Maintenance
4.2. Minimum Inhibitory Concentration (MIC)
4.3. Staphylococcus aureus Transformation
4.4. In Vitro Growth Analysis of S. aureus
4.5. Preparation of Bacterial Microinjection Stock Solutions
4.6. Microinjection of Zebrafish Embryos
4.7. Toxicity of Antimicrobials on Zebrafish Embryos
4.8. Determination of Bacterial Burden
4.9. Imaging of Infected Zebrafish Embryos
4.10. Statistical Analysis
4.11. In Vitro Pharmacokinetic Profiling
4.12. In Vivo Pharmacokinetics in Mice
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Robles Aguilar, G.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Aslam, B.; Wang, W.; Arshad, M.I.; Khurshid, M.; Muzammil, S.; Rasool, M.H.; Nisar, M.A.; Alvi, R.F.; Aslam, M.A.; Qamar, M.U.; et al. Antibiotic resistance: A rundown of a global crisis. Infect. Drug Resist. 2018, 11, 1645–1658. [Google Scholar] [CrossRef]
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations; Review on Antimicrobial Resistance: London, UK, 2014. [Google Scholar]
- de Kraker, M.E.A.; Stewardson, A.J.; Harbarth, S. Will 10 Million People Die a Year due to Antimicrobial Resistance by 2050? PLoS Med. 2016, 13, e1002184. [Google Scholar] [CrossRef] [PubMed]
- Pendleton, J.N.; Gorman, S.P.; Gilmore, B.F. Clinical relevance of the ESKAPE pathogens. Expert. Rev. Anti-Infect. Ther. 2013, 11, 297–308. [Google Scholar] [CrossRef]
- Santajit, S.; Indrawattana, N. Mechanisms of Antimicrobial Resistance in ESKAPE Pathogens. Biomed. Res. Int. 2016, 2016, 2475067. [Google Scholar] [CrossRef]
- Coates, A.R.; Halls, G.; Hu, Y. Novel classes of antibiotics or more of the same?: New antibiotic classes are urgently needed. Br. J. Pharmacol. 2011, 163, 184–194. [Google Scholar] [CrossRef]
- Venkatesan, P. WHO 2020 Report on the Antibacterial Production and Development Pipeline. Lancet Microbe 2021, 2, e239. [Google Scholar] [CrossRef] [PubMed]
- Beyer, P.; Paulin, S. The Antibacterial Research and Development Pipeline Needs Urgent Solutions. ACS Infect. Dis. 2020, 6, 1289–1291. [Google Scholar] [CrossRef]
- Tacconelli, E.; Carrara, E.; Savoldi, A.; Harbarth, S.; Mendelson, M.; Monnet, D.L.; Pulcini, C.; Kahlmeter, G.; Kluytmans, J.; Carmeli, Y.; et al. Discovery, research, and development of new antibiotics: The WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 2018, 18, 318–327. [Google Scholar] [CrossRef]
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Fernández Guerrero, M.L.; González López, J.J.; Goyenechea, A.; Fraile, J.; de Górgolas, M. Endocarditis caused by Staphylococcus aureus: A reappraisal of the epidemiologic, clinical, and pathologic manifestations with analysis of factors determining outcome. Medicine 2009, 88, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Al-Mebairik, N.F.; El-Kersh, T.A.; Al-Sheikh, Y.A.; Marie, M.A.M. A review of virulence factors, pathogenesis, and antibiotic resistance in Staphylococcus aureus. Rev. Med. Microbiol. 2016, 27, 50–56. [Google Scholar] [CrossRef]
- John, J., Jr. The treatment of resistant staphylococcal infections. F1000Res 2020, 9, 150. [Google Scholar] [CrossRef]
- Bukharie, H. A review of community-acquired methicillin-resistant Staphylococcus aureus for primary care physicians. J. Fam. Community Med. 2010, 17, 117. [Google Scholar] [CrossRef]
- Rasmussen, R.V.; Fowler, V.G., Jr.; Skov, R.; Bruun, N.E. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 2011, 6, 43–56. [Google Scholar] [CrossRef]
- Chambers, H.F.; DeLeo, F.R. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol. 2009, 7, 629–641. [Google Scholar] [CrossRef]
- Hassoun, A.; Linden, P.K.; Friedman, B. Incidence, prevalence, and management of MRSA bacteremia across patient populations—A review of recent developments in MRSA management and treatment. Crit. Care 2017, 21, 211. [Google Scholar] [CrossRef]
- Cong, Y.; Yang, S.; Rao, X. Vancomycin resistant Staphylococcus aureus infections: A review of case updating and clinical features. J. Adv. Res. 2020, 21, 169–176. [Google Scholar] [CrossRef]
- Shariati, A.; Dadashi, M.; Chegini, Z.; van Belkum, A.; Mirzaii, M.; Khoramrooz, S.S.; Darban-Sarokhalil, D. The global prevalence of Daptomycin, Tigecycline, Quinupristin/Dalfopristin, and Linezolid-resistant Staphylococcus aureus and coagulase–negative staphylococci strains: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2020, 9, 56. [Google Scholar] [CrossRef]
- Hughes, J.; Rees, S.; Kalindjian, S.; Philpott, K. Principles of early drug discovery: Principles of early drug discovery. Br. J. Pharmacol. 2011, 162, 1239–1249. [Google Scholar] [CrossRef]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef]
- Rasheed, S.; Fries, F.; Müller, R.; Herrmann, J. Zebrafish: An Attractive Model to Study Staphylococcus aureus Infection and Its Use as a Drug Discovery Tool. Pharmaceuticals 2021, 14, 594. [Google Scholar] [CrossRef] [PubMed]
- Prajsnar, T.K.; Cunliffe, V.T.; Foster, S.J.; Renshaw, S.A. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell. Microbiol. 2008, 10, 2312–2325. [Google Scholar] [CrossRef] [PubMed]
- Prajsnar, T.K.; Hamilton, R.; Garcia-Lara, J.; McVicker, G.; Williams, A.; Boots, M.; Foster, S.J.; Renshaw, S.A. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol. 2012, 14, 1600–1619. [Google Scholar] [CrossRef]
- Li, Y.; Hu, B. Establishment of multi-site infection model in zebrafish larvae for studying Staphylococcus aureus infectious disease. J. Genet. Genom. 2012, 39, 521–534. [Google Scholar] [CrossRef]
- Irschik, H.; Jansen, R.; Gerth, K.; Höfle, G.; Reichenbach, H. The sorangicins, novel and powerful inhibitors of eubacterial RNA polymerase isolated from myxobacteria. J. Antibiot. 1987, 40, 7–13. [Google Scholar] [CrossRef]
- de Jong, N.W.M.; van der Horst, T.; van Strijp, J.A.G.; Nijland, R. Fluorescent reporters for markerless genomic integration in Staphylococcus aureus. Sci. Rep. 2017, 7, 43889. [Google Scholar] [CrossRef]
- Bolcome, R.E.; Sullivan, S.E.; Zeller, R.; Barker, A.P.; Collier, R.J.; Chan, J. Anthrax lethal toxin induces cell death-independent permeability in zebrafish vasculature. Proc. Natl. Acad. Sci. USA 2008, 105, 2439–2444. [Google Scholar] [CrossRef]
- Bernut, A.; Le Moigne, V.; Lesne, T.; Lutfalla, G.; Herrmann, J.-L.; Kremer, L. In Vivo Assessment of Drug Efficacy against Mycobacterium abscessus Using the Embryonic Zebrafish Test System. Antimicrob. Agents Chemother. 2014, 58, 4054–4063. [Google Scholar] [CrossRef]
- Hashemian, S.M.R.; Farhadi, T.; Ganjparvar, M. Linezolid: A review of its properties, function, and use in critical care. Drug Des. Devel Ther. 2018, 12, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Choo, E.J.; Chambers, H.F. Treatment of Methicillin-Resistant Staphylococcus aureus Bacteremia. Infect. Chemother. 2016, 48, 267. [Google Scholar] [CrossRef]
- Campbell, E.A.; Pavlova, O.; Zenkin, N.; Leon, F.; Irschik, H.; Jansen, R.; Severinov, K.; Darst, S.A. Structural, functional, and genetic analysis of sorangicin inhibition of bacterial RNA polymerase. EMBO J. 2005, 24, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B.; Dong, S.; Fox, R.J.; Brenneman, J.B.; Vanecko, J.A.; Maegawa, T. (+)-Sorangicin A: Evolution of a viable synthetic strategy. Tetrahedron 2011, 67, 9809–9828. [Google Scholar] [CrossRef]
- Lilic, M.; Chen, J.; Boyaci, H.; Braffman, N.; Hubin, E.A.; Herrmann, J.; Müller, R.; Mooney, R.; Landick, R.; Darst, S.A.; et al. The antibiotic sorangicin A inhibits promoter DNA unwinding in a Mycobacterium tuberculosis rifampicin-resistant RNA polymerase. Proc. Natl. Acad. Sci. USA 2020, 117, 30423–30432. [Google Scholar] [CrossRef]
- van der Sar, A.M.; Musters, R.J.P.; van Eeden, F.J.M.; Appelmelk, B.J.; Vandenbroucke-Grauls, C.M.J.E.; Bitter, W. Zebrafish embryos as a model host for the real time analysis of Salmonella typhimurium infections. Cell Microbiol. 2003, 5, 601–611. [Google Scholar] [CrossRef]
- Varas, M.; Ortíz-Severín, J.; Marcoleta, A.E.; Díaz-Pascual, F.; Allende, M.L.; Santiviago, C.A.; Chávez, F.P. Salmonella typhimurium induces cloacitis-like symptomsin zebrafish larvae. Microb. Pathog. 2017, 107, 317–320. [Google Scholar] [CrossRef]
- Varas, M.; Fariña, A.; Díaz-Pascual, F.; Ortíz-Severín, J.; Marcoleta, A.E.; Allende, M.L.; Santiviago, C.A.; Chávez, F.P. Live-cell imaging of Salmonella typhimurium interaction with zebrafish larvae after injection and immersion delivery methods. J. Microbiol. Methods 2017, 135, 20–25. [Google Scholar] [CrossRef]
- Tannenbaum, J.; Bennett, B.T. Russell and Burch’s 3Rs then and now: The need for clarity in definition and purpose. J. Am. Assoc. Lab. Anim. Sci. 2015, 54, 120–132. [Google Scholar]
- Kaito, C.; Murakami, K.; Imai, L.; Furuta, K. Animal infection models using non-mammals. Microbiol. Immunol. 2020, 64, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Prajsnar, T.K.; Renshaw, S.A.; Ogryzko, N.V.; Foster, S.J.; Serror, P.; Mesnage, S. Zebrafish as a novel vertebrate model to dissect enterococcal pathogenesis. Infect. Immun. 2013, 81, 4271–4279. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, M.; Suzuki, T.; Pasquier, L.D. On the origins of the adaptive immune system: Novel insights from invertebrates and cold-blooded vertebrates. Trends Immunol. 2004, 25, 105–111. [Google Scholar] [CrossRef]
- Stevens, C.S.; Rosado, H.; Harvey, R.J.; Taylor, P.W. Epicatechin gallate, a naturally occurring polyphenol, alters the course of infection with β-lactam-resistant Staphylococcus aureus in the zebrafish embryo. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Ordas, A.; Raterink, R.-J.; Cunningham, F.; Jansen, H.J.; Wiweger, M.I.; Jong-Raadsen, S.; Bos, S.; Bates, R.H.; Barros, D.; Meijer, A.H.; et al. Testing Tuberculosis Drug Efficacy in a Zebrafish High-Throughput Translational Medicine Screen. Antimicrob. Agents Chemother. 2015, 59, 753–762. [Google Scholar] [CrossRef]
- Heman-Ackah, S.M. Comparison of Tetracycline Action on Staphylococcus aureus and Escherichia coli by Microbial Kinetics. Antimicrob. Agents Chemother. 1976, 10, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Brox, S.; Seiwert, B.; Küster, E.; Reemtsma, T. Toxicokinetics of Polar Chemicals in Zebrafish Embryo (Danio rerio): Influence of Physicochemical Properties and of Biological Processes. Environ. Sci. Technol. 2016, 50, 10264–10272. [Google Scholar] [CrossRef]
- Kristofco, L.A.; Haddad, S.P.; Chambliss, C.K.; Brooks, B.W. Differential uptake of and sensitivity to diphenhydramine in embryonic and larval zebrafish. Environ. Toxicol. Chem. 2018, 37, 1175–1181. [Google Scholar] [CrossRef]
- Brox, S.; Ritter, A.P.; Küster, E.; Reemtsma, T. A quantitative HPLC–MS/MS method for studying internal concentrations and toxicokinetics of 34 polar analytes in zebrafish (Danio rerio) embryos. Anal. Bioanal. Chem. 2014, 406, 4831–4840. [Google Scholar] [CrossRef] [PubMed]
- Lantz-McPeak, S.; Guo, X.; Cuevas, E.; Dumas, M.; Newport, G.D.; Ali, S.F.; Paule, M.G.; Kanungo, J. Developmental toxicity assay using high content screening of zebrafish embryos. J. Appl. Toxicol. 2015, 35, 261–272. [Google Scholar] [CrossRef]
- Kämmer, N.; Erdinger, L.; Braunbeck, T. The onset of active gill respiration in post-embryonic zebrafish (Danio rerio) larvae triggers an increased sensitivity to neurotoxic compounds. Aquat. Toxicol. 2022, 249, 106240. [Google Scholar] [CrossRef]
- Rao, S.; Kupfer, Y.; Pagala, M.; Chapnick, E.; Tessler, S. Systemic absorption of oral vancomycin in patients with Clostridium difficile infection. Scand. J. Infect. Dis. 2011, 43, 386–388. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Meyer, M.R.; Müller, R.; Herrmann, J. Drug Administration Routes Impact the Metabolism of a Synthetic Cannabinoid in the Zebrafish Larvae Model. Molecules 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Pham, D.-H.; Zhang, C.; Yin, C. Using Zebrafish to Model Liver Diseases-Where Do We Stand? Curr. Pathobiol. Rep. 2017, 5, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, R.C.; Krekels, E.H.J.; Kantae, V.; Ordas, A.; Kreling, T.; Harms, A.C.; Hankemeier, T.; Spaink, H.P.; van der Graaf, P.H. Mechanistic and Quantitative Understanding of Pharmacokinetics in Zebrafish Larvae through Nanoscale Blood Sampling and Metabolite Modeling of Paracetamol. J. Pharmacol. Exp. Ther. 2019, 371, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Strähle, U.; Scholz, S.; Geisler, R.; Greiner, P.; Hollert, H.; Rastegar, S.; Schumacher, A.; Selderslaghs, I.; Weiss, C.; Witters, H.; et al. Zebrafish embryos as an alternative to animal experiments—A commentary on the definition of the onset of protected life stages in animal welfare regulations. Reprod. Toxicol. 2012, 33, 128–132. [Google Scholar] [CrossRef]
- Patel, J.B. Performance Standards for Antimicrobial Susceptibility Testing; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017; ISBN 978-1-56238-804-1. [Google Scholar]
- Schneewind, O.; Missiakas, D. Genetic manipulation of Staphylococcus aureus. Curr. Protoc. Microbiol. 2014, 32. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Metabolic Stability | Plasma Stability | PPB (%) | |
|---|---|---|---|---|
| Liver Microsomes t1/2 (min) | CLint (µL/mg/min) | t1/2 (min) | ||
| mouse | 3.4 ± 2.6 | 557 ± 297 | 17.5 ± 7.1 | 87.9 ± 4.8 |
| rat | 4.4 ± 0.5 | 239 ± 155 | > 240 | 88.8 ± 1.7 |
| human | 15.7 ± 2.8 | 90.4 ± 16.5 | > 240 | 87.0 ± 0.4 |
| PK Parameters: 5 mg/kg i.v. | |
|---|---|
| c0 (ng/mL) | 1833.6 |
| cZ (ng/mL) | 7.2 |
| t1/2 (h) | 0.87 |
| AUC(0-tz) (ng·h/mL) | 340.9 |
| Vz (mL/kg) | 17,961.8 |
| CL (mL/h/kg) | 14,287.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fries, F.; Kany, A.M.; Rasheed, S.; Hirsch, A.K.H.; Müller, R.; Herrmann, J. Impact of Drug Administration Routes on the In Vivo Efficacy of the Natural Product Sorangicin A Using a Staphylococcus aureus Infection Model in Zebrafish Embryos. Int. J. Mol. Sci. 2023, 24, 12791. https://doi.org/10.3390/ijms241612791
Fries F, Kany AM, Rasheed S, Hirsch AKH, Müller R, Herrmann J. Impact of Drug Administration Routes on the In Vivo Efficacy of the Natural Product Sorangicin A Using a Staphylococcus aureus Infection Model in Zebrafish Embryos. International Journal of Molecular Sciences. 2023; 24(16):12791. https://doi.org/10.3390/ijms241612791
Chicago/Turabian StyleFries, Franziska, Andreas M. Kany, Sari Rasheed, Anna K. H. Hirsch, Rolf Müller, and Jennifer Herrmann. 2023. "Impact of Drug Administration Routes on the In Vivo Efficacy of the Natural Product Sorangicin A Using a Staphylococcus aureus Infection Model in Zebrafish Embryos" International Journal of Molecular Sciences 24, no. 16: 12791. https://doi.org/10.3390/ijms241612791