
The Molecular Mechanism of Positive Allosteric Modulation at the Dopamine D1 Receptor

Abstract
:1. Introduction
2. Results and Discussion
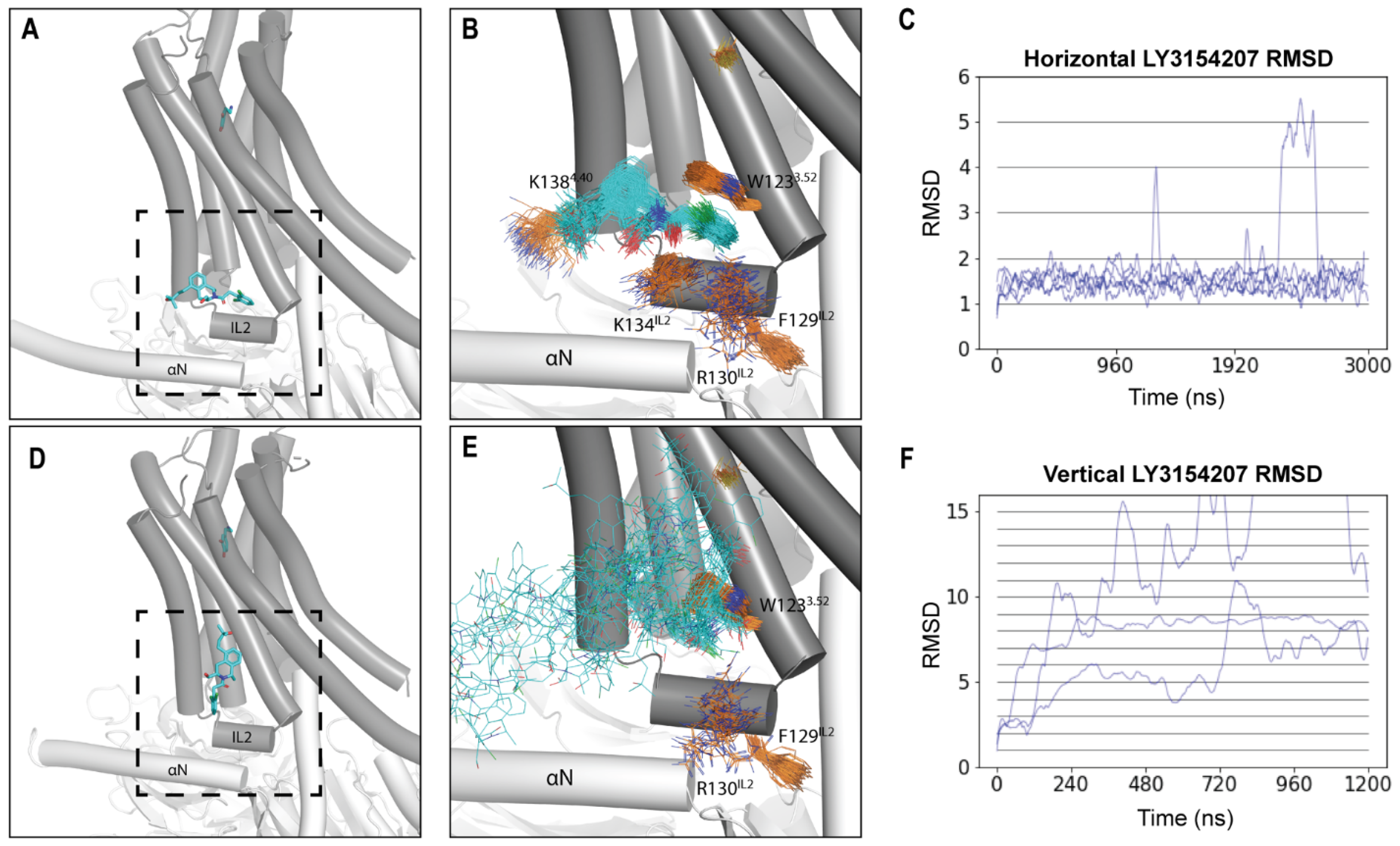
2.1. LY3154207 Is Stable in the Horizontal Orientation
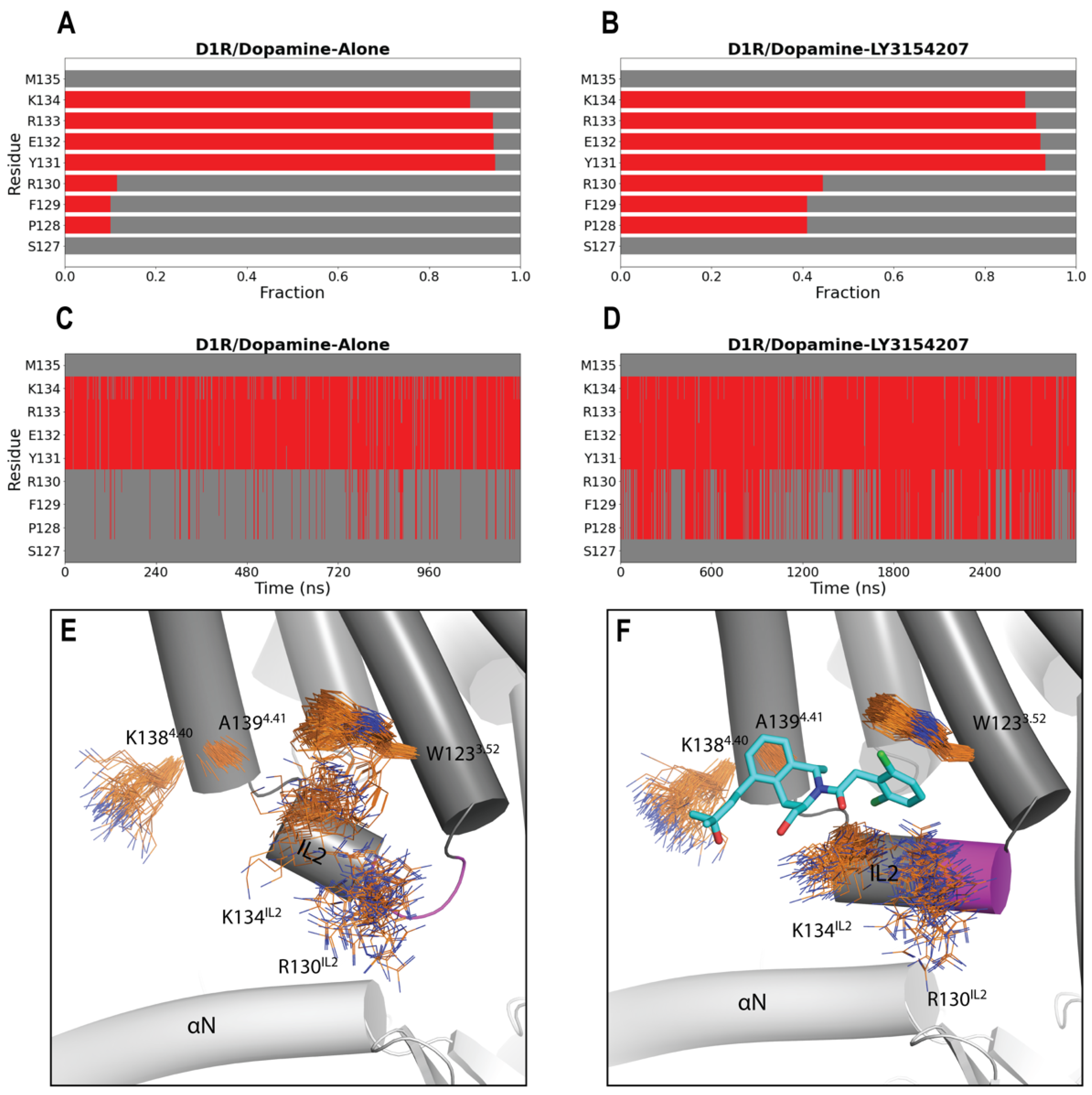
2.2. LY3154207 Binding Stabilizes Intracellular Loop 2 (IL2) in a Helical Conformation
2.3. LY3154207 Has No Strong Impact on the Orthosteric Agonist Binding
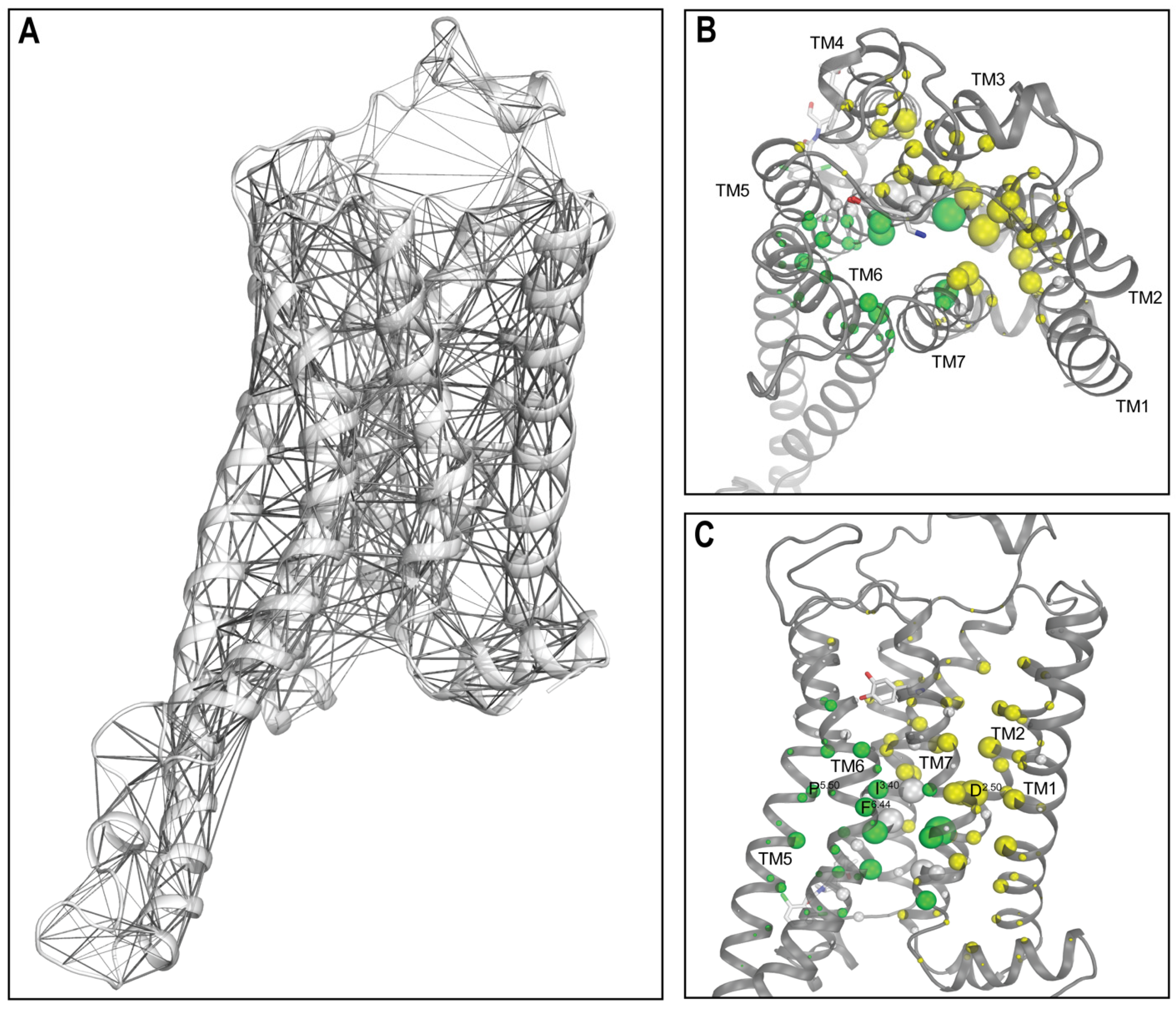
2.4. Network Analysis Identified the Significant Impact of LY3154207 Binding on the Key Structural Motifs
2.5. LY3154207 Compacts and Loosens the Extracellular and Intracellular Regions of D1R, Respectively
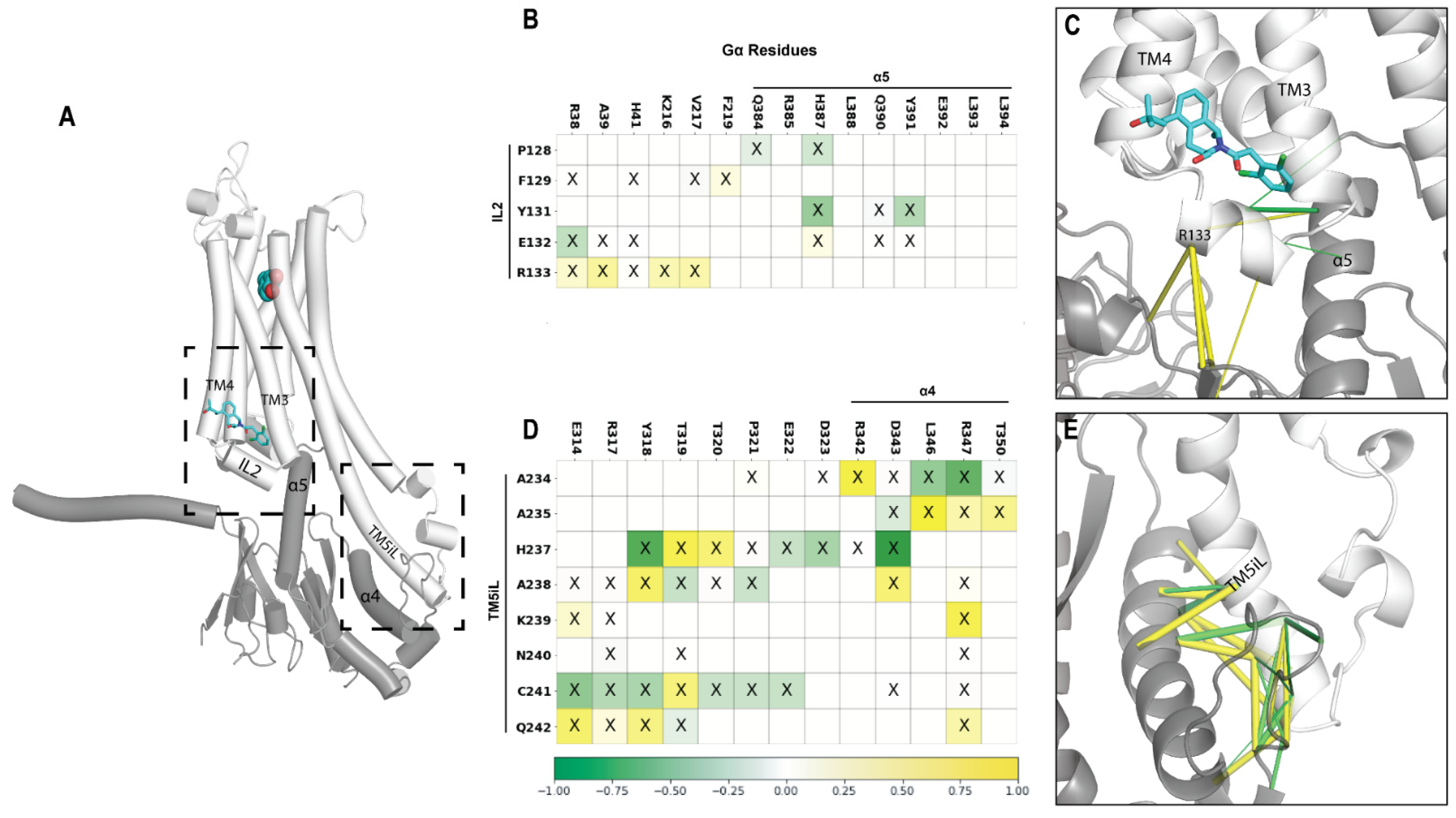
2.6. A Potential Allosteric Pathway at the Receptor–G Protein Interface
3. Methods and Materials
3.1. Homology Modeling
3.2. Molecular Dynamics Simulations
3.3. DSSP Structure Prediction
3.4. Ligand RMSD Calculation
3.5. MM/GBSA Calculation
3.6. Protein Interaction Analyzer (PIA)
3.7. Eigenvector Centrality
3.8. Contact Frequency Difference Analysis of the D1R–Gα Interface
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schioth, H.B.; Gloriam, D.E. Trends in GPCR drug discovery: New agents, targets and indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Insel, P.A.; Sriram, K.; Gorr, M.W.; Wiley, S.Z.; Michkov, A.; Salmeron, C.; Chinn, A.M. GPCRomics: An Approach to Discover GPCR Drug Targets. Trends Pharmacol. Sci. 2019, 40, 378–387. [Google Scholar] [CrossRef]
- Svensson, K.A.; Hao, J.; Bruns, R.F. Positive allosteric modulators of the dopamine D1 receptor: A new mechanism for the treatment of neuropsychiatric disorders. Adv. Pharmacol. 2019, 86, 273–305. [Google Scholar] [PubMed]
- Beaulieu, J.M.; Gainetdinov, R.R. The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev. 2011, 63, 182–217. [Google Scholar] [CrossRef] [PubMed]
- Sibley, D.R. New Insights into Dopaminergic Receptor Function Using Antisense and Genetically Altered Animals. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 313–341. [Google Scholar] [CrossRef] [PubMed]
- Arnsten, A.F.; Girgis, R.R.; Gray, D.L.; Mailman, R.B. Novel Dopamine Therapeutics for Cognitive Deficits in Schizophrenia. Biol. Psychiatry 2017, 81, 67–77. [Google Scholar] [CrossRef]
- Felsing, D.E.; Jain, M.K.; Allen, J.A. Advances in Dopamine D1 Receptor Ligands for Neurotherapeutics. Curr. Top. Med. Chem. 2019, 19, 1365–1380. [Google Scholar] [CrossRef]
- Brogden, R.; Markham, A. Fenoldopam A Review of its Pharmacodynamic and Pharmacokinetic Properties and Intravenous Clinical Potential in the Management of Hypertensive Urgencies and Emergencies. Drugs 1997, 54, 634–650. [Google Scholar] [CrossRef]
- Wootten, D.; Christopoulos, A.; Sexton, P.M. Emerging paradigms in GPCR allostery: Implications for drug discovery. Nat. Rev. Drug Discov. 2013, 12, 630–644. [Google Scholar] [CrossRef]
- Thal, D.M.; Glukhova, A.; Sexton, P.M.; Christopoulos, A. Structural insights into G-protein-coupled receptor allostery. Nature 2018, 559, 45–53. [Google Scholar] [CrossRef]
- Wang, X.; Hembre, E.J.; Goldsmith, P.J.; Beck, J.P.; Svensson, K.A.; Willard, F.S.; Bruns, R.F. Mutual Cooperativity of Three Allosteric Sites on the Dopamine D1 Receptor. Mol. Pharmacol. 2023, 103, 176–187. [Google Scholar] [CrossRef]
- Svensson, K.A.; Heinz, B.A.; Schaus, J.M.; Beck, J.P.; Hao, J.; Krushinski, J.H.; Reinhard, M.R.; Cohen, M.P.; Hellman, S.L.; Getman, B.G.; et al. An Allosteric Potentiator of the Dopamine D1 Receptor Increases Locomotor Activity in Human D1 Knock-In Mice without Causing Stereotypy or Tachyphylaxis. J. Pharmacol. Exp. Ther. 2017, 360, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hao, J.; Beck, J.P.; Schaus, J.M.; Krushinski, J.H.; Chen, Q.; Beadle, C.D.; Vidal, P.; Reinhard, M.R.; Dressman, B.A.; Massey, S.M.; et al. Synthesis and Pharmacological Characterization of 2-(2,6-Dichlorophenyl)-1-((1S,3R)-5-(3-hydroxy-3-methylbutyl)-3-(hydroxymethyl)-1 -methyl-3,4-dihydroisoquinolin-2(1H)-yl)ethan-1-one (LY3154207), a Potent, Subtype Selective, and Orally Available Positive Allosteric Modulator of the Human Dopamine D1 Receptor. J. Med. Chem. 2019, 62, 8711–8732. [Google Scholar]
- Bruns, R.F.; Mitchell, S.N.; Wafford, K.A.; Harper, A.J.; Shanks, E.A.; Carter, G.; O’Neill, M.J.; Murray, T.K.; Eastwood, B.J.; Schaus, J.M.; et al. Preclinical profile of a dopamine D1 potentiator suggests therapeutic utility in neurological and psychiatric disorders. Neuropharmacology 2018, 128, 351–365. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.; Provins, L.; Valade, A. Novel Strategies To Activate the Dopamine D1 Receptor: Recent Advances in Orthosteric Agonism and Positive Allosteric Modulation. J. Med. Chem. 2019, 62, 128–140. [Google Scholar] [CrossRef]
- Wilbraham, D.; Biglan, K.M.; Svensson, K.A.; Tsai, M.; Kielbasa, W. Safety, Tolerability, and Pharmacokinetics of Mevidalen (LY3154207), a Centrally Acting Dopamine D1 Receptor-Positive Allosteric Modulator (D1PAM), in Healthy Subjects. Clin. Pharmacol. Drug Dev. 2021, 10, 393–403. [Google Scholar] [CrossRef]
- Biglan, K.; Munsie, L.; Svensson, K.A.; Ardayfio, P.; Pugh, M.; Sims, J.; Brys, M. Safety and Efficacy of Mevidalen in Lewy Body Dementia: A Phase 2, Randomized, Placebo-Controlled Trial. Mov. Disord. 2022, 37, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Feng, D.; Chu, M.L.; Fish, I.; Lovera, S.; Sands, Z.A.; Kelm, S.; Valade, A.; Wood, M.; Ceska, T.; et al. Crystal structure of dopamine D1 receptor in complex with G protein and a non-catechol agonist. Nat. Commun. 2021, 12, 3305. [Google Scholar] [CrossRef]
- Teng, X.; Chen, S.; Nie, Y.; Xiao, P.; Yu, X.; Shao, Z.; Zheng, S. Ligand recognition and biased agonism of the D1 dopamine receptor. Nat. Commun. 2022, 13, 3186. [Google Scholar] [CrossRef]
- Xiao, P.; Yan, W.; Gou, L.; Zhong, Y.N.; Kong, L.; Wu, C.; Wen, X.; Yuan, Y.; Cao, S.; Qu, C.; et al. Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell 2021, 184, 943–956.e18. [Google Scholar] [CrossRef]
- Zhuang, Y.; Xu, P.; Mao, C.; Wang, L.; Krumm, B.; Zhou, X.E.; Huang, S.; Liu, H.; Cheng, X.; Huang, X.P.; et al. Structural insights into the human D1 and D2 dopamine receptor signaling complexes. Cell 2021, 184, 931–942. [Google Scholar] [CrossRef]
- Zhuang, Y.; Krumm, B.; Zhang, H.; Zhou, X.E.; Wang, Y.; Huang, X.P.; Liu, Y.; Cheng, X.; Jiang, Y.; Jiang, H.; et al. Mechanism of dopamine binding and allosteric modulation of the human D1 dopamine receptor. Cell Res. 2021, 31, 593–596. [Google Scholar] [CrossRef] [PubMed]
- Sibley, D.R.; Luderman, K.D.; Free, R.B.; Shi, L. Novel Cryo-EM structures of the D1 dopamine receptor unlock its therapeutic potential. Signal Transduct. Target. Ther. 2021, 6, 205. [Google Scholar] [CrossRef]
- Xu, P.; Huang, S.; Krumm, B.E.; Zhuang, Y.; Mao, C.; Zhang, Y.; Wang, Y.; Huang, X.P.; Liu, Y.F.; He, X.; et al. Structural genomics of the human dopamine receptor system. Cell Res. 2023, 33, 604–616. [Google Scholar] [CrossRef]
- Wang, X.; Heinz, B.A.; Qian, Y.W.; Carter, J.H.; Gadski, R.A.; Beavers, L.S.; Little, S.P.; Yang, C.R.; Beck, J.P.; Hao, J.; et al. Intracellular Binding Site for a Positive Allosteric Modulator of the Dopamine D1 Receptor. Mol. Pharmacol. 2018, 94, 1232–1245. [Google Scholar] [CrossRef] [PubMed]
- Luderman, K.D.; Conroy, J.L.; Free, R.B.; Southall, N.; Ferrer, M.; Sanchez-Soto, M.; Moritz, A.E.; Willette, B.K.A.; Fyfe, T.J.; Jain, P.; et al. Identification of Positive Allosteric Modulators of the D1 Dopamine Receptor That Act at Diverse Binding Sites. Mol. Pharmacol. 2018, 94, 1197–1209. [Google Scholar] [CrossRef] [PubMed]
- Luderman, K.D.; Jain, P.; Benjamin Free, R.; Conroy, J.L.; Aube, J.; Sibley, D.R.; Frankowski, K.J. Development of pyrimidone D1 dopamine receptor positive allosteric modulators. Bioorg. Med. Chem. Lett. 2021, 31, 127696. [Google Scholar] [CrossRef]
- Liu, X.; Masoudi, A.; Kahsai, A.W.; Huang, L.Y.; Pani, B.; Staus, D.P.; Shim, P.J.; Hirata, K.; Simhal, R.K.; Schwalb, A.M.; et al. Mechanism of beta2AR regulation by an intracellular positive allosteric modulator. Science 2019, 364, 1283–1287. [Google Scholar] [CrossRef]
- Ho, J.D.; Chau, B.; Rodgers, L.; Lu, F.; Wilbur, K.L.; Otto, K.A.; Chen, Y.; Song, M.; Riley, J.P.; Yang, H.C.; et al. Structural basis for GPR40 allosteric agonism and incretin stimulation. Nat. Commun. 2018, 9, 1645. [Google Scholar] [CrossRef]
- Nussinov, R.; Tsai, C.J. Allostery without a conformational change? Revisiting the paradigm. Curr. Opin. Struct. Biol. 2015, 30, 17–24. [Google Scholar] [CrossRef]
- Ballesteros, J.; Weinstein, H. Integrated methods for the construction of three-dimensional models of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Abramyan, A.M.; Yano, H.; Xu, M.; Liu, L.; Naing, S.; Fant, A.D.; Shi, L. The Glu102 mutation disrupts higher-order oligomerization of the sigma 1 receptor. Comput. Struct. Biotechnol. J. 2020, 18, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.G.; Choi, H.J.; Fung, J.J.; Pardon, E.; Casarosa, P.; Chae, P.S.; Devree, B.T.; Rosenbaum, D.M.; Thian, F.S.; Kobilka, T.S.; et al. Structure of a nanobody-stabilized active state of the beta(2) adrenoceptor. Nature 2011, 469, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Fu, D.; Ballesteros, J.A.; Weinstein, H.; Chen, J.; Javitch, J.A. Residues in the seventh membrane-spanning segment of the dopamine D2 receptor accessible in the binding-site crevice. Biochemistry 1996, 35, 11278–11285. [Google Scholar] [CrossRef]
- Michino, M.; Free, R.B.; Doyle, T.B.; Sibley, D.R.; Shi, L. Structural basis for Na(+)-sensitivity in dopamine D2 and D3 receptors. Chem. Commun. (Camb.) 2015, 51, 8618–8621. [Google Scholar] [CrossRef]
- Lane, J.R.; Abramyan, A.M.; Adhikari, P.; Keen, A.C.; Lee, K.H.; Sanchez, J.; Verma, R.K.; Lim, H.D.; Yano, H.; Javitch, J.A.; et al. Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism. Elife 2020, 9, e52189. [Google Scholar] [CrossRef]
- Liu, W.; Chun, E.; Thompson, A.A.; Chubukov, P.; Xu, F.; Katritch, V.; Han, G.W.; Roth, C.B.; Heitman, L.H.; AP, I.J.; et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science 2012, 337, 232–236. [Google Scholar] [CrossRef]
- Picard, L.P.; Schonegge, A.M.; Bouvier, M. Structural Insight into G Protein-Coupled Receptor Signaling Efficacy and Bias between Gs and beta-Arrestin. ACS Pharmacol. Transl. Sci. 2019, 2, 148–154. [Google Scholar] [CrossRef]
- Katritch, V.; Fenalti, G.; Abola, E.E.; Roth, B.L.; Cherezov, V.; Stevens, R.C. Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci. 2014, 39, 233–244. [Google Scholar] [CrossRef]
- Zhou, Q.; Yang, D.; Wu, M.; Guo, Y.; Guo, W.; Zhong, L.; Cai, X.; Dai, A.; Jang, W.; Shakhnovich, E.I.; et al. Common activation mechanism of class A GPCRs. Elife 2019, 8, e50279. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Shi, L.; Javitch, J.A. Structural mimicry in G protein-coupled receptors: Implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol. Pharmacol. 2001, 60, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg, S.; Michino, M.; LeVine, M.V.; Weinstein, H.; Shi, L. Computational approaches to detect allosteric pathways in transmembrane molecular machines. Biochim. Biophys. Acta 2016, 1858 Pt B, 1652–1662. [Google Scholar] [CrossRef]
- Michino, M.; Boateng, C.A.; Donthamsetti, P.; Yano, H.; Bakare, O.M.; Bonifazi, A.; Ellenberger, M.P.; Keck, T.M.; Kumar, V.; Zhu, C.; et al. Toward Understanding the Structural Basis of Partial Agonism at the Dopamine D3 Receptor. J. Med. Chem. 2017, 60, 580–593. [Google Scholar] [CrossRef]
- Huang, S.; Xu, P.; Shen, D.D.; Simon, I.A.; Mao, C.; Tan, Y.; Zhang, H.; Harpsoe, K.; Li, H.; Zhang, Y.; et al. GPCRs steer G(i) and G(s) selectivity via TM5-TM6 switches as revealed by structures of serotonin receptors. Mol. Cell 2022, 82, 2681–2695.e6. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.; Madhusudhan, M.; Eramian, D.; Shen, M.; Pieper, U.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2014, 15, 5.6.1–5.6.37. [Google Scholar]
- Teng, X.; Chen, S.; Wang, Q.; Chen, Z.; Wang, X.; Huang, N.; Zheng, S. Structural insights into G protein activation by D1 dopamine receptor. Sci. Adv. 2022, 8, eabo4158. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.; Kortemme, T. Improvements to robotics-inspired conformational sampling in rosetta. PLoS ONE 2013, 8, e63090. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Dror, R.O.; Arlow, D.H.; Maragakis, P.; Mildorf, T.J.; Pan, A.C.; Xu, H.; Borhani, D.W.; Shaw, D.E. Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 18684–18689. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theory Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Xie, B.; Goldberg, A.; Shi, L. A comprehensive evaluation of the potential binding poses of fentanyl and its analogs at the micro-opioid receptor. Comput. Struct. Biotechnol. J. 2022, 20, 2309–2321. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Le Rouzic, V.P.; Goldberg, A.; Tsai, M.M.; Chen, L.; Zhang, T.; Sinha, A.; Pan, Y.X.; Baumann, M.H.; Shi, L. Binding preference at the mu-opioid receptor underlies distinct pharmacology of cyclopropyl versus valeryl analogs of fentanyl. Neuropharmacology 2023, 227, 109442. [Google Scholar] [CrossRef] [PubMed]
- McGibbon, R.T.; Beauchamp, K.A.; Harrigan, M.P.; Klein, C.; Swails, J.M.; Hernandez, C.X.; Schwantes, C.R.; Wang, L.P.; Lane, T.J.; Pande, V.S. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J. 2015, 109, 1528–1532. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Li, J.; Abel, R.; Zhu, K.; Cao, Y.; Zhao, S.; Friesner, R.A. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins 2011, 79, 2794–2812. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Condition | LY3154207 Orientation | No. Trajs | Length (μs) |
|---|---|---|---|---|
| dopamine | D1R/dopamine-alone | - | 15 | 24.9 |
| D1R/dopamine-LY3154207 | horizontal | 9 | 21.3 | |
| vertical | 3 | 3.6 | ||
| apomorphine | D1R/apomorphine-alone | - | 15 | 23.8 |
| D1R/apomorphine-LY3154207 | horizontal | 9 | 21.0 |
| Residue | BW Index | D1R/DA | D1R/DA-LY | DA—DA-LY | abs Diff |
|---|---|---|---|---|---|
| 37 | TM1.46 | 0.115 | 0.132 | −0.017 | 0.017 |
| 41 | TM1.50 | 0.118 | 0.140 | −0.022 | 0.022 |
| 62 | TM2.42 | 0.169 | 0.169 | 0.000 | 0.000 |
| 63 | TM2.43 | 0.114 | 0.111 | 0.003 | 0.003 |
| 65 | TM2.45 | 0.115 | 0.120 | −0.005 | 0.005 |
| 66 | TM2.46 | 0.217 | 0.204 | 0.013 | 0.013 |
| 67 | TM2.47 | 0.088 | 0.100 | −0.012 | 0.012 |
| 69 | TM2.49 | 0.162 | 0.168 | −0.006 | 0.006 |
| 70 | TM2.50 | 0.165 | 0.186 | −0.021 | 0.021 |
| 72 | TM2.52 | 0.096 | 0.105 | −0.009 | 0.009 |
| 73 | TM2.53 | 0.137 | 0.147 | −0.010 | 0.010 |
| 77 | TM2.57 | 0.097 | 0.107 | −0.010 | 0.010 |
| 78 | TM2.58 | 0.098 | 0.108 | −0.010 | 0.010 |
| 105 | TM3.34 | 0.093 | 0.106 | −0.013 | 0.013 |
| 106 | TM3.35 | 0.115 | 0.124 | −0.009 | 0.009 |
| 107 | TM3.36 | 0.082 | 0.083 | −0.001 | 0.001 |
| 109 | TM3.38 | 0.116 | 0.123 | −0.007 | 0.007 |
| 110 | TM3.39 | 0.172 | 0.172 | 0.000 | 0.000 |
| 111 | TM3.40 | 0.150 | 0.133 | 0.017 | 0.017 |
| 113 | TM3.42 | 0.174 | 0.177 | −0.003 | 0.003 |
| 114 | TM3.43 | 0.202 | 0.167 | 0.035 | 0.035 |
| 117 | TM3.46 | 0.163 | 0.146 | 0.017 | 0.017 |
| 118 | TM3.47 | 0.109 | 0.086 | 0.023 | 0.023 |
| 148 | TM4.50 | 0.135 | 0.144 | −0.009 | 0.009 |
| 151 | TM4.53 | 0.094 | 0.103 | −0.009 | 0.009 |
| 203 | TM5.47 | 0.111 | 0.094 | 0.017 | 0.017 |
| 206 | TM5.50 | 0.104 | 0.093 | 0.011 | 0.011 |
| 210 | TM5.54 | 0.142 | 0.116 | 0.026 | 0.026 |
| 214 | TM5.58 | 0.103 | 0.071 | 0.032 | 0.032 |
| 281 | TM6.44 | 0.151 | 0.125 | 0.026 | 0.026 |
| 285 | TM6.48 | 0.123 | 0.108 | 0.015 | 0.015 |
| 321 | TM7.43 | 0.117 | 0.126 | −0.009 | 0.009 |
| 323 | TM7.45 | 0.113 | 0.090 | 0.023 | 0.023 |
| 324 | TM7.46 | 0.128 | 0.142 | −0.014 | 0.014 |
| 327 | TM7.49 | 0.154 | 0.149 | 0.005 | 0.005 |
| 328 | TM7.50 | 0.090 | 0.115 | −0.025 | 0.025 |
| 331 | TM7.53 | 0.160 | 0.130 | 0.030 | 0.030 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goldberg, A.; Xie, B.; Shi, L. The Molecular Mechanism of Positive Allosteric Modulation at the Dopamine D1 Receptor. Int. J. Mol. Sci. 2023, 24, 12848. https://doi.org/10.3390/ijms241612848
Goldberg A, Xie B, Shi L. The Molecular Mechanism of Positive Allosteric Modulation at the Dopamine D1 Receptor. International Journal of Molecular Sciences. 2023; 24(16):12848. https://doi.org/10.3390/ijms241612848
Chicago/Turabian StyleGoldberg, Alexander, Bing Xie, and Lei Shi. 2023. "The Molecular Mechanism of Positive Allosteric Modulation at the Dopamine D1 Receptor" International Journal of Molecular Sciences 24, no. 16: 12848. https://doi.org/10.3390/ijms241612848