

The Roles of Periodontal Bacteria in Atherosclerosis
 , ,
, ,
Abstract
:1. Introduction
2. Porphyromonas gingivalis
2.1. The Association between Pg Exposure and AS in Humans and Animals
2.2. Mechanistic Investigations on the Effect of Pg in Different Courses of AS
2.2.1. Endothelial Barrier Disruption
2.2.2. Monocyte Adherence and Aggregation
2.2.3. Foam Cell Formation
2.2.4. Calcification and Angiogenesis in Plaque
2.2.5. Plaque Destabilization
3. Aggregatibacter actinomycetemcomitans
3.1. The Association between Aa Exposure and AS in Humans and Animals
3.2. Mechanistic Investigations on the Effect of Aa in AS
4. Fusobacterium nucleatum
4.1. The Association between Fn Exposure and AS in Humans and Animals
4.2. Mechanistic Investigations on the Effect of Fn in AS
5. Prevotella intermedia
5.1. The Association between Pi Exposure and AS in Humans and Animals
5.2. Mechanistic Investigations on the Effect of Pi in AS
6. Tannerella forsythia
6.1. The Association between Tf Exposure and AS in Humans and Animals
6.2. Mechanistic Investigations on the Effect of Tf in AS
7. Treponema denticola
7.1. The Association between Td Exposure and AS in Humans and Animals
7.2. Mechanistic Investigations on the Effect of Td in AS
8. Campylobacter rectus
8.1. The Association between Cr Exposure and AS in Humans and Animals
8.2. Mechanistic Investigations on the Effect of Cr in AS
9. Interactions between Bacteria
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Murray, C.J.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990–2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2197–2223. [Google Scholar] [CrossRef] [PubMed]
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Frostegard, J. Immunity, atherosclerosis and cardiovascular disease. BMC Med. 2013, 11, 117. [Google Scholar] [CrossRef]
- Silvestre-Roig, C.; de Winther, M.P.; Weber, C.; Daemen, M.J.; Lutgens, E.; Soehnlein, O. Atherosclerotic plaque destabilization: Mechanisms, models, and therapeutic strategies. Circ. Res. 2014, 114, 214–226. [Google Scholar] [CrossRef]
- Sanz, M.; Del Castillo, A.M.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and Cardiovascular Diseases. Consensus Report. Glob. Heart 2020, 15, 1. [Google Scholar] [CrossRef]
- Fabricant, C.G.; Fabricant, J.; Minick, C.R.; Litrenta, M.M. Herpesvirus-induced atherosclerosis in chickens. Fed. Proc. 1983, 42, 2476–2479. [Google Scholar]
- Ibrahim, A.I.; Obeid, M.T.; Jouma, M.J.; Moasis, G.A.; Al-Richane, W.L.; Kindermann, I.; Boehm, M.; Roemer, K.; Mueller-Lantzsch, N.; Gartner, B.C. Detection of herpes simplex virus, cytomegalovirus and Epstein-Barr virus DNA in atherosclerotic plaques and in unaffected bypass grafts. J. Clin. Virol. 2005, 32, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.A.; Kuo, C.C. Chlamydia pneumoniae—An infectious risk factor for atherosclerosis? Nat. Rev. Microbiol. 2004, 2, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Seymour, G.J.; Ford, P.J.; Cullinan, M.P.; Leishman, S.; Yamazaki, K. Relationship between periodontal infections and systemic disease. Clin. Microbiol. Infect. 2007, 13 (Suppl. S4), 3–10. [Google Scholar] [CrossRef]
- Franceschi, F.; Sepulveda, A.R.; Gasbarrini, A.; Pola, P.; Silveri, N.G.; Gasbarrini, G.; Graham, D.Y.; Genta, R.M. Cross-reactivity of anti-CagA antibodies with vascular wall antigens: Possible pathogenic link between Helicobacter pylori infection and atherosclerosis. Circulation 2002, 106, 430–434. [Google Scholar] [CrossRef]
- Gurevich, V.S.; Pleskov, V.M.; Levaia, M.V.; Bannikov, A.I.; Mitrofanova, L.B.; Urazgil’deeva, S.A. Influenza virus infection in progressing atherosclerosis. Kardiologiia 2002, 42, 21–24. [Google Scholar] [PubMed]
- Hsu, Y.H.; Muo, C.H.; Liu, C.Y.; Tsai, W.C.; Hsu, C.C.; Sung, F.C.; Kao, C.H. Hepatitis C virus infection increases the risk of developing peripheral arterial disease: A 9-year population-based cohort study. J. Hepatol. 2015, 62, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Adam, E.; Melnick, J.L.; Probtsfield, J.L.; Petrie, B.L.; Burek, J.; Bailey, K.R.; McCollum, C.H.; DeBakey, M.E. High levels of cytomegalovirus antibody in patients requiring vascular surgery for atherosclerosis. Lancet 1987, 2, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Hsue, P.Y.; Waters, D.D. HIV infection and coronary heart disease: Mechanisms and management. Nat. Rev. Cardiol. 2019, 16, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, Y.; Takeshita, T. The oral microbiome and human health. J. Oral Sci. 2017, 59, 201–206. [Google Scholar] [CrossRef]
- Parahitiyawa, N.B.; Scully, C.; Leung, W.K.; Yam, W.C.; Jin, L.J.; Samaranayake, L.P. Exploring the oral bacterial flora: Current status and future directions. Oral Dis. 2010, 16, 136–145. [Google Scholar] [CrossRef]
- Darveau, R.P.; Tanner, A.; Page, R.C. The microbial challenge in periodontitis. Periodontol. 2000 1997, 14, 12–32. [Google Scholar] [CrossRef]
- Bui, F.Q.; Almeida-da-Silva, C.L.C.; Huynh, B.; Trinh, A.; Liu, J.; Woodward, J.; Asadi, H.; Ojcius, D.M. Association between periodontal pathogens and systemic disease. Biomed. J. 2019, 42, 27–35. [Google Scholar] [CrossRef]
- Kassebaum, N.J.; Bernabe, E.; Dahiya, M.; Bhandari, B.; Murray, C.J.; Marcenes, W. Global burden of severe periodontitis in 1990–2010: A systematic review and meta-regression. J. Dent. Res. 2014, 93, 1045–1053. [Google Scholar] [CrossRef]
- Lockhart, P.B.; Bolger, A.F.; Papapanou, P.N.; Osinbowale, O.; Trevisan, M.; Levison, M.E.; Taubert, K.A.; Newburger, J.W.; Gornik, H.L.; Gewitz, M.H.; et al. Periodontal disease and atherosclerotic vascular disease: Does the evidence support an independent association?: A scientific statement from the American Heart Association. Circulation 2012, 125, 2520–2544. [Google Scholar] [CrossRef]
- Zeng, X.T.; Leng, W.D.; Lam, Y.Y.; Yan, B.P.; Wei, X.M.; Weng, H.; Kwong, J.S. Periodontal disease and carotid atherosclerosis: A meta-analysis of 17,330 participants. Int. J. Cardiol. 2016, 203, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, M.; Kimura, Y.; Yamaga, T.; Yamamoto, N.; Ishikawa, M.; Wada, T.; Sakamoto, R.; Ishimoto, Y.; Fujisawa, M.; Okumiya, K.; et al. A population-based cross-sectional study of the association between periodontitis and arterial stiffness among the older Japanese population. J. Periodontal Res. 2021, 56, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Berent, R.; Auer, J.; Schmid, P.; Krennmair, G.; Crouse, S.F.; Green, J.S.; Sinzinger, H.; von Duvillard, S.P. Periodontal and coronary heart disease in patients undergoing coronary angiography. Metabolism 2011, 60, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Ryden, L.; Buhlin, K.; Ekstrand, E.; de Faire, U.; Gustafsson, A.; Holmer, J.; Kjellstrom, B.; Lindahl, B.; Norhammar, A.; Nygren, A.; et al. Periodontitis Increases the Risk of a First Myocardial Infarction: A Report From the PAROKRANK Study. Circulation 2016, 133, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Weintraub, W.S.; Ghazzal, Z.M.; Douglas, J.S., Jr.; Liberman, H.A.; Morris, D.C.; Cohen, C.L.; King, S.B., 3rd. Long-term clinical follow-up in patients with angiographic restudy after successful angioplasty. Circulation 1993, 87, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Du, L.; Fan, M.; Chen, X.; Tang, Y.; Wang, Y.; Wang, K.; Wang, S.; Li, G. Association between oral infections, triglyceride-glucose index, and in-stent restenosis. Oral Dis. 2022. [Google Scholar] [CrossRef]
- Osugue, R.; Castro dos Santos, N.C.; Araujo, C.F.; de Almeida, F.X.; Feres, M.; Santamaria, M.P. Periodontitis Is Associated With Risk of Conventional Stent Restenosis: Pilot Case-Control Study. Front. Dent. Med. 2021, 2, 673626. [Google Scholar] [CrossRef]
- Nagy, F.T.; Gheorghita, D.; Dharmarajan, L.; Braunitzer, G.; Achim, A.; Ruzsa, Z.; Antal, M.A. Oral Health of Patients Undergoing Percutaneous Coronary Intervention-A Possible Link between Periodontal Disease and In-Stent Restenosis. J. Pers. Med. 2023, 13, 760. [Google Scholar] [CrossRef]
- van Dam-Nolen, D.H.K.; Truijman, M.T.B.; van der Kolk, A.G.; Liem, M.I.; Schreuder, F.; Boersma, E.; Daemen, M.; Mess, W.H.; van Oostenbrugge, R.J.; van der Steen, A.F.W.; et al. Carotid Plaque Characteristics Predict Recurrent Ischemic Stroke and TIA: The PARISK (Plaque At RISK) Study. JACC Cardiovasc. Imaging 2022, 15, 1715–1726. [Google Scholar] [CrossRef]
- Achim, A.; Lacko, D.; Huttl, A.; Csobay-Novak, C.; Csavajda, A.; Sotonyi, P.; Merkely, B.; Nemes, B.; Ruzsa, Z. Impact of Diabetes Mellitus on Early Clinical Outcome and Stent Restenosis after Carotid Artery Stenting. J. Diabetes Res. 2022, 2022, 4196195. [Google Scholar] [CrossRef]
- Desvarieux, M.; Demmer, R.T.; Rundek, T.; Boden-Albala, B.; Jacobs, D.R., Jr.; Sacco, R.L.; Papapanou, P.N. Periodontal microbiota and carotid intima-media thickness: The Oral Infections and Vascular Disease Epidemiology Study (INVEST). Circulation 2005, 111, 576–582. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, D.; Orho-Melander, M.; Demmer, R.T.; Engstrom, G.; Melander, O.; Klinge, B.; Nilsson, P.M. Periodontal disease is associated with carotid plaque area: The Malmo Offspring Dental Study (MODS). J. Intern. Med. 2020, 287, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, N.; Ahlqvist, J.; Naslund, U.; Buhlin, K.; Gustafsson, A.; Kjellstrom, B.; Klinge, B.; Ryden, L.; Levring Jaghagen, E. Associations among Periodontitis, Calcified Carotid Artery Atheromas, and Risk of Myocardial Infarction. J. Dent. Res. 2020, 99, 60–68. [Google Scholar] [CrossRef]
- Morley, R.L.; Sharma, A.; Horsch, A.D.; Hinchliffe, R.J. Peripheral artery disease. BMJ 2018, 360, j5842. [Google Scholar] [CrossRef]
- Achim, A.; Stanek, A.; Homorodean, C.; Spinu, M.; Onea, H.L.; Lazar, L.; Marc, M.; Ruzsa, Z.; Olinic, D.M. Approaches to Peripheral Artery Disease in Diabetes: Are There Any Differences? Int. J. Environ. Res. Public Health 2022, 19, 9801. [Google Scholar] [CrossRef]
- Aday, A.W.; Matsushita, K. Epidemiology of Peripheral Artery Disease and Polyvascular Disease. Circ. Res. 2021, 128, 1818–1832. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.B.; Shin, M.S.; Han, D.H.; Sukhbaatar, M.; Kim, M.S.; Shin, H.S.; Kim, H.D. Periodontitis is associated with the risk of subclinical atherosclerosis and peripheral arterial disease in Korean adults. Atherosclerosis 2016, 251, 311–318. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, L.S.; Cai, C.; Shi, Q.; Wen, N.; Xu, J. Association between periodontitis and peripheral artery disease: A systematic review and meta-analysis. BMC Cardiovasc. Disord. 2018, 18, 141. [Google Scholar] [CrossRef]
- Lu, B.; Parker, D.; Eaton, C.B. Relationship of periodontal attachment loss to peripheral vascular disease: An analysis of NHANES 1999–2002 data. Atherosclerosis 2008, 200, 199–205. [Google Scholar] [CrossRef]
- Calapkorur, M.U.; Alkan, B.A.; Tasdemir, Z.; Akcali, Y.; Saatci, E. Association of peripheral arterial disease with periodontal disease: Analysis of inflammatory cytokines and an acute phase protein in gingival crevicular fluid and serum. J. Periodontal Res. 2017, 52, 532–539. [Google Scholar] [CrossRef]
- Chen, Y.W.; Umeda, M.; Nagasawa, T.; Takeuchi, Y.; Huang, Y.; Inoue, Y.; Iwai, T.; Izumi, Y.; Ishikawa, I. Periodontitis may increase the risk of peripheral arterial disease. Eur. J. Vasc. Endovasc. Surg. 2008, 35, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Soto-Barreras, U.; Olvera-Rubio, J.O.; Loyola-Rodriguez, J.P.; Reyes-Macias, J.F.; Martinez-Martinez, R.E.; Patino-Marin, N.; Martinez-Castanon, G.A.; Aradillas-Garcia, C.; Little, J.W. Peripheral arterial disease associated with caries and periodontal disease. J. Periodontol. 2013, 84, 486–494. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Willett, W.; Merchant, A.; Rosner, B.A.; Ascherio, A.; Joshipura, K.J. Oral health and peripheral arterial disease. Circulation 2003, 107, 1152–1157. [Google Scholar] [CrossRef]
- Munoz-Torres, F.J.; Mukamal, K.J.; Pai, J.K.; Willett, W.; Joshipura, K.J. Relationship between tooth loss and peripheral arterial disease among women. J. Clin. Periodontol. 2017, 44, 989–995. [Google Scholar] [CrossRef]
- Shanker, J.; Setty, P.; Arvind, P.; Nair, J.; Bhasker, D.; Balakrishna, G.; Kakkar, V.V. Relationship between periodontal disease, Porphyromonas gingivalis, peripheral vascular resistance markers and coronary artery disease in Asian Indians. Thromb. Res. 2013, 132, e8–e14. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Tang, Q.; Nie, J.; Zhang, C.; Zhou, X.; Yu, S.; Sun, J.; Cheng, X.; Dong, N.; Hu, Y.; et al. BMAL1-Downregulation Aggravates Porphyromonas Gingivalis-Induced Atherosclerosis by Encouraging Oxidative Stress. Circ. Res. 2020, 126, e15–e29. [Google Scholar] [CrossRef] [PubMed]
- Kannosh, I.; Staletovic, D.; Toljic, B.; Radunovic, M.; Pucar, A.; Matic Petrovic, S.; Grubisa, I.; Lazarevic, M.; Brkic, Z.; Knezevic Vukcevic, J.; et al. The presence of periopathogenic bacteria in subgingival and atherosclerotic plaques—An age related comparative analysis. J. Infect. Dev. Ctries. 2018, 12, 1088–1095. [Google Scholar] [CrossRef]
- Fernandes, C.P.; Oliveira, F.A.F.; Silva, P.G.d.B.; Alves, A.P.N.N.; Mota, M.R.L.; Montenegro, R.C.; Burbano, R.M.R.; Seabra, A.D.; Lobo Filho, J.G.; Lima, D.L.F.; et al. Molecular analysis of oral bacteria in dental biofilm and atherosclerotic plaques of patients with vascular disease. Int. J. Cardiol. 2014, 174, 710–712. [Google Scholar] [CrossRef]
- Datta, H.K.; Ng, W.F.; Walker, J.A.; Tuck, S.P.; Varanasi, S.S. The cell biology of bone metabolism. J. Clin. Pathol. 2008, 61, 577–587. [Google Scholar] [CrossRef]
- Sanz, M.; Lau, L.; Herrera, D.; Morillo, J.M.; Silva, A. Methods of detection of Actinobacillus actinomycetemcomitans, Porphyromonas gingivalis and Tannerella forsythensis in periodontal microbiology, with special emphasis on advanced molecular techniques: A review. J. Clin. Periodontol. 2004, 31, 1034–1047. [Google Scholar] [CrossRef]
- Jepsen, K.; Falk, W.; Brune, F.; Fimmers, R.; Jepsen, S.; Bekeredjian-Ding, I. Prevalence and antibiotic susceptibility trends of periodontal pathogens in the subgingival microbiota of German periodontitis patients: A retrospective surveillance study. J. Clin. Periodontol. 2021, 48, 1216–1227. [Google Scholar] [CrossRef] [PubMed]
- Figuero, E.; Sanchez-Beltran, M.; Cuesta-Frechoso, S.; Tejerina, J.M.; del Castro, J.A.; Gutierrez, J.M.; Herrera, D.; Sanz, M. Detection of periodontal bacteria in atheromatous plaque by nested polymerase chain reaction. J. Periodontol. 2011, 82, 1469–1477. [Google Scholar] [CrossRef] [PubMed]
- Gaetti-Jardim, E.; Marcelino, S.L.; Feitosa, A.C.R.; Romito, G.A.; Avila-Campos, M.J. Quantitative detection of periodontopathic bacteria in atherosclerotic plaques from coronary arteries. J. Med. Microbiol. 2009, 58, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Brun, A.; Range, H.; Prouvost, B.; Mazighi, M.; Kapila, Y.; Bouchard, P.; Michel, J.B. Innovative application of nested PCR for detection of Porphyromonas gingivalis in human highly calcified atherothrombotic plaques. J. Oral Microbiol. 2020, 12, 1742523. [Google Scholar] [CrossRef]
- Choi, J.I.; Chung, S.W.; Kang, H.S.; Rhim, B.Y.; Park, Y.M.; Kim, U.S.; Kim, S.J. Epitope mapping of Porphyromonas gingivalis heat-shock protein and human heat-shock protein in human atherosclerosis. J. Dent. Res. 2004, 83, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, A.; Nishimura, F.; Murayama, Y.; Nagasaka, S.; Fukushima, M.; Sakai, M.; Yoshii, S.; Kuroe, A.; Suzuki, H.; Iwamoto, Y.; et al. Porphyromonas gingivalis infection is associated with carotid atherosclerosis in non-obese Japanese type 2 diabetic patients. Metabolism 2003, 52, 142–145. [Google Scholar] [CrossRef]
- Salhi, L.; Sakalihasan, N.; Okroglic, A.G.; Labropoulos, N.; Seidel, L.; Albert, A.; Teughels, W.; Defraigne, J.O.; Lambert, F. Further evidence on the relationship between abdominal aortic aneurysm and periodontitis: A cross-sectional study. J. Periodontol. 2020, 91, 1453–1464. [Google Scholar] [CrossRef]
- Pussinen, P.J.; Alfthan, G.; Jousilahti, P.; Paju, S.; Tuomilehto, J. Systemic exposure to Porphyromonas gingivalis predicts incident stroke. Atherosclerosis 2007, 193, 222–228. [Google Scholar] [CrossRef]
- Li, L.; Messas, E.; Batista, E.L.; Levine, R.A.; Amar, S. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation 2002, 105, 861–867. [Google Scholar] [CrossRef]
- Lalla, E.; Lamster, I.B.; Hofmann, M.A.; Bucciarelli, L.; Jerud, A.P.; Tucker, S.; Lu, Y.; Papapanou, P.N.; Schmidt, A.M. Oral infection with a periodontal pathogen accelerates early atherosclerosis in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1405–1411. [Google Scholar] [CrossRef]
- Ford, P.J.; Gemmell, E.; Timms, P.; Chan, A.; Preston, F.M.; Seymour, G.J. Anti-P. gingivalis response correlates with atherosclerosis. J. Dent. Res. 2007, 86, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, Y.; Kurita-Ochiai, T.; Oguchi, S.; Yamamoto, M. Nasal immunization with Porphyromonas gingivalis outer membrane protein decreases P. gingivalis-induced atherosclerosis and inflammation in spontaneously hyperlipidemic mice. Infect. Immun. 2008, 76, 2958–2965. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Wu, J.; Zhang, R.; Yao, M.; Liu, Y.; Miao, L.; Sun, W. Porphyromonas gingivalis oral infection promote T helper 17/Treg imbalance in the development of atherosclerosis. J. Dent. Sci. 2017, 12, 60–69. [Google Scholar] [CrossRef]
- Miyauchi, S.; Maekawa, T.; Aoki, Y.; Miyazawa, H.; Tabeta, K.; Nakajima, T.; Yamazaki, K. Oral infection with Porphyromonas gingivalis and systemic cytokine profile in C57BL/6.KOR-ApoE shl mice. J. Periodontal Res. 2012, 47, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.S.; Kim, S.; Bostrom, K.I.; Wang, C.Y.; Kim, R.H.; Park, N.H. Periodontitis-induced systemic inflammation exacerbates atherosclerosis partly via endothelial-mesenchymal transition in mice. Int. J. Oral Sci. 2019, 11, 21. [Google Scholar] [CrossRef]
- Shiheido, Y.; Maejima, Y.; Suzuki, J.I.; Aoyama, N.; Kaneko, M.; Watanabe, R.; Sakamaki, Y.; Wakayama, K.; Ikeda, Y.; Akazawa, H.; et al. Porphyromonas gingivalis, a periodontal pathogen, enhances myocardial vulnerability, thereby promoting post-infarct cardiac rupture. J. Mol. Cell. Cardiol. 2016, 99, 123–137. [Google Scholar] [CrossRef]
- Komatsu, T.; Nagano, K.; Sugiura, S.; Hagiwara, M.; Tanigawa, N.; Abiko, Y.; Yoshimura, F.; Furuichi, Y.; Matsushita, K. E-selectin mediates Porphyromonas gingivalis adherence to human endothelial cells. Infect. Immun. 2012, 80, 2570–2576. [Google Scholar] [CrossRef]
- Song, H.; Belanger, M.; Whitlock, J.; Kozarov, E.; Progulske-Fox, A. Hemagglutinin B is involved in the adherence of Porphyromonas gingivalis to human coronary artery endothelial cells. Infect. Immun. 2005, 73, 7267–7273. [Google Scholar] [CrossRef]
- Walter, C.; Zahlten, J.; Schmeck, B.; Schaudinn, C.; Hippenstiel, S.; Frisch, E.; Hocke, A.C.; Pischon, N.; Kuramitsu, H.K.; Bernimoulin, J.P.; et al. Porphyromonas gingivalis strain-dependent activation of human endothelial cells. Infect. Immun. 2004, 72, 5910–5918. [Google Scholar] [CrossRef]
- Nassar, H.; Chou, H.H.; Khlgatian, M.; Gibson, F.C., 3rd; Van Dyke, T.E.; Genco, C.A. Role for fimbriae and lysine-specific cysteine proteinase gingipain K in expression of interleukin-8 and monocyte chemoattractant protein in Porphyromonas gingivalis-infected endothelial cells. Infect. Immun. 2002, 70, 268–276. [Google Scholar] [CrossRef]
- Khlgatian, M.; Nassar, H.; Chou, H.H.; Gibson, F.C., 3rd; Genco, C.A. Fimbria-dependent activation of cell adhesion molecule expression in Porphyromonas gingivalis-infected endothelial cells. Infect. Immun. 2002, 70, 257–267. [Google Scholar] [CrossRef]
- Hayashi, C.; Madrigal, A.G.; Liu, X.; Ukai, T.; Goswami, S.; Gudino, C.V.; Gibson, F.C., 3rd; Genco, C.A. Pathogen-mediated inflammatory atherosclerosis is mediated in part via Toll-like receptor 2-induced inflammatory responses. J. Innate Immun. 2010, 2, 334–343. [Google Scholar] [CrossRef]
- Belanger, M.; Rodrigues, P.H.; Dunn, W.A., Jr.; Progulske-Fox, A. Autophagy: A highway for Porphyromonas gingivalis in endothelial cells. Autophagy 2006, 2, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Dorn, B.R.; Dunn, W.A., Jr.; Progulske-Fox, A. Porphyromonas gingivalis traffics to autophagosomes in human coronary artery endothelial cells. Infect. Immun. 2001, 69, 5698–5708. [Google Scholar] [CrossRef] [PubMed]
- Madrigal, A.G.; Barth, K.; Papadopoulos, G.; Genco, C.A. Pathogen-mediated proteolysis of the cell death regulator RIPK1 and the host defense modulator RIPK2 in human aortic endothelial cells. PLoS Pathog. 2012, 8, e1002723. [Google Scholar] [CrossRef]
- Xie, M.; Tang, Q.; Yu, S.; Sun, J.; Mei, F.; Zhao, J.; Chen, L. Porphyromonas gingivalis disrupts vascular endothelial homeostasis in a TLR-NF-kappaB axis dependent manner. Int. J. Oral Sci. 2020, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Sheets, S.M.; Potempa, J.; Travis, J.; Casiano, C.A.; Fletcher, H.M. Gingipains from Porphyromonas gingivalis W83 induce cell adhesion molecule cleavage and apoptosis in endothelial cells. Infect. Immun. 2005, 73, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Aoudjit, F.; Vuori, K. Matrix attachment regulates Fas-induced apoptosis in endothelial cells: A role for c-flip and implications for anoikis. J. Cell. Biol. 2001, 152, 633–643. [Google Scholar] [CrossRef]
- Viafara-Garcia, S.M.; Morantes, S.J.; Chacon-Quintero, Y.; Castillo, D.M.; Lafaurie, G.I.; Buitrago, D.M. Repeated Porphyromonas gingivalis W83 exposure leads to release pro-inflammatory cytokynes and angiotensin II in coronary artery endothelial cells. Sci. Rep. 2019, 9, 19379. [Google Scholar] [CrossRef]
- Forstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Sampath, C.; Okoro, E.U.; Gipson, M.J.; Chukkapalli, S.S.; Farmer-Dixon, C.M.; Gangula, P.R. Porphyromonas gingivalis infection alters Nrf2-phase II enzymes and nitric oxide in primary human aortic endothelial cells. J. Periodontol. 2021, 92, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Xu, T.; Dong, Q.; Luo, Y.; Liu, Y.; Gao, L.; Pan, Y.; Zhang, D. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int. J. Oral Sci. 2021, 13, 28. [Google Scholar] [CrossRef]
- Hashizume, T.; Kurita-Ochiai, T.; Yamamoto, M. Porphyromonas gingivalis stimulates monocyte adhesion to human umbilical vein endothelial cells. FEMS Immunol. Med. Microbiol. 2011, 62, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Liu, J.; Ouyang, X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappaB pathway. J. Periodontal Res. 2015, 50, 189–196. [Google Scholar] [CrossRef]
- Takahashi, Y.; Davey, M.; Yumoto, H.; Gibson, F.C., 3rd; Genco, C.A. Fimbria-dependent activation of pro-inflammatory molecules in Porphyromonas gingivalis infected human aortic endothelial cells. Cell. Microbiol. 2006, 8, 738–757. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Liu, J.; Liu, W.; Chu, Y.; Zhong, J.; Xie, Y.; Lou, X.; Ouyang, X. LOX-1 Regulates -Induced Monocyte Migration and Adhesion to Human Umbilical Vein Endothelial Cells. Front. Cell. Dev. Biol. 2020, 8, 596. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Pan, Y.; Xu, Q.; Wu, Y.; Pan, J.; Hou, J.; Lin, L.; Tang, X.; Li, C.; Liu, J.; et al. Porphyromonas gingivalis ATCC 33277 promotes intercellular adhesion molecule-1 expression in endothelial cells and monocyte-endothelial cell adhesion through macrophage migration inhibitory factor. BMC Microbiol. 2018, 18, 16. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, H.; Zhao, J.; Lin, L.; Li, C.; Liu, J.; Pan, Y. Porphorymonas gingivalis induces intracellular adhesion molecule-1 expression in endothelial cells through the nuclear factor-kappaB pathway, but not through the p38 MAPK pathway. J. Periodontal Res. 2011, 46, 31–38. [Google Scholar] [CrossRef]
- Wang, X.; Liu, Y.; Zhang, S.; Ouyang, X.; Wang, Y.; Jiang, Y.; An, N. Crosstalk between Akt and NF-kappaB pathway mediates inhibitory effect of gas6 on monocytes-endothelial cells interactions stimulated by P. gingivalis-LPS. J. Cell. Mol. Med. 2020, 24, 7979–7990. [Google Scholar] [CrossRef]
- Wu, Y.; Xu, W.; Hou, J.; Liu, Y.; Li, R.; Liu, J.; Li, C.; Tang, X.; Lin, L.; Pan, Y.; et al. Porphyromonas gingivalis-Induced MIF Regulates Intercellular Adhesion Molecule-1 Expression in EA.hy926 Cells and Monocyte-Endothelial Cell Adhesion Through the Receptors CD74 and CXCR4. Inflammation 2019, 42, 874–883. [Google Scholar] [CrossRef]
- Hayashi, C.; Gudino, C.V.; Gibson, F.C., 3rd; Genco, C.A. Review: Pathogen-induced inflammation at sites distant from oral infection: Bacterial persistence and induction of cell-specific innate immune inflammatory pathways. Mol. Oral Microbiol. 2010, 25, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Li, Z.; Zhang, Y.; Luo, K.; Xu, X.; Li, J.; Peng, X.; Zhou, X. Cyclic di-AMP Rescues Porphyromonas gingivalis-Aggravated Atherosclerosis. J. Dent. Res. 2023, 102, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Qi, M.; Miyakawa, H.; Kuramitsu, H.K. Porphyromonas gingivalis induces murine macrophage foam cell formation. Microb. Pathog. 2003, 35, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Wang, C.; Xiang, X.R.; Chen, F.C.; Yang, C.M.; Wu, J. Porphyromonas gingivalis lipopolysaccharide increases lipid accumulation by affecting CD36 and ATP-binding cassette transporter A1 in macrophages. Oncol. Rep. 2013, 30, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.Y.; Liu, F.; Chen, J.X.; He, X.L.; Zhou, Y.L.; Ge, B.X.; Luo, L.J. Porphyromonas gingivalis infected macrophages upregulate CD36 expression via ERK/NF-kappaB pathway. Cell. Signal. 2016, 28, 1292–1303. [Google Scholar] [CrossRef]
- Yang, Y.; He, X.; Xia, S.; Liu, F.; Luo, L. Porphyromonas gingivalis facilitated the foam cell formation via lysosomal integral membrane protein 2 (LIMP2). J. Periodontal Res. 2021, 56, 265–274. [Google Scholar] [CrossRef]
- Ruan, Q.; Guan, P.; Qi, W.; Li, J.; Xi, M.; Xiao, L.; Zhong, S.; Ma, D.; Ni, J. Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front. Immunol. 2023, 14, 1103592. [Google Scholar] [CrossRef]
- Kim, D.J.; Rho, J.H.; Woo, B.H.; Joo, J.Y.; Lee, J.Y.; Song, J.M.; Lee, J.H.; Park, H.R. Periodontal Pathogens Modulate Lipid Flux via Fatty Acid Binding Protein 4. J. Dent. Res. 2019, 98, 1511–1520. [Google Scholar] [CrossRef]
- Kim, H.J.; Cha, G.S.; Kim, H.J.; Kwon, E.Y.; Lee, J.Y.; Choi, J.; Joo, J.Y. Porphyromonas gingivalis accelerates atherosclerosis through oxidation of high-density lipoprotein. J. Periodontal Implant. Sci. 2018, 48, 60–68. [Google Scholar] [CrossRef]
- Park, H.J.; Kim, Y.; Kim, M.K.; Park, H.R.; Kim, H.J.; Bae, S.K.; Bae, M.K. Infection of Porphyromonas gingivalis Increases Phosphate-Induced Calcification of Vascular Smooth Muscle Cells. Cells 2020, 9, 2694. [Google Scholar] [CrossRef]
- Hokamura, K.; Inaba, H.; Nakano, K.; Nomura, R.; Yoshioka, H.; Taniguchi, K.; Ooshima, T.; Wada, K.; Amano, A.; Umemura, K. Molecular analysis of aortic intimal hyperplasia caused by Porphyromonas gingivalis infection in mice with endothelial damage. J. Periodontal Res. 2010, 45, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Hokamura, K.; Nakano, K.; Nomura, R.; Katayama, K.; Nakajima, A.; Yoshioka, H.; Taniguchi, K.; Kamisaki, Y.; Ooshima, T.; et al. Upregulation of S100 calcium-binding protein A9 is required for induction of smooth muscle cell proliferation by a periodontal pathogen. Febs Lett. 2009, 583, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Elmabsout, A.A.; Khalaf, H.; Basic, V.T.; Jayaprakash, K.; Kruse, R.; Bengtsson, T.; Sirsjo, A. The periodontal pathogen Porphyromonas gingivalis changes the gene expression in vascular smooth muscle cells involving the TGFbeta/Notch signalling pathway and increased cell proliferation. BMC Genom. 2013, 14, 770. [Google Scholar] [CrossRef]
- Yang, W.W.; Guo, B.; Jia, W.Y.; Jia, Y. Porphyromonas gingivalis-derived outer membrane vesicles promote calcification of vascular smooth muscle cells through ERK1/2-RUNX2. FEBS Open Bio 2016, 6, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Deng, J.; Shang, S.; Liu, G.; Song, W.; Sun, P.; Jiang, W.; Pan, K. Effect of Porphyromonas gingivalis lipopolysaccharide on calcification of human umbilical artery smooth muscle cells co-cultured with human periodontal ligament cells. Exp. Ther. Med. 2021, 21, 655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Sirsjo, A.; Khalaf, H.; Bengtsson, T. Transcriptional profiling of human smooth muscle cells infected with gingipain and fimbriae mutants of Porphyromonas gingivalis. Sci. Rep. 2016, 6, 21911. [Google Scholar] [CrossRef]
- Zhang, B.; Khalaf, H.; Sirsjo, A.; Bengtsson, T. Gingipains from the Periodontal Pathogen Porphyromonas gingivalis Play a Significant Role in Regulation of Angiopoietin 1 and Angiopoietin 2 in Human Aortic Smooth Muscle Cells. Infect. Immun. 2015, 83, 4256–4265. [Google Scholar] [CrossRef]
- Roth, G.A.; Aumayr, K.; Giacona, M.B.; Papapanou, P.N.; Schmidt, A.M.; Lalla, E. Porphyromonas gingivalis infection and prothrombotic effects in human aortic smooth muscle cells. Thromb. Res. 2009, 123, 780–784. [Google Scholar] [CrossRef]
- Mubarokah, S.N.; Susilawati, I.D.A.; Sumarno, S.; Muliartha, I.K.G.; Sargowo, D. Porphyromonas gingivalis Induced Fragmentation of Type IV Collagen Through Macrophage-Activated MMP-9: (In Vitro Study of Collagenolytic Mechanism in Pathogenesis of Atherosclerotic Plaque Rupture). Indones. Biomed. J. 2009, 1, 88. [Google Scholar] [CrossRef]
- Shah, P.K. Inflammation and plaque vulnerability. Cardiovasc. Drugs Ther. 2009, 23, 31–40. [Google Scholar] [CrossRef]
- Naito, M.; Sakai, E.; Shi, Y.; Ideguchi, H.; Shoji, M.; Ohara, N.; Yamamoto, K.; Nakayama, K. Porphyromonas gingivalis-induced platelet aggregation in plasma depends on Hgp44 adhesin but not Rgp proteinase. Mol. Microbiol. 2006, 59, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Herbert, B.A.; Novince, C.M.; Kirkwood, K.L. Aggregatibacter actinomycetemcomitans, a potent immunoregulator of the periodontal host defense system and alveolar bone homeostasis. Mol. Oral Microbiol. 2016, 31, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Akhi, R.; Nissinen, A.E.; Wang, C.; Kyrklund, M.; Paju, S.; Mantyla, P.; Buhlin, K.; Sinisalo, J.; Pussinen, P.J.; Horkko, S. Salivary IgA antibody to malondialdehyde-acetaldehyde associates with mild periodontal pocket depth. Oral Dis. 2022, 28, 2285–2293. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Honma, K.; Miura, T.; Kato, T.; Okuda, K. Cloning and sequence analysis of the fimbriae associated protein (fap) gene from Actinobacillus actinomycetemcomitans. Microb. Pathog. 1997, 23, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Fine, D.H.; Furgang, D.; Kaplan, J.; Charlesworth, J.; Figurski, D.H. Tenacious adhesion of Actinobacillus actinomycetemcomitans strain CU1000 to salivary-coated hydroxyapatite. Arch. Oral Biol. 1999, 44, 1063–1076. [Google Scholar] [CrossRef]
- Schreiner, H.C.; Sinatra, K.; Kaplan, J.B.; Furgang, D.; Kachlany, S.C.; Planet, P.J.; Perez, B.A.; Figurski, D.H.; Fine, D.H. Tight-adherence genes of Actinobacillus actinomycetemcomitans are required for virulence in a rat model. Proc. Natl. Acad. Sci. USA 2003, 100, 7295–7300. [Google Scholar] [CrossRef]
- Razeghian-Jahromi, I.; Elyaspour, Z.; Zibaeenezhad, M.J.; Hassanipour, S. Prevalence of Microorganisms in Atherosclerotic Plaques of Coronary Arteries: A Systematic Review and Meta-Analysis. Evid. Based Complement. Alternat Med. 2022, 2022, 8678967. [Google Scholar] [CrossRef]
- Kozarov, E.; Sweier, D.; Shelburne, C.; Progulske-Fox, A.; Lopatin, D. Detection of bacterial DNA in atheromatous plaques by quantitative PCR. Microbes Infect. 2006, 8, 687–693. [Google Scholar] [CrossRef]
- Giles, J.T.; Reinholdt, J.; Andrade, F.; Konig, M.F. Associations of Antibodies Targeting Periodontal Pathogens With Subclinical Coronary, Carotid, and Peripheral Arterial Atherosclerosis in Rheumatoid Arthritis. Arthritis Rheumatol. 2021, 73, 568–575. [Google Scholar] [CrossRef]
- Pyysalo, M.J.; Pyysalo, L.M.; Pessi, T.; Karhunen, P.J.; Ohman, J.E. The connection between ruptured cerebral aneurysms and odontogenic bacteria. J. Neurol. Neurosurg. Psychiatry 2013, 84, 1214–1218. [Google Scholar] [CrossRef]
- Hyvarinen, K.; Mantyla, P.; Buhlin, K.; Paju, S.; Nieminen, M.S.; Sinisalo, J.; Pussinen, P.J. A common periodontal pathogen has an adverse association with both acute and stable coronary artery disease. Atherosclerosis 2012, 223, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Tsiantoulas, D.; Diehl, C.J.; Witztum, J.L.; Binder, C.J. B cells and humoral immunity in atherosclerosis. Circ. Res. 2014, 114, 1743–1756. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, J.B.; Perry, M.B.; MacLean, L.L.; Furgang, D.; Wilson, M.E.; Fine, D.H. Structural and genetic analyses of O polysaccharide from Actinobacillus actinomycetemcomitans serotype f. Infect. Immun. 2001, 69, 5375–5384. [Google Scholar] [CrossRef]
- Saarela, M.; Asikainen, S.; Alaluusua, S.; Pyhala, L.; Lai, C.H.; Jousimies-Somer, H. Frequency and stability of mono- or poly-infection by Actinobacillus actinomycetemcomitans serotypes a, b, c, d or e. Oral Microbiol. Immunol. 1992, 7, 277–279. [Google Scholar] [CrossRef]
- Takada, K.; Saito, M.; Tsuzukibashi, O.; Kawashima, Y.; Ishida, S.; Hirasawa, M. Characterization of a new serotype g isolate of Aggregatibacter actinomycetemcomitans. Mol. Oral Microbiol. 2010, 25, 200–206. [Google Scholar] [CrossRef]
- Zambon, J.J.; Slots, J.; Genco, R.J. Serology of oral Actinobacillus actinomycetemcomitans and serotype distribution in human periodontal disease. Infect. Immun. 1983, 41, 19–27. [Google Scholar] [CrossRef]
- Pietiainen, M.; Kopra, K.A.E.; Vuorenkoski, J.; Salminen, A.; Paju, S.; Mantyla, P.; Buhlin, K.; Liljestrand, J.M.; Nieminen, M.S.; Sinisalo, J.; et al. Aggregatibacter actinomycetemcomitans serotypes associate with periodontal and coronary artery disease status. J. Clin. Periodontol. 2018, 45, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Jia, R.; Kurita-Ochiai, T.; Oguchi, S.; Yamamoto, M. Periodontal pathogen accelerates lipid peroxidation and atherosclerosis. J. Dent. Res. 2013, 92, 247–252. [Google Scholar] [CrossRef]
- Tuomainen, A.M.; Jauhiainen, M.; Kovanen, P.T.; Metso, J.; Paju, S.; Pussinen, P.J. Aggregatibacter actinomycetemcomitans induces MMP-9 expression and proatherogenic lipoprotein profile in apoE-deficient mice. Microb. Pathog. 2008, 44, 111–117. [Google Scholar] [CrossRef]
- Zhang, T.; Kurita-Ochiai, T.; Hashizume, T.; Du, Y.; Oguchi, S.; Yamamoto, M. Aggregatibacter actinomycetemcomitans accelerates atherosclerosis with an increase in atherogenic factors in spontaneously hyperlipidemic mice. FEMS Immunol. Med. Microbiol. 2010, 59, 143–151. [Google Scholar] [CrossRef]
- Kieba, I.R.; Fong, K.P.; Tang, H.Y.; Hoffman, K.E.; Speicher, D.W.; Klickstein, L.B.; Lally, E.T. Aggregatibacter actinomycetemcomitans leukotoxin requires beta-sheets 1 and 2 of the human CD11a beta-propeller for cytotoxicity. Cell. Microbiol. 2007, 9, 2689–2699. [Google Scholar] [CrossRef] [PubMed]
- Dileepan, T.; Kachlany, S.C.; Balashova, N.V.; Patel, J.; Maheswaran, S.K. Human CD18 is the functional receptor for Aggregatibacter actinomycetemcomitans leukotoxin. Infect. Immun. 2007, 75, 4851–4856. [Google Scholar] [CrossRef] [PubMed]
- Lally, E.T.; Kieba, I.R.; Sato, A.; Green, C.L.; Rosenbloom, J.; Korostoff, J.; Wang, J.F.; Shenker, B.J.; Ortlepp, S.; Robinson, M.K.; et al. RTX toxins recognize a beta2 integrin on the surface of human target cells. J. Biol. Chem. 1997, 272, 30463–30469. [Google Scholar] [CrossRef] [PubMed]
- Shenker, B.J.; Ojcius, D.M.; Walker, L.P.; Zekavat, A.; Scuron, M.D.; Boesze-Battaglia, K. Aggregatibacter actinomycetemcomitans cytolethal distending toxin activates the NLRP3 inflammasome in human macrophages, leading to the release of proinflammatory cytokines. Infect. Immun. 2015, 83, 1487–1496. [Google Scholar] [CrossRef]
- Dietmann, A.; Millonig, A.; Combes, V.; Couraud, P.O.; Kachlany, S.C.; Grau, G.E. Effects of Aggregatibacter actinomycetemcomitans leukotoxin on endothelial cells. Microb. Pathog. 2013, 61–62, 43–50. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nakashima, K.; Isoda, T.; Yokota, M.; Nishihara, T. Involvement of adhesion molecule in in vitro plaque-like formation of macrophages stimulated with Aggregatibacter actinomycetemcomitans lipopolysaccharide. J. Periodontal Res. 2010, 45, 550–556. [Google Scholar] [CrossRef]
- Oksaharju, A.; Lappalainen, J.; Tuomainen, A.M.; Pussinen, P.J.; Puolakkainen, M.; Kovanen, P.T.; Lindstedt, K.A. Pro-atherogenic lung and oral pathogens induce an inflammatory response in human and mouse mast cells. J. Cell. Mol. Med. 2009, 13, 103–113. [Google Scholar] [CrossRef]
- Lakio, L.; Lehto, M.; Tuomainen, A.M.; Jauhiainen, M.; Malle, E.; Asikainen, S.; Pussinen, P.J. Pro-atherogenic properties of lipopolysaccharide from the periodontal pathogen Actinobacillus actinomycetemcomitans. J. Endotoxin Res. 2006, 12, 57–64. [Google Scholar] [CrossRef]
- Jia, R.; Hashizume-Takizawa, T.; Du, Y.; Yamamoto, M.; Kurita-Ochiai, T. Aggregatibacter actinomycetemcomitans induces Th17 cells in atherosclerotic lesions. Pathog. Dis. 2015, 73, ftu027. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K.; Dhingra, A.; Walker, L.M.; Zekavat, A.; Shenker, B.J. Internalization and Intoxication of Human Macrophages by the Active Subunit of the Aggregatibacter actinomycetemcomitans Cytolethal Distending Toxin Is Dependent Upon Cellugyrin (Synaptogyrin-2). Front. Immunol. 2020, 11, 1262. [Google Scholar] [CrossRef]
- Kaplan, C.W.; Ma, X.; Paranjpe, A.; Jewett, A.; Lux, R.; Kinder-Haake, S.; Shi, W. Fusobacterium nucleatum outer membrane proteins Fap2 and RadD induce cell death in human lymphocytes. Infect. Immun. 2010, 78, 4773–4778. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.; Tan, K.S. Fusobacterium nucleatum activates the immune response through retinoic acid-inducible gene I. J. Dent. Res. 2014, 93, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Fang, J.Y. Fusobacterium nucleatum, a key pathogenic factor and microbial biomarker for colorectal cancer. Trends Microbiol. 2023, 31, 159–172. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Li, Z.; Liu, H.; Zheng, S.; Zhang, F.; Zhu, J.; Shi, H.; Ye, H.; Chou, Z.; Gao, L.; et al. Fusobacterium nucleatum aggravates rheumatoid arthritis through FadA-containing outer membrane vesicles. Cell. Host Microbe 2023, 31, 798–810.e7. [Google Scholar] [CrossRef]
- Han, Y.W.; Wang, X. Mobile microbiome: Oral bacteria in extra-oral infections and inflammation. J. Dent. Res. 2013, 92, 485–491. [Google Scholar] [CrossRef]
- Fan, Z.; Tang, P.; Li, C.; Yang, Q.; Xu, Y.; Su, C.; Li, L. Fusobacterium nucleatum and its associated systemic diseases: Epidemiologic studies and possible mechanisms. J. Oral Microbiol. 2023, 15, 2145729. [Google Scholar] [CrossRef]
- Ford, P.J.; Gemmell, E.; Chan, A.; Carter, C.L.; Walker, P.J.; Bird, P.S.; West, M.J.; Cullinan, M.P.; Seymour, G.J. Inflammation, heat shock proteins and periodontal pathogens in atherosclerosis: An immunohistologic study. Oral Microbiol. Immunol. 2006, 21, 206–211. [Google Scholar] [CrossRef]
- Elkaim, R.; Dahan, M.; Kocgozlu, L.; Werner, S.; Kanter, D.; Kretz, J.G.; Tenenbaum, H. Prevalence of periodontal pathogens in subgingival lesions, atherosclerotic plaques and healthy blood vessels: A preliminary study. J. Periodontal Res. 2008, 43, 224–231. [Google Scholar] [CrossRef]
- Liljestrand, J.M.; Paju, S.; Pietiainen, M.; Buhlin, K.; Persson, G.R.; Nieminen, M.S.; Sinisalo, J.; Mantyla, P.; Pussinen, P.J. Immunologic burden links periodontitis to acute coronary syndrome. Atherosclerosis 2018, 268, 177–184. [Google Scholar] [CrossRef]
- Zhou, L.J.; Lin, W.Z.; Meng, X.Q.; Zhu, H.; Liu, T.; Du, L.J.; Bai, X.B.; Chen, B.Y.; Liu, Y.; Xu, Y.; et al. Periodontitis exacerbates atherosclerosis through Fusobacterium nucleatum-promoted hepatic glycolysis and lipogenesis. Cardiovasc. Res. 2023, 119, 1706–1717. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, L.; Wu, P.; Zhao, L.; Wu, Y. Fusobacterium nucleatum Accelerates Atherosclerosis via Macrophage-Driven Aberrant Proinflammatory Response and Lipid Metabolism. Front. Microbiol. 2022, 13, 798685. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Jun, H.K.; Kim, H.D.; Lee, S.H.; Choi, B.K. Fusobacterium nucleatum GroEL induces risk factors of atherosclerosis in human microvascular endothelial cells and ApoE(-/-) mice. Mol. Oral Microbiol. 2012, 27, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Farrugia, C.; Stafford, G.P.; Gains, A.F.; Cutts, A.R.; Murdoch, C. Fusobacterium nucleatum mediates endothelial damage and increased permeability following single species and polymicrobial infection. J. Periodontol. 2022, 93, 1421–1433. [Google Scholar] [CrossRef] [PubMed]
- Mendes, R.T.; Nguyen, D.; Stephens, D.; Pamuk, F.; Fernandes, D.; Van Dyke, T.E.; Kantarci, A. Endothelial Cell Response to Fusobacterium nucleatum. Infect. Immun. 2016, 84, 2141–2148. [Google Scholar] [CrossRef]
- Wang, Q.; Zhao, L.; Xu, C.; Zhou, J.; Wu, Y. Fusobacterium nucleatum stimulates monocyte adhesion to and transmigration through endothelial cells. Arch. Oral Biol. 2019, 100, 86–92. [Google Scholar] [CrossRef]
- Shen, S.; Sun, T.; Ding, X.; Gu, X.; Wang, Y.; Ma, X.; Li, Z.; Gao, H.; Ge, S.; Feng, Q. The exoprotein Gbp of Fusobacterium nucleatum promotes THP-1 cell lipid deposition by binding to CypA and activating PI3K-AKT/MAPK/NF-kappaB pathways. J. Adv. Res. 2023. [Google Scholar] [CrossRef]
- Slots, J.; Bragd, L.; Wikstrom, M.; Dahlen, G. The occurrence of Actinobacillus actinomycetemcomitans, Bacteroides gingivalis and Bacteroides intermedius in destructive periodontal disease in adults. J. Clin. Periodontol. 1986, 13, 570–577. [Google Scholar] [CrossRef]
- Tanner, A.C.; Haffer, C.; Bratthall, G.T.; Visconti, R.A.; Socransky, S.S. A study of the bacteria associated with advancing periodontitis in man. J. Clin. Periodontol. 1979, 6, 278–307. [Google Scholar] [CrossRef]
- Lie, M.A.; van der Weijden, G.A.; Timmerman, M.F.; Loos, B.G.; van Steenbergen, T.J.M.; van der Velden, U. Occurrence of Prevotella intermedia and Prevotella nigrescens in relation to gingivitis and gingival health. J. Clin. Periodontol. 2001, 28, 189–193. [Google Scholar] [CrossRef]
- Shah, H.N.; Gharbia, S.E. Biochemical and chemical studies on strains designated Prevotella intermedia and proposal of a new pigmented species, Prevotella nigrescens sp. nov. Int. J. Syst. Bacteriol. 1992, 42, 542–546. [Google Scholar] [CrossRef]
- Van der Velden, U.; Van Winkelhoff, A.J.; Abbas, F.; De Graaff, J. The habitat of periodontopathic micro-organisms. J. Clin. Periodontol. 1986, 13, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Van Winkelhoff, A.J.; Van der Velden, U.; Winkel, E.G.; de Graaff, J. Black-pigmented Bacteroides and motile organisms on oral mucosal surfaces in individuals with and without periodontal breakdown. J. Periodontal Res. 1986, 21, 434–439. [Google Scholar] [CrossRef]
- Gharbia, S.E.; Haapasalo, M.; Shah, H.N.; Kotiranta, A.; Lounatmaa, K.; Pearce, M.A.; Devine, D.A. Characterization of Prevotella intermedia and Prevotella nigrescens isolates from periodontic and endodontic infections. J. Periodontol. 1994, 65, 56–61. [Google Scholar] [CrossRef]
- Dahlen, G.; Wikstrom, M.; Renvert, S.; Gmur, R.; Guggenheim, B. Biochemical and serological characterization of Bacteroides intermedius strains isolated from the deep periodontal pocket. J. Clin. Microbiol. 1990, 28, 2269–2274. [Google Scholar] [CrossRef] [PubMed]
- Matto, J.; Saarela, M.; von Troil-Linden, B.; Kononen, E.; Jousimies-Somer, H.; Torkko, H.; Alaluusua, S.; Asikainen, S. Distribution and genetic analysis of oral Prevotella intermedia and Prevotella nigrescens. Oral Microbiol. Immunol. 1996, 11, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Lo, C.H.; Wu, D.C.; Jao, S.W.; Wu, C.C.; Lin, C.Y.; Chuang, C.H.; Lin, Y.B.; Chen, C.H.; Chen, Y.T.; Chen, J.H.; et al. Enrichment of Prevotella intermedia in human colorectal cancer and its additive effects with Fusobacterium nucleatum on the malignant transformation of colorectal adenomas. J. Biomed. Sci. 2022, 29, 88. [Google Scholar] [CrossRef]
- Dong, T.; Xu, S.; Chen, Z.Y.; Liang, Y.J.; Meng, X.Q.; Niu, C.G.; Yuan, K.Y.; Li, P.L.; Duan, S.Z.; Huang, Z.W. Prevotella intermedia Aggravates Subclinical Hypothyroidism. J. Dent. Res. 2023, 102, 814–824. [Google Scholar] [CrossRef]
- Boukobza, M.; Raffoul, R.; Duval, X.; Laissy, J.P. First report of prosthetic aortic valve Infective Endocarditis due to Prevotella Intermedia. Ann. Cardiol. Angeiol. 2022, 71, 240–242. [Google Scholar] [CrossRef]
- Akhi, R.; Wang, C.; Nissinen, A.E.; Kankaanpaa, J.; Bloigu, R.; Paju, S.; Mantyla, P.; Buhlin, K.; Sinisalo, J.; Pussinen, P.J.; et al. Salivary IgA to MAA-LDL and Oral Pathogens Are Linked to Coronary Disease. J. Dent. Res. 2019, 98, 296–303. [Google Scholar] [CrossRef]
- Yakob, M.; Soder, B.; Meurman, J.H.; Jogestrand, T.; Nowak, J.; Soder, P.O. Prevotella nigrescens and Porphyromonas gingivalis are associated with signs of carotid atherosclerosis in subjects with and without periodontitis. J. Periodontal Res. 2011, 46, 749–755. [Google Scholar] [CrossRef]
- Beck, J.D.; Eke, P.; Heiss, G.; Madianos, P.; Couper, D.; Lin, D.; Moss, K.; Elter, J.; Offenbacher, S. Periodontal disease and coronary heart disease: A reappraisal of the exposure. Circulation 2005, 112, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hosomi, N.; Aoki, S.; Matsuo, K.; Deguchi, K.; Masugata, H.; Murao, K.; Ichihara, N.; Ohyama, H.; Dobashi, H.; Nezu, T.; et al. Association of serum anti-periodontal pathogen antibody with ischemic stroke. Cerebrovasc. Dis. 2012, 34, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Asai, Y.; Tamai, R.; Jinno, T.; Umatani, K.; Ogawa, T. Chemical structure and immunobiological activity of lipid A from Prevotella intermedia ATCC 25611 lipopolysaccharide. Febs Lett. 2003, 543, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.Y.; Jin, J.Y.; Lee, J.Y.; Choi, J.I.; Choi, I.S.; Kim, S.J. Melatonin inhibits Prevotella intermedia lipopolysaccharide-induced production of nitric oxide and interleukin-6 in murine macrophages by suppressing NF-kappaB and STAT1 activity. J. Pineal Res. 2011, 50, 197–206. [Google Scholar] [CrossRef]
- Iki, K.; Kawahara, K.; Sawamura, S.; Arakaki, R.; Sakuta, T.; Sugiyama, A.; Tamura, H.; Sueda, T.; Hamada, S.; Takada, H. A novel component different from endotoxin extracted from Prevotella intermedia ATCC 25611 activates lymphoid cells from C3H/HeJ mice and gingival fibroblasts from humans. Infect. Immun. 1997, 65, 4531–4538. [Google Scholar] [CrossRef]
- Sugawara, S.; Yang, S.; Iki, K.; Hatakeyama, J.; Tamai, R.; Takeuchi, O.; Akashi, S.; Espevik, T.; Akira, S.; Takada, H. Monocytic cell activation by Nonendotoxic glycoprotein from Prevotella intermedia ATCC 25611 is mediated by toll-like receptor 2. Infect. Immun. 2001, 69, 4951–4957. [Google Scholar] [CrossRef]
- Dorn, B.R.; Dunn, W.A., Jr.; Progulske-Fox, A. Invasion of human coronary artery cells by periodontal pathogens. Infect. Immun. 1999, 67, 5792–5798. [Google Scholar] [CrossRef]
- Bodet, C.; Chandad, F.; Grenier, D. Inflammatory responses of a macrophage/epithelial cell co-culture model to mono and mixed infections with Porphyromonas gingivalis, Treponema denticola, and Tannerella forsythia. Microbes Infect. 2006, 8, 27–35. [Google Scholar] [CrossRef]
- Sekot, G.; Posch, G.; Messner, P.; Matejka, M.; Rausch-Fan, X.; Andrukhov, O.; Schaffer, C. Potential of the Tannerella forsythia S-layer to delay the immune response. J. Dent. Res. 2011, 90, 109–114. [Google Scholar] [CrossRef]
- Cecil, J.D.; O’Brien-Simpson, N.M.; Lenzo, J.C.; Holden, J.A.; Singleton, W.; Perez-Gonzalez, A.; Mansell, A.; Reynolds, E.C. Outer Membrane Vesicles Prime and Activate Macrophage Inflammasomes and Cytokine Secretion In Vitro and In Vivo. Front. Immunol. 2017, 8, 1017. [Google Scholar] [CrossRef]
- Lee, H.R.; Jun, H.K.; Choi, B.K. Tannerella forsythia BspA increases the risk factors for atherosclerosis in ApoE(-/-) mice. Oral Dis. 2014, 20, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, S.S.; Rivera-Kweh, M.F.; Velsko, I.M.; Chen, H.; Zheng, D.; Bhattacharyya, I.; Gangula, P.R.; Lucas, A.R.; Kesavalu, L. Chronic oral infection with major periodontal bacteria Tannerella forsythia modulates systemic atherosclerosis risk factors and inflammatory markers. Pathog. Dis. 2015, 73, ftv009. [Google Scholar] [CrossRef]
- Range, H.; Labreuche, J.; Louedec, L.; Rondeau, P.; Planesse, C.; Sebbag, U.; Bourdon, E.; Michel, J.B.; Bouchard, P.; Meilhac, O. Periodontal bacteria in human carotid atherothrombosis as a potential trigger for neutrophil activation. Atherosclerosis 2014, 236, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Okuda, T.; Kimizuka, R.; Miyamoto, M.; Kato, T.; Yamada, S.; Okuda, K.; Ishihara, K. Treponema denticola induces interleukin-8 and macrophage chemoattractant protein 1 production in human umbilical vein epithelial cells. Microbes Infect. 2007, 9, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Chukkapalli, S.S.; Rivera, M.F.; Velsko, I.M.; Lee, J.Y.; Chen, H.; Zheng, D.; Bhattacharyya, I.; Gangula, P.R.; Lucas, A.R.; Kesavalu, L. Invasion of oral and aortic tissues by oral spirochete Treponema denticola in ApoE(-/-) mice causally links periodontal disease and atherosclerosis. Infect. Immun. 2014, 82, 1959–1967. [Google Scholar] [CrossRef] [PubMed]
- Ardila, C.M.; Olarte-Sossa, M.; Ariza-Garces, A.A. Association between the presence of Treponema denticola and reduced levels of antiatherogenic high density lipoprotein in periodontitis. Quintessence Int. 2015, 46, 207–215. [Google Scholar] [CrossRef]
- Marchesan, J.; Jiao, Y.; Schaff, R.A.; Hao, J.; Morelli, T.; Kinney, J.S.; Gerow, E.; Sheridan, R.; Rodrigues, V.; Paster, B.J.; et al. TLR4, NOD1 and NOD2 mediate immune recognition of putative newly identified periodontal pathogens. Mol. Oral Microbiol. 2016, 31, 243–258. [Google Scholar] [CrossRef]
- Beck, J.D.; Eke, P.; Lin, D.; Madianos, P.; Couper, D.; Moss, K.; Elter, J.; Heiss, G.; Offenbacher, S. Associations between IgG antibody to oral organisms and carotid intima-medial thickness in community-dwelling adults. Atherosclerosis 2005, 183, 342–348. [Google Scholar] [CrossRef]
- Sakamoto, M.; Suzuki, M.; Umeda, M.; Ishikawa, I.; Benno, Y. Reclassification of Bacteroides forsythus (Tanner et al. 1986) as Tannerella forsythensis corrig., gen. nov., comb. nov. Int. J. Syst. Evol. Microbiol. 2002, 52, 841–849. [Google Scholar] [CrossRef]
- Maiden, M.F.; Cohee, P.; Tanner, A.C. Proposal to conserve the adjectival form of the specific epithet in the reclassification of Bacteroides forsythus Tanner et al. 1986 to the genus Tannerella Sakamoto et al. 2002 as Tannerella forsythia corrig., gen. nov., comb. nov. Request for an Opinion. Int. J. Syst. Evol. Microbiol. 2003, 53, 2111–2112. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X.; Li, N.; Shen, J.; Cheng, J. A propeptide-independent protease from Tannerella sp.6_1_58FAA_CT1 displays trypsin-like specificity. J. Basic. Microbiol. 2017, 57, 50–56. [Google Scholar] [CrossRef]
- Hamlet, S.M.; Taiyeb-Ali, T.B.; Cullinan, M.P.; Westerman, B.; Palmer, J.E.; Seymour, G.J. Tannerella forsythensis prtH genotype and association with periodontal status. J. Periodontol. 2007, 78, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Thompson, H.; Homer, K.A.; Rao, S.; Booth, V.; Hosie, A.H. An orthologue of Bacteroides fragilis NanH is the principal sialidase in Tannerella forsythia. J. Bacteriol. 2009, 191, 3623–3628. [Google Scholar] [CrossRef]
- Sharma, A.; Inagaki, S.; Honma, K.; Sfintescu, C.; Baker, P.J.; Evans, R.T. Tannerella forsythia-induced alveolar bone loss in mice involves leucine-rich-repeat BspA protein. J. Dent. Res. 2005, 84, 462–467. [Google Scholar] [CrossRef]
- Hughes, C.V.; Malki, G.; Loo, C.Y.; Tanner, A.C.; Ganeshkumar, N. Cloning and expression of alpha-D-glucosidase and N-acetyl-beta-glucosaminidase from the periodontal pathogen, Tannerella forsythensis (Bacteroides forsythus). Oral Microbiol. Immunol. 2003, 18, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Retamal, I.N.; Hernandez, R.; Gonzalez-Rivas, C.; Caceres, M.; Arancibia, R.; Romero, A.; Martinez, C.; Tobar, N.; Martinez, J.; Smith, P.C. Methylglyoxal and methylglyoxal-modified collagen as inducers of cellular injury in gingival connective tissue cells. J. Periodontal Res. 2016, 51, 812–821. [Google Scholar] [CrossRef]
- Settem, R.P.; Honma, K.; Nakajima, T.; Phansopa, C.; Roy, S.; Stafford, G.P.; Sharma, A. A bacterial glycan core linked to surface (S)-layer proteins modulates host immunity through Th17 suppression. Mucosal Immunol. 2013, 6, 415–426. [Google Scholar] [CrossRef]
- Tanabe, S.; Bodet, C.; Grenier, D. Treponema denticola lipooligosaccharide activates gingival fibroblasts and upregulates inflammatory mediator production. J. Cell. Physiol. 2008, 216, 727–731. [Google Scholar] [CrossRef]
- Ishihara, K. Virulence factors of Treponema denticola. Periodontol. 2000 2010, 54, 117–135. [Google Scholar] [CrossRef] [PubMed]
- Vandamme, P.; Falsen, E.; Rossau, R.; Hoste, B.; Segers, P.; Tytgat, R.; De Ley, J. Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: Emendation of generic descriptions and proposal of Arcobacter gen. nov. Int. J. Syst. Bacteriol. 1991, 41, 88–103. [Google Scholar] [CrossRef]
- Lam, J.Y.; Wu, A.K.; Ngai, D.C.; Teng, J.L.; Wong, E.S.; Lau, S.K.; Lee, R.A.; Woo, P.C. Three cases of severe invasive infections caused by Campylobacter rectus and first report of fatal C. rectus infection. J. Clin. Microbiol. 2011, 49, 1687–1691. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Jun, H.K.; Choi, B.K. Gingipain-dependent augmentation by Porphyromonas gingivalis of phagocytosis of Tannerella forsythia. Mol. Oral Microbiol. 2016, 31, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Inagaki, S.; Kimizuka, R.; Okuda, K.; Hosaka, Y.; Nakagawa, T.; Ishihara, K. Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol. Med. Microbiol. 2008, 54, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Grenier, D.; Mayrand, D. Nutritional relationships between oral bacteria. Infect. Immun. 1986, 53, 616–620. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.K.; Bhattacharyya, I.; Sevilla, A.; Lieberman, I.; Pola, S.; Nair, M.; Wallet, S.M.; Aukhil, I.; Kesavalu, L. Virulence of major periodontal pathogens and lack of humoral immune protection in a rat model of periodontal disease. Oral Dis. 2010, 16, 686–695. [Google Scholar] [CrossRef] [PubMed]
- Settem, R.P.; El-Hassan, A.T.; Honma, K.; Stafford, G.P.; Sharma, A. Fusobacterium nucleatum and Tannerella forsythia induce synergistic alveolar bone loss in a mouse periodontitis model. Infect. Immun. 2012, 80, 2436–2443. [Google Scholar] [CrossRef]
- Chukkapalli, S.S.; Velsko, I.M.; Rivera-Kweh, M.F.; Zheng, D.; Lucas, A.R.; Kesavalu, L. Polymicrobial Oral Infection with Four Periodontal Bacteria Orchestrates a Distinct Inflammatory Response and Atherosclerosis in ApoE null Mice. PLoS ONE 2015, 10, e0143291. [Google Scholar] [CrossRef] [PubMed]
- Velsko, I.M.; Chukkapalli, S.S.; Rivera-Kweh, M.F.; Zheng, D.; Aukhil, I.; Lucas, A.R.; Larjava, H.; Kesavalu, L. Periodontal pathogens invade gingiva and aortic adventitia and elicit inflammasome activation in alphavbeta6 integrin-deficient mice. Infect. Immun. 2015, 83, 4582–4593. [Google Scholar] [CrossRef]
- Tan, B.K.; Chalouni, M.; Ceron, D.S.; Cinaud, A.; Esterle, L.; Loko, M.A.; Katlama, C.; Poizot-Martin, I.; Neau, D.; Chas, J.; et al. Atherosclerotic Cardiovascular Events in Patients Infected With Human Immunodeficiency Virus and Hepatitis C Virus. Clin. Infect. Dis. 2021, 72, e215–e223. [Google Scholar] [CrossRef]
- Genebat, M.; Tarancon-Diez, L.; Pulido, I.; Alvarez-Rios, A.I.; Munoz-Fernandez, M.A.; Ruiz-Mateos, E.; Leal, M. Hepatitis C virus and cumulative infections are associated with atherogenic cardiovascular events in HIV-infected subjects. Antiviral Res. 2019, 169, 104527. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Hwang, B.O.; Lim, M.; Ok, S.H.; Lee, S.K.; Chun, K.S.; Park, K.K.; Hu, Y.; Chung, W.Y.; Song, N.Y. Oral-Gut Microbiome Axis in Gastrointestinal Disease and Cancer. Cancers 2021, 13, 2124. [Google Scholar] [CrossRef] [PubMed]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed]
- Kitamoto, S.; Nagao-Kitamoto, H.; Jiao, Y.; Gillilland, M.G., 3rd; Hayashi, A.; Imai, J.; Sugihara, K.; Miyoshi, M.; Brazil, J.C.; Kuffa, P.; et al. The Intermucosal Connection between the Mouth and Gut in Commensal Pathobiont-Driven Colitis. Cell 2020, 182, 447–462.e14. [Google Scholar] [CrossRef]
- Chen, B.Y.; Lin, W.Z.; Li, Y.L.; Bi, C.; Du, L.J.; Liu, Y.; Zhou, L.J.; Liu, T.; Xu, S.; Shi, C.J.; et al. Roles of oral microbiota and oral-gut microbial transmission in hypertension. J. Adv. Res. 2023, 43, 147–161. [Google Scholar] [CrossRef]
- Acharya, C.; Sahingur, S.E.; Bajaj, J.S. Microbiota, cirrhosis, and the emerging oral-gut-liver axis. JCI Insight 2017, 2, e94416. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, S.; Huang, Y.; Qian, J.; Tan, B.; Qian, X.; Zhuang, J.; Zou, X.; Li, Y.; Yan, F. Periodontitis-related salivary microbiota aggravates Alzheimer’s disease via gut-brain axis crosstalk. Gut Microbes 2022, 14, 2126272. [Google Scholar] [CrossRef]
- Koren, O.; Spor, A.; Felin, J.; Fak, F.; Stombaugh, J.; Tremaroli, V.; Behre, C.J.; Knight, R.; Fagerberg, B.; Ley, R.E.; et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4592–4598. [Google Scholar] [CrossRef]
- Jonsson, A.L.; Backhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 79–87. [Google Scholar] [CrossRef]
- Ma, S.R.; Tong, Q.; Lin, Y.; Pan, L.B.; Fu, J.; Peng, R.; Zhang, X.F.; Zhao, Z.X.; Li, Y.; Yu, J.B.; et al. Berberine treats atherosclerosis via a vitamine-like effect down-regulating Choline-TMA-TMAO production pathway in gut microbiota. Signal. Transduct. Target. Ther. 2022, 7, 207. [Google Scholar] [CrossRef]
- Haghikia, A.; Zimmermann, F.; Schumann, P.; Jasina, A.; Roessler, J.; Schmidt, D.; Heinze, P.; Kaisler, J.; Nageswaran, V.; Aigner, A.; et al. Propionate attenuates atherosclerosis by immune-dependent regulation of intestinal cholesterol metabolism. Eur. Heart J. 2022, 43, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.; Lu, B.; Zhang, R.; Luo, Y.; Chen, S.; Lei, H.; Li, Y.; Cai, Z.; Huang, X. Chronic apical periodontitis exacerbates atherosclerosis in apolipoprotein E-deficient mice and leads to changes in the diversity of gut microbiota. Int. Endod. J. 2022, 55, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.; Zhang, R.; Lu, B.; Luo, Y.; Chen, S.; Lei, H.; Li, Y.; Cai, Z.; Huang, X. Gut microbiota may mediate the impact of chronic apical periodontitis on atherosclerosis in apolipoprotein E-deficient mice. Int. Endod. J. 2023, 56, 53–68. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeting Cell | Effection on Cells | Pathways | Association with AS of Mice | Association with AS of Human |
|---|---|---|---|---|
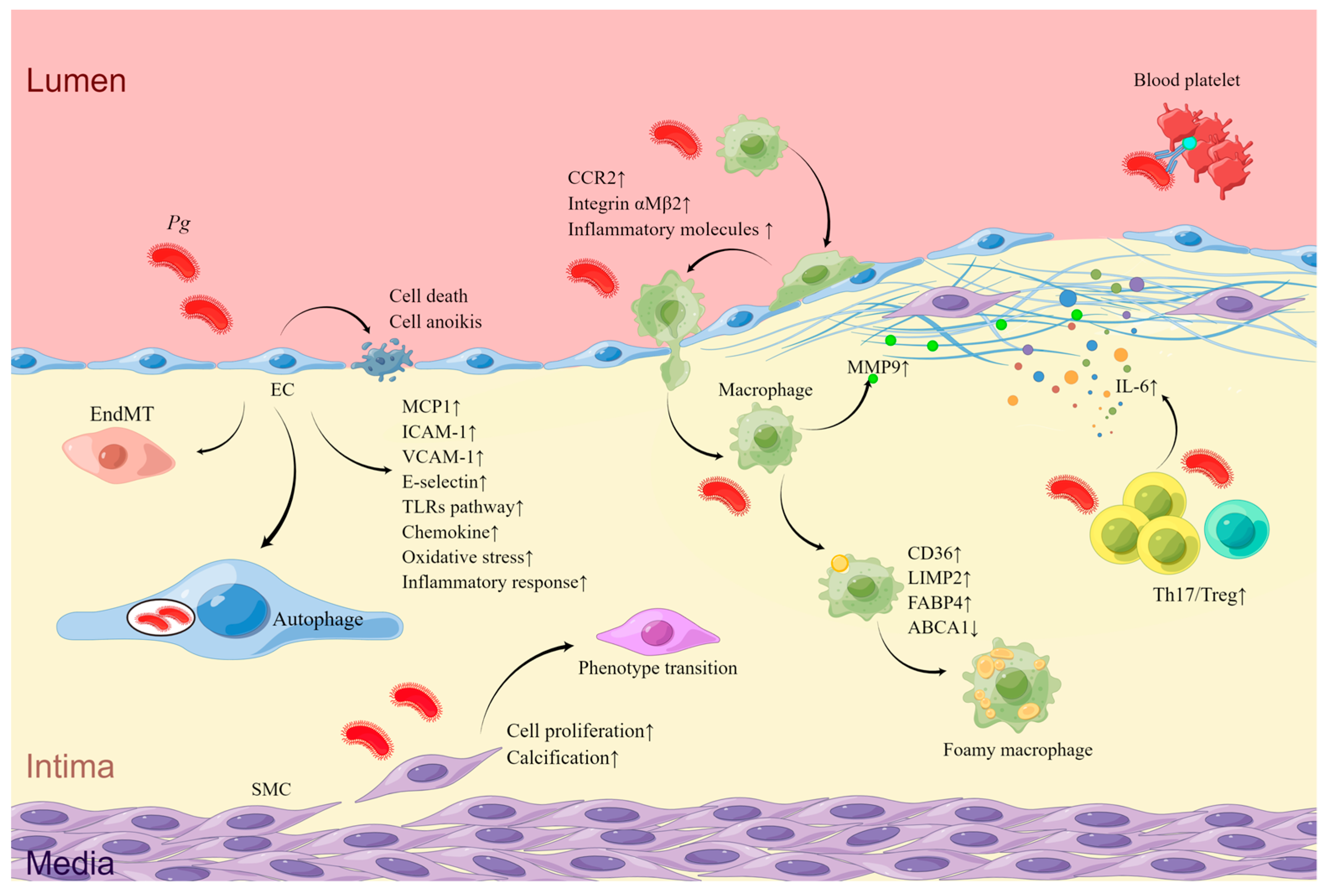
| ECs | Upregulation of adhesion factors and chemokine | TLRs signaling pathway [70,71,72]; NF-κB pathway [83,87,88]; MIF-CD47-CXCR4 receptor–ligand complex [90] | Plaque growth [59,60,61,62]; increased serum IgG against Pg and systemic pro-inflammatory cytokines [60,65]; plaque instability [58,59]; cardiac rupture [66] | High detective rate in AS plaques [45,51,52,53]; high serum IgG to Pg and IgG to Pg HSP60 [55]; more serious carotid stenosis for type 2 DB with more serum anti-Pg IgG [56]; patients; abdominal aortic aneurysm [57]; stroke [58] |
| Pg intracellular survival and immune escape | Autophagy [73,74] | |||
| TNF-mediated cell death | Kgp initiates proteolysis of RIPK1, RIPK2, and PARP in ECs [75] | |||
| Suppression of cell proliferation | [76] | |||
| Cell anoikis | Neural cadherin and vascular endothelial cadherin cleavage and integrin β1 degradation by gingipains [77,78] | |||
| EndMT | Inflammation-induced by Pg [65] | |||
| Oxidative stress and inflammatory response | TLRs-NF-κB-Bmal1- NF-κB pathway [46]; NOS/BH4/Nrf2/GSK-3β pathway [81]; mitochondrial fission and dysfunction via phosphorylation and recruitment of Drp1 [82] | |||
| Macrophages | Adhesion and aggregation to the endothelium | Through CCR2 and integrin αMβ2 [83,84,85,86,87] | ||
| Release of inflammation molecules | [65,91,92] | |||
| Lipid accumulation | modification of LDL [93] Upregulation of CD36 via the c-Jun-AP-1 pathway [94] or ERK/NF-κB [95] Upregulation of LIMP2 [96,97] and FABP4 [98] Degradation of ABCA1 [94]; oxidation of HDL [99] | |||
| MMP9 activation | [109] | |||
| VSMCs | Proliferation | TGF-β and Notch signaling pathways and so on [103]; upregulation of the Angpt2/Angpt1 ratio [107] | ||
| Differentiation and calcification | Upregulation of Runx2 via ERK1/2 signaling by OMV of Pg [104] | |||
| Phenotype transition | Upregulation of the Angpt2/Angpt1 ratio [100,106,107] | |||
| Th17 and Treg cells | Th17/Treg imbalance | Upregulation of IL-6 [63] | ||
| Platelets | Aggregation | Interaction between Hgp44 of Pg, reactive IgG, Fc γRIIa receptors, and GPIb α receptors on platelet surface [111] |
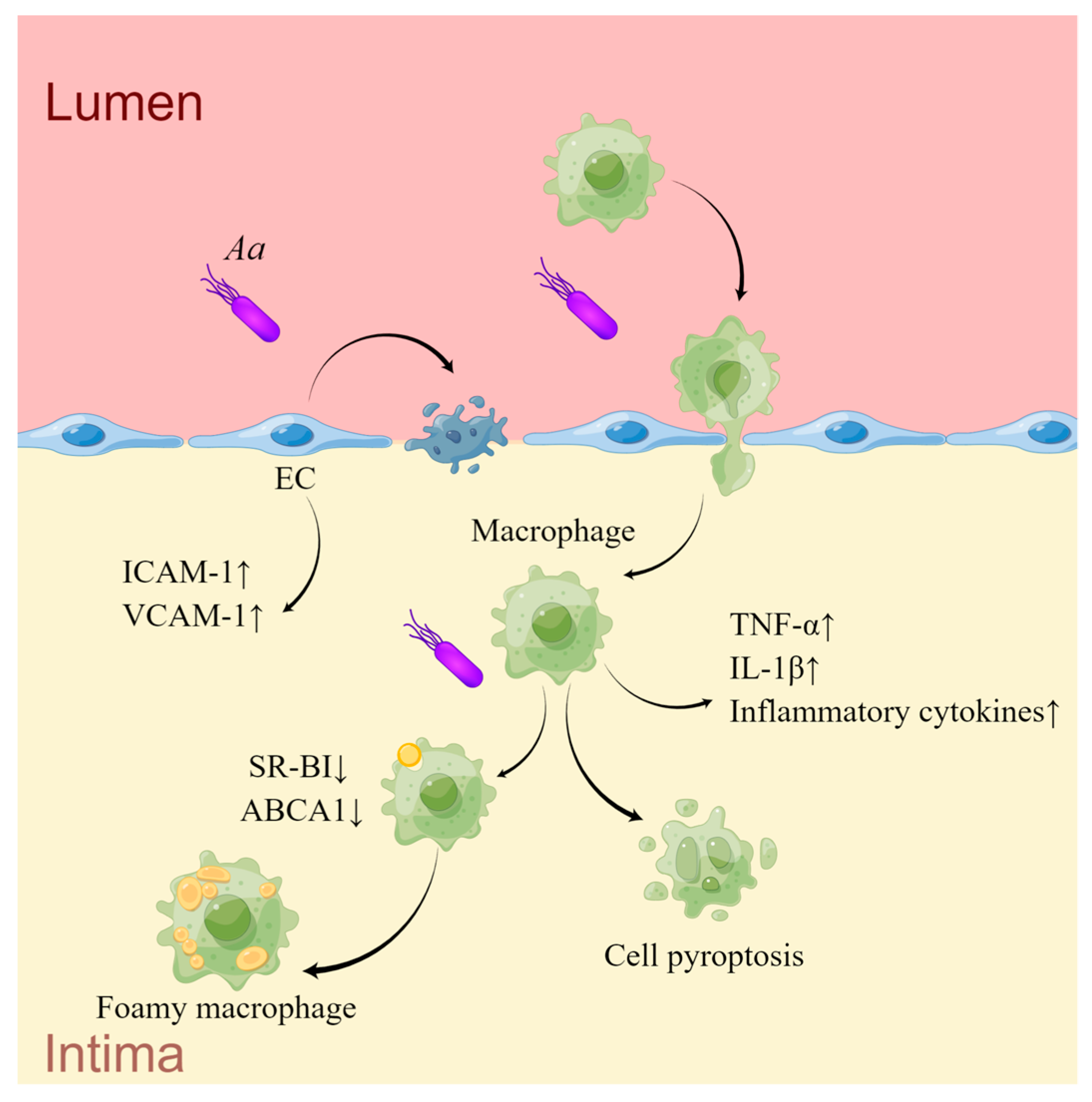
| Targeting Cell | Effection on Cells | Pathways | Association with AS of Mice | Association with AS of Human |
|---|---|---|---|---|
| ECs | Upregulation of ICAM-1 and VCAM-1; | [135] | Plaque lipid deposition [128]; proatherogenic lipoprotein profiles [129]; increased serum ox-LDL and oxidative stress [128]; elevated serum inflammatory factors and chemokines [130], upregulation of innate immune signaling molecules, adhesion molecules, chemokines, LOX-1, and hsCRP in the aorta [130] | A 55.5% positivity in plaques among elderly AS patients [118]; higher coronary artery calcification, thicker carotid IMT, and lower ABI [119]; ruptured cerebral aneurysms [120]; stable CAD [121]; ACS [121] |
| suppression of cell proliferation; promotion of cell apoptosis | LtxA arrests G2/M phase of cell cycle [135] | |||
| Monocytes | Pyroptosis, release of inflammatory cytokines | LtxA of Aa binds with LFA-1 and activates the inflammasome [134] | ||
| aggregation and adhesion | Upregulation of ICAM-1 [136,137] | |||
| Release of inflammatory cytokines, increased cholesterol accumulation | Downregulation of SR-BI and ABCA1 [138] |
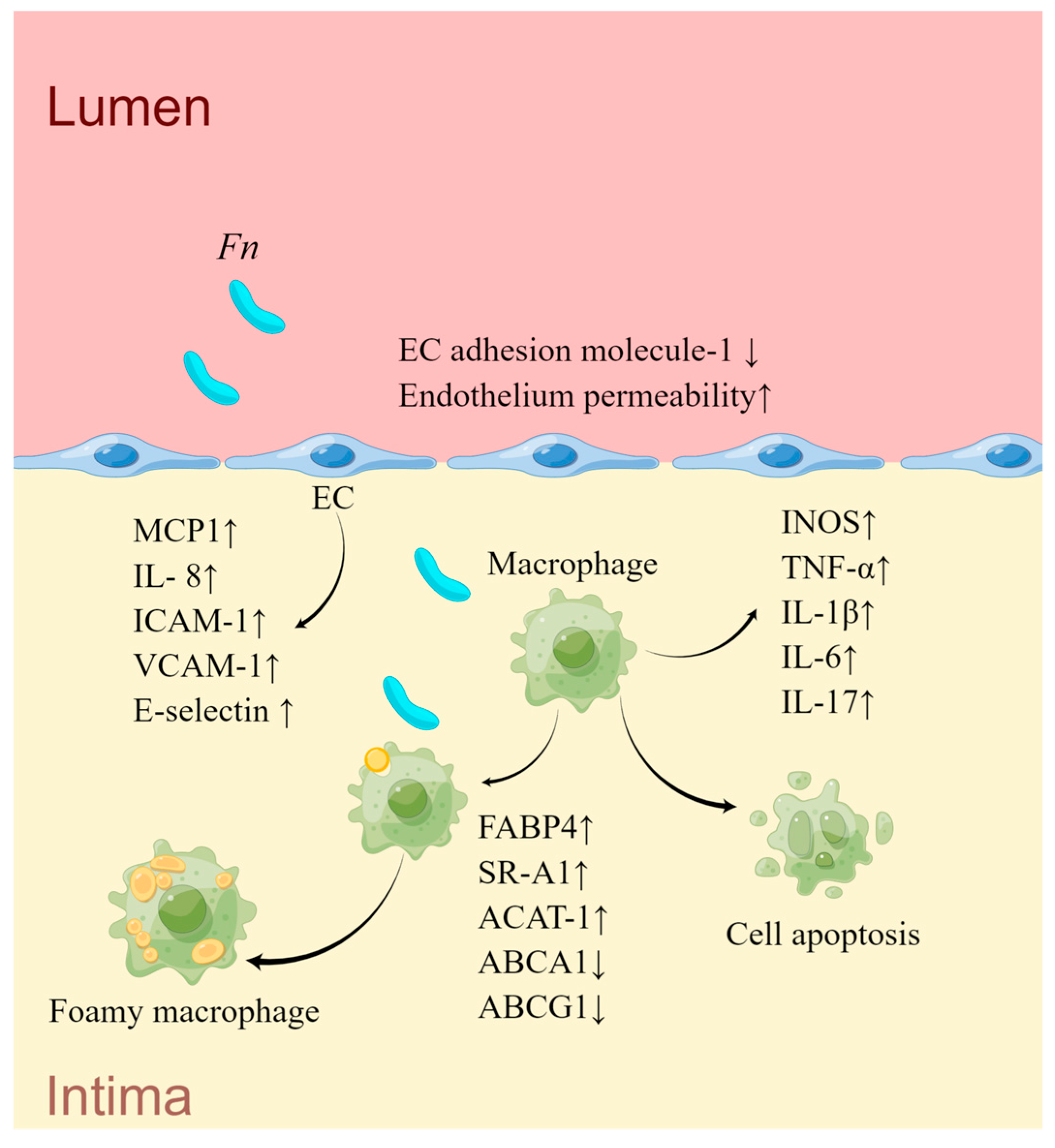
| Targeting Cell | Effection on Cells | Pathways | Association with AS of Mice | Association with AS of Human |
|---|---|---|---|---|
| ECs | Increased permeability Impaired proliferation and induced apoptosis | Reduction of EC adhesion molecule-1 [153] [154,155] | Increased plasma TG and cholesterol levels [150,151]; elevated serum levels of IL-6, CRP, and LDL, decreased levels of HDL [152] | A positivity rate of 34% in carotid endarterectomy specimens [147]; ruptured cerebral aneurysms [120]; stable CAD [149] |
| Upregulated chemotactic factors and cell adhesion molecules | [152] | |||
| Hepatic cells | Glycolysis and lipid synthesis | PI3K/Akt/mTOR signaling pathway [150] | ||
| Monocytes | Inflammatory responses | PI3K-AKT/MAPK/NF-κB signaling pathway [156] | ||
| M1 polarization; cell apoptosis cholesterol accumulation | modulation of lipid metabolism-related genes including SR-A1, ACAT-1, ABCA1 and ABCG1 [151]; upregulation of FABP4 via a JNK-dependent mechanism [98] |
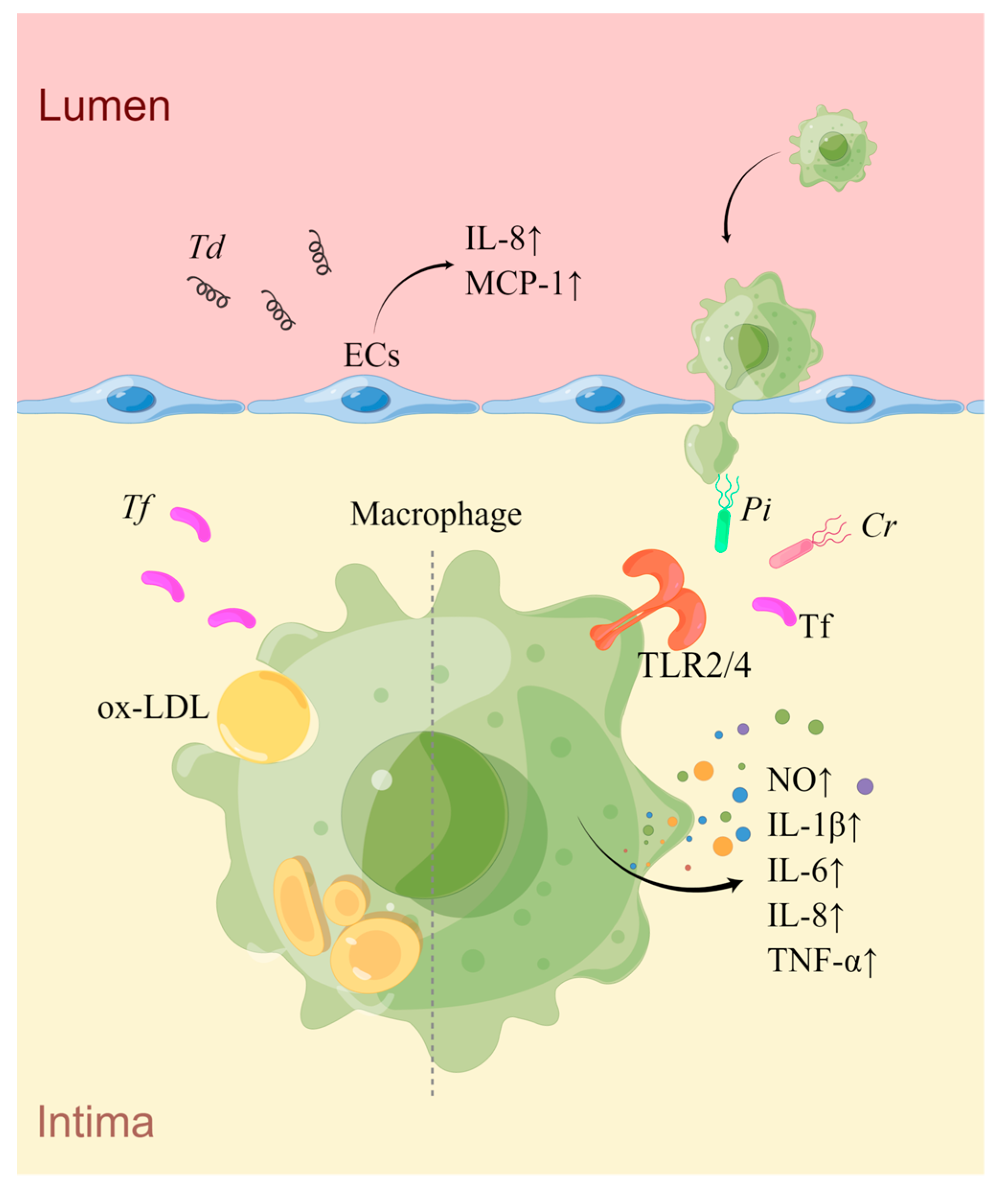
| Bacteria | Targeting Cell | Effection on Cells | Pathways | Association with AS of Mice | Association with AS of Human |
|---|---|---|---|---|---|
| Pi | Macrophages | Release of inflammatory cytokines | TLR4 signaling pathway [174]; | - | Stable CAD and ACS [169]; thicker intimamedia [170]; CHD in smokers [171]; stroke [172] |
| IL-8 production | CD14 and TLR2 signaling pathway activated by PCG of Pi [176] | ||||
| Tf | Macrophages | Release of pro-inflammatory mediators | [178,179,180] | Plaque enlargement, increased serum levels of CRP and LDL, decreased serum level of HDL, and expression of cholesterol efflux-related gene expression in liver [181]; lowered serum NO level and increased SAA [182]; intraplaque hemorrhage [183] | CAD and ACS [169] |
| Foam cell formation | [181] | ||||
| Td | ECs | Facilitating chemotaxis and aggregation of monocytes to the subendothelium | Upregulation of IL-8 and MCP-1 [184] | Enlarged arterial plaques, decreased serum NO levels, and increased serum VLDL and ox-LDL levels [185] | CHD in ever smokers [171]; elevated levels of TG and reduced levels of HDL [186] |
| Cr | Macrophages | IL-6 secretion | TLR4 signaling pathway [187] | - | Stable CAD and ACS [149]; thick carotid arterial walls [188] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, X.; Xie, M.; Lu, X.; Mei, F.; Song, W.; Liu, Y.; Chen, L. The Roles of Periodontal Bacteria in Atherosclerosis. Int. J. Mol. Sci. 2023, 24, 12861. https://doi.org/10.3390/ijms241612861
Huang X, Xie M, Lu X, Mei F, Song W, Liu Y, Chen L. The Roles of Periodontal Bacteria in Atherosclerosis. International Journal of Molecular Sciences. 2023; 24(16):12861. https://doi.org/10.3390/ijms241612861
Chicago/Turabian StyleHuang, Xiaofei, Mengru Xie, Xiaofeng Lu, Feng Mei, Wencheng Song, Yang Liu, and Lili Chen. 2023. "The Roles of Periodontal Bacteria in Atherosclerosis" International Journal of Molecular Sciences 24, no. 16: 12861. https://doi.org/10.3390/ijms241612861
APA StyleHuang, X., Xie, M., Lu, X., Mei, F., Song, W., Liu, Y., & Chen, L. (2023). The Roles of Periodontal Bacteria in Atherosclerosis. International Journal of Molecular Sciences, 24(16), 12861. https://doi.org/10.3390/ijms241612861