
MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis



 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
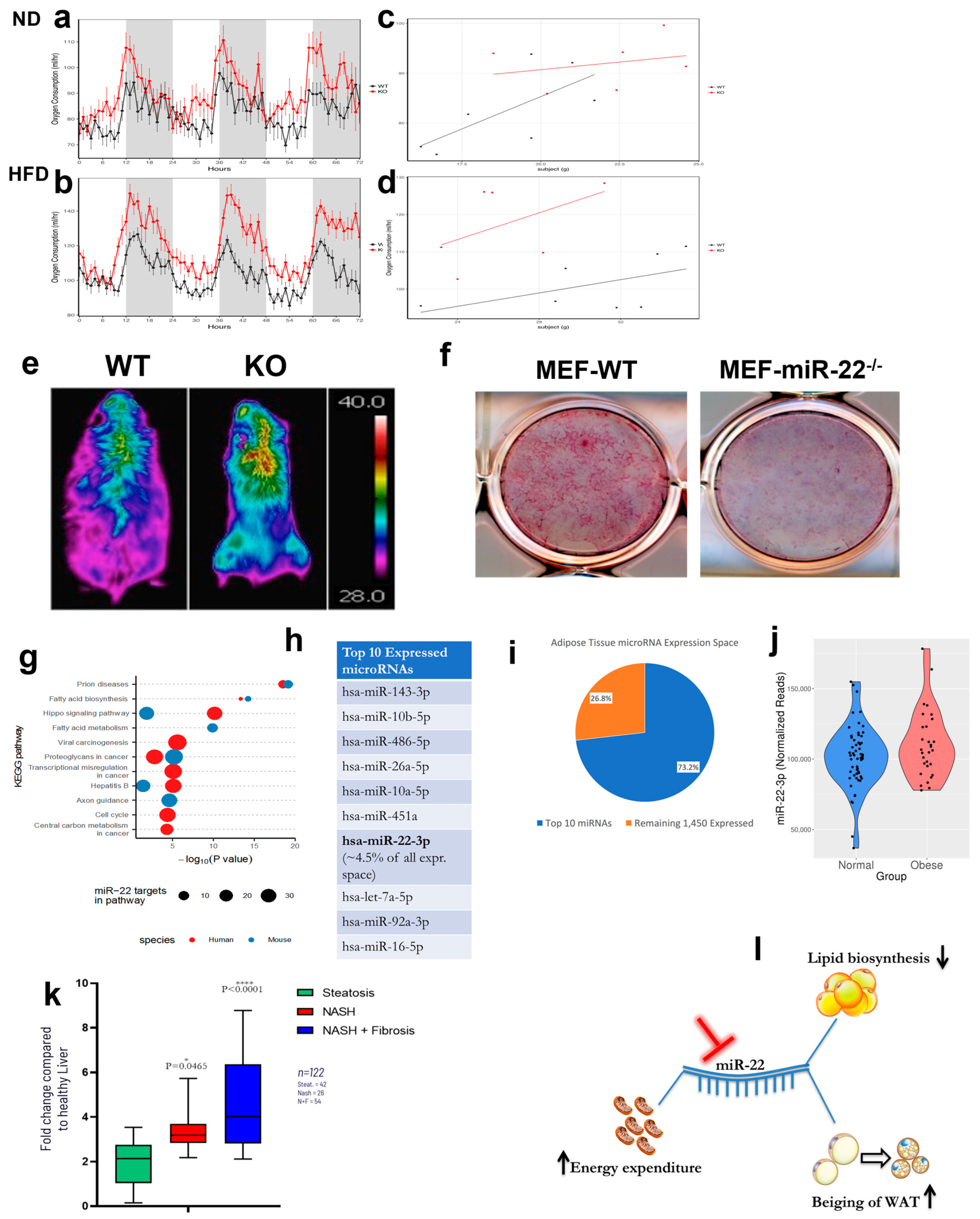
2. Results
3. Discussion
4. Materials and Methods
4.1. Data Analysis
4.2. Mouse Models
4.3. Indirect Calorimetry, Body Composition and Thermal Imaging
4.4. Tissue Harvesting and Manipulation
4.5. H&E and HIC Staining
4.6. Statistical Analysis
4.7. Isolation and Manipulation of Primary Mouse Cells
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of Obesity among Adults and Youth: United States, 2015–2016; NCHS Data Brief No. 288; United States Department of Health and Human Services: Washington, DC, USA, 2017.
- GHO Database. Wordwire Obesity Incidence. GHO Database. 2017. Available online: https://www.who.int/data/gho (accessed on 1 December 2018).
- Rice, G.M.; Steiner, R.D. Inborn Errors of Metabolism (Metabolic Disorders). Pediatr. Rev. 2016, 37, 3–15, quiz 16–17, 47. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.G. Prader-Willi Syndrome: Obesity due to Genomic Imprinting. Curr. Genom. 2011, 12, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Esteve Rafols, M. Adipose tissue: Cell heterogeneity and functional diversity. Endocrinol. Nutr. 2014, 61, 100–112. [Google Scholar] [CrossRef] [PubMed]
- Fenzl, A.; Kiefer, F.W. Brown adipose tissue and thermogenesis. Horm. Mol. Biol. Clin. Investig. 2014, 19, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target recognition and regulatory functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Song, S.J.; Ito, K.; Ala, U.; Kats, L.; Webster, K.; Sun, S.M.; Jongen-Lavrencic, M.; Manova-Todorova, K.; Teruya-Feldstein, J.; Avigan, D.E.; et al. The oncogenic microRNA miR-22 targets the TET2 tumor suppressor to promote hematopoietic stem cell self-renewal and transformation. Cell Stem Cell 2013, 13, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Song, S.J.; Poliseno, L.; Song, M.S.; Ala, U.; Webster, K.; Ng, C.; Beringer, G.; Brikbak, N.J.; Yuan, X.; Cantley, L.C.; et al. MicroRNA-antagonism regulates breast cancer stemness and metastasis via TET-family-dependent chromatin remodeling. Cell 2013, 154, 311–324. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.P.; Ilter, D.; Low, V.; Drapela, S.; Schild, T.; Mullarky, E.; Han, J.; Elia, I.; Broekaert, D.; Rosenzweig, A.; et al. Altered propionate metabolism contributes to tumour progression and aggressiveness. Nat. Metab. 2022, 4, 435–443. [Google Scholar] [CrossRef]
- Bergers, G.; Fendt, S.M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 2021, 21, 162–180. [Google Scholar] [CrossRef]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef]
- Chen, M.; Zhang, J.; Sampieri, K.; Clohessy, J.G.; Mendez, L.; Gonzalez-Billalabeitia, E.; Liu, X.S.; Lee, Y.R.; Fung, J.; Katon, J.M.; et al. An aberrant SREBP-dependent lipogenic program promotes metastatic prostate cancer. Nat. Genet. 2018, 50, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Thibonnier, M.; Esau, C. Metabolic Benefits of MicroRNA-22 Inhibition. Nucleic Acid Ther. 2020, 30, 104–116. [Google Scholar] [CrossRef]
- Thibonnier, M.; Esau, C.; Ghosh, S.; Wargent, E.; Stocker, C. Metabolic and energetic benefits of microRNA-22 inhibition. BMJ Open Diabetes Res. Care 2020, 8, e001478. [Google Scholar] [CrossRef] [PubMed]
- Karsenty, G.; Park, R.W. Regulation of type I collagen genes expression. Int. Rev. Immunol. 1995, 12, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Rossert, J.; Terraz, C.; Dupont, S. Regulation of type I collagen genes expression. Nephrol. Dial. Transplant. 2000, 15 (Suppl. S6), 66–68. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.L.; Liska, D.J.; Bornstein, P. Regulation of expression of the type I collagen genes. Am. J. Med. Genet. 1993, 45, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Mina, A.I.; LeClair, R.A.; LeClair, K.B.; Cohen, D.E.; Lantier, L.; Banks, A.S. CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell Metab. 2018, 28, 656–666.e651. [Google Scholar] [CrossRef] [PubMed]
- Civelek, M.; Hagopian, R.; Pan, C.; Che, N.; Yang, W.P.; Kayne, P.S.; Saleem, N.K.; Cederberg, H.; Kuusisto, J.; Gargalovic, P.S.; et al. Genetic regulation of human adipose microRNA expression and its consequences for metabolic traits. Hum. Mol. Genet. 2013, 22, 3023–3037. [Google Scholar] [CrossRef]
- Chen, M.; Wan, L.; Zhang, J.; Zhang, J.; Mendez, L.; Clohessy, J.G.; Berry, K.; Victor, J.; Yin, Q.; Zhu, Y.; et al. Deregulated PP1alpha phosphatase activity towards MAPK activation is antagonized by a tumor suppressive failsafe mechanism. Nat. Commun. 2018, 9, 159. [Google Scholar] [CrossRef]
- Moore, P.W.; Malone, K.; VanValkenburg, D.; Rando, L.L.; Williams, B.C.; Matejowsky, H.G.; Ahmadzadeh, S.; Shekoohi, S.; Cornett, E.M.; Kaye, A.D. GLP-1 Agonists for Weight Loss: Pharmacology and Clinical Implications. Adv. Ther. 2022, 40, 723–742. [Google Scholar] [CrossRef]
- Pories, W.J. Bariatric surgery: Risks and rewards. J. Clin. Endocrinol. Metab. 2008, 93, S89–S96. [Google Scholar] [CrossRef] [PubMed]
- Arterburn, D.E.; Telem, D.A.; Kushner, R.F.; Courcoulas, A.P. Benefits and Risks of Bariatric Surgery in Adults: A Review. JAMA 2020, 324, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Beekley, A.; Johnson, D.C.; Davis, K.A. Early and late complications of bariatric operation. Trauma. Surg. Acute Care Open 2018, 3, e000219. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Wang, M.; Wen, Z.; Lu, Z.; Cui, L.; Fu, C.; Xue, H.; Liu, Y.; Zhang, Y. GLP-1 Receptor Agonists: Beyond Their Pancreatic Effects. Front. Endocrinol. 2021, 12, 721135. [Google Scholar] [CrossRef] [PubMed]
- Filippatos, T.D.; Panagiotopoulou, T.V.; Elisaf, M.S. Adverse Effects of GLP-1 Receptor Agonists. Rev. Diabet. Stud. 2014, 11, 202–230. [Google Scholar] [CrossRef] [PubMed]
- Coskun, T.; Urva, S.; Roell, W.C.; Qu, H.; Loghin, C.; Moyers, J.S.; O’Farrell, L.S.; Briere, D.A.; Sloop, K.W.; Thomas, M.K.; et al. LY3437943, a novel triple glucagon, GIP, and GLP-1 receptor agonist for glycemic control and weight loss: From discovery to clinical proof of concept. Cell Metab. 2022, 34, 1234–1247.e1239. [Google Scholar] [CrossRef]
- Somm, E.; Montandon, S.A.; Loizides-Mangold, U.; Gaia, N.; Lazarevic, V.; De Vito, C.; Perroud, E.; Bochaton-Piallat, M.L.; Dibner, C.; Schrenzel, J.; et al. The GLP-1R agonist liraglutide limits hepatic lipotoxicity and inflammatory response in mice fed a methionine-choline deficient diet. Transl. Res. 2021, 227, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Storey, J.D. The positive false discovery rate: A Bayesian interpretation and the q-value. Ann. Stat. 2003, 31, 2013–2035. [Google Scholar] [CrossRef]
- Ritchie, M.E.; Phipson, B.; Wu, D.; Hu, Y.; Law, C.W.; Shi, W.; Smyth, G.K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015, 43, e47. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef]
- Huang, Z.P.; Chen, J.; Seok, H.Y.; Zhang, Z.; Kataoka, M.; Hu, X.; Wang, D.Z. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ. Res. 2013, 112, 1234–1243. [Google Scholar] [CrossRef]
- Douris, N.; Desai, B.N.; Fisher, F.M.; Cisu, T.; Fowler, A.J.; Zarebidaki, E.; Nguyen, N.L.T.; Morgan, D.A.; Bartness, T.J.; Rahmouni, K.; et al. Beta-adrenergic receptors are critical for weight loss but not for other metabolic adaptations to the consumption of a ketogenic diet in male mice. Mol. Metab. 2017, 6, 854–862. [Google Scholar] [CrossRef]
- Crane, J.D.; Mottillo, E.P.; Farncombe, T.H.; Morrison, K.M.; Steinberg, G.R. A standardized infrared imaging technique that specifically detects UCP1-mediated thermogenesis in vivo. Mol. Metab. 2014, 3, 490–494. [Google Scholar] [CrossRef]
- Guarnerio, J.; Riccardi, L.; Taulli, R.; Maeda, T.; Wang, G.; Hobbs, R.M.; Song, M.S.; Sportoletti, P.; Bernardi, R.; Bronson, R.T.; et al. A genetic platform to model sarcomagenesis from primary adult mesenchymal stem cells. Cancer Discov. 2015, 5, 396–409. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panella, R.; Petri, A.; Desai, B.N.; Fagoonee, S.; Cotton, C.A.; Nguyen, P.K.; Lundin, E.M.; Wagshal, A.; Wang, D.-Z.; Näär, A.M.; et al. MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis. Int. J. Mol. Sci. 2023, 24, 12870. https://doi.org/10.3390/ijms241612870
Panella R, Petri A, Desai BN, Fagoonee S, Cotton CA, Nguyen PK, Lundin EM, Wagshal A, Wang D-Z, Näär AM, et al. MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis. International Journal of Molecular Sciences. 2023; 24(16):12870. https://doi.org/10.3390/ijms241612870
Chicago/Turabian StylePanella, Riccardo, Andreas Petri, Bhavna N. Desai, Sharmila Fagoonee, Cody A. Cotton, Piercen K. Nguyen, Eric M. Lundin, Alexandre Wagshal, Da-Zhi Wang, Anders M. Näär, and et al. 2023. "MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis" International Journal of Molecular Sciences 24, no. 16: 12870. https://doi.org/10.3390/ijms241612870
APA StylePanella, R., Petri, A., Desai, B. N., Fagoonee, S., Cotton, C. A., Nguyen, P. K., Lundin, E. M., Wagshal, A., Wang, D.-Z., Näär, A. M., Vlachos, I. S., Maratos-Flier, E., Altruda, F., Kauppinen, S., & Paolo Pandolfi, P. (2023). MicroRNA-22 Is a Key Regulator of Lipid and Metabolic Homeostasis. International Journal of Molecular Sciences, 24(16), 12870. https://doi.org/10.3390/ijms241612870