
Gene Immunotherapy of Colon Carcinoma with IL-2 and IL-12 Using Gene Electrotransfer


 , , , ,
, , , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
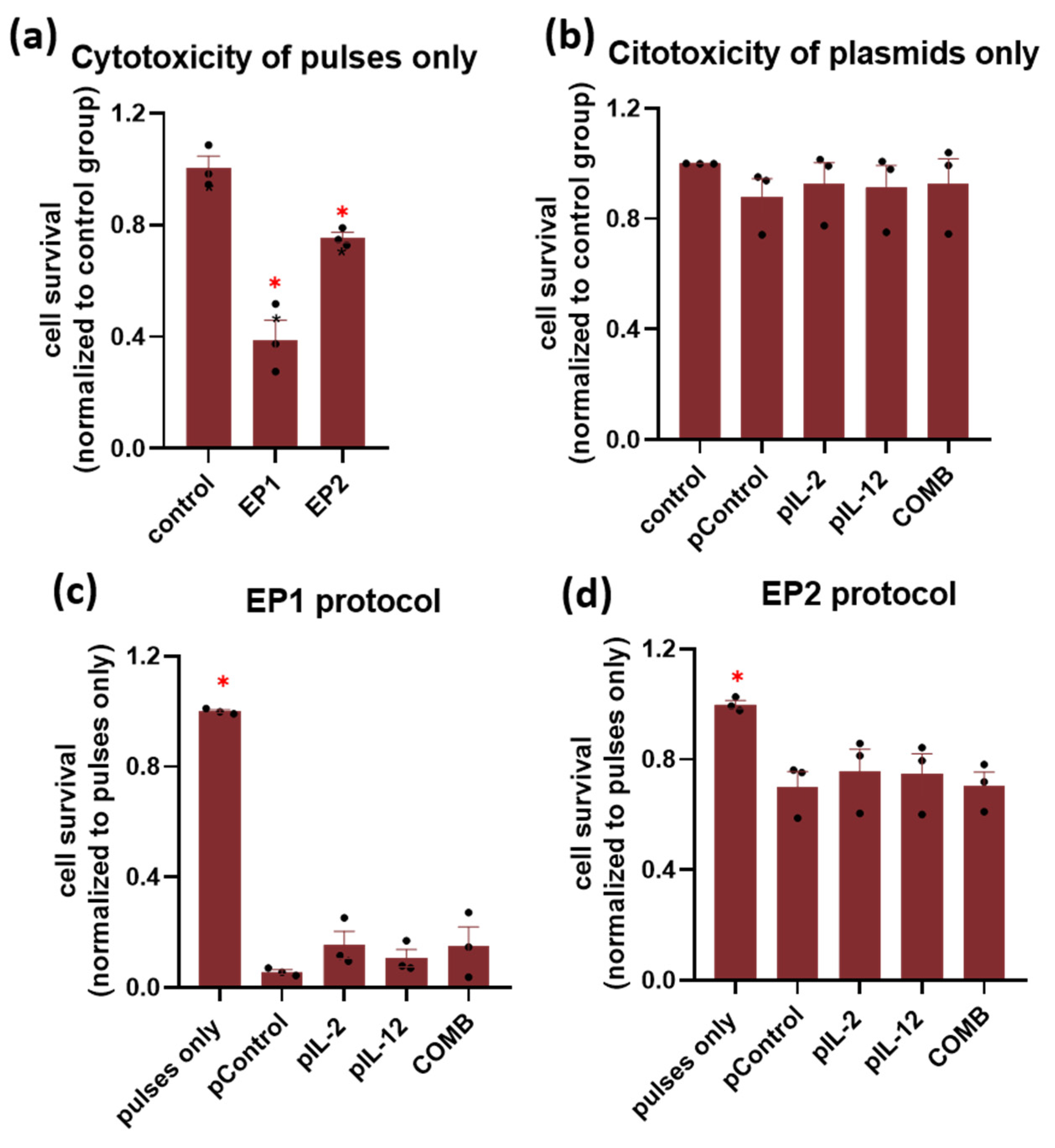
2.1. In Vitro Survival after Electroporation
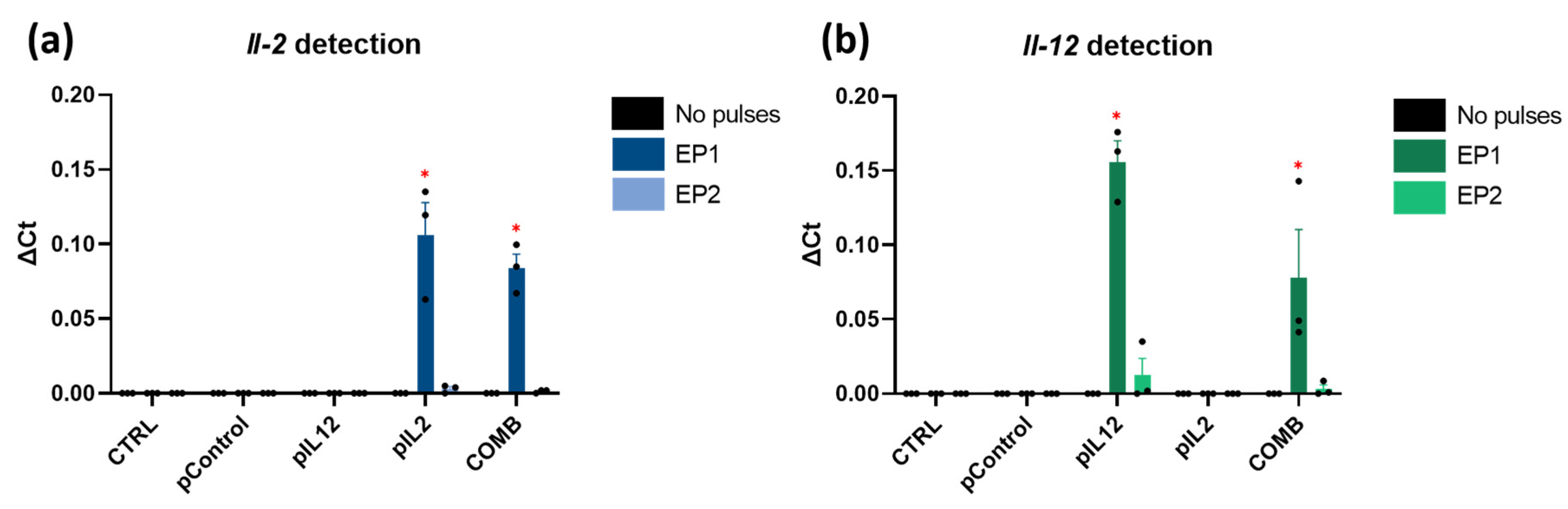
2.2. In Vitro Il-2 and Il-12 Transcripts Expression
2.3. In Vitro IL-2 and IL-12 Protein Expression
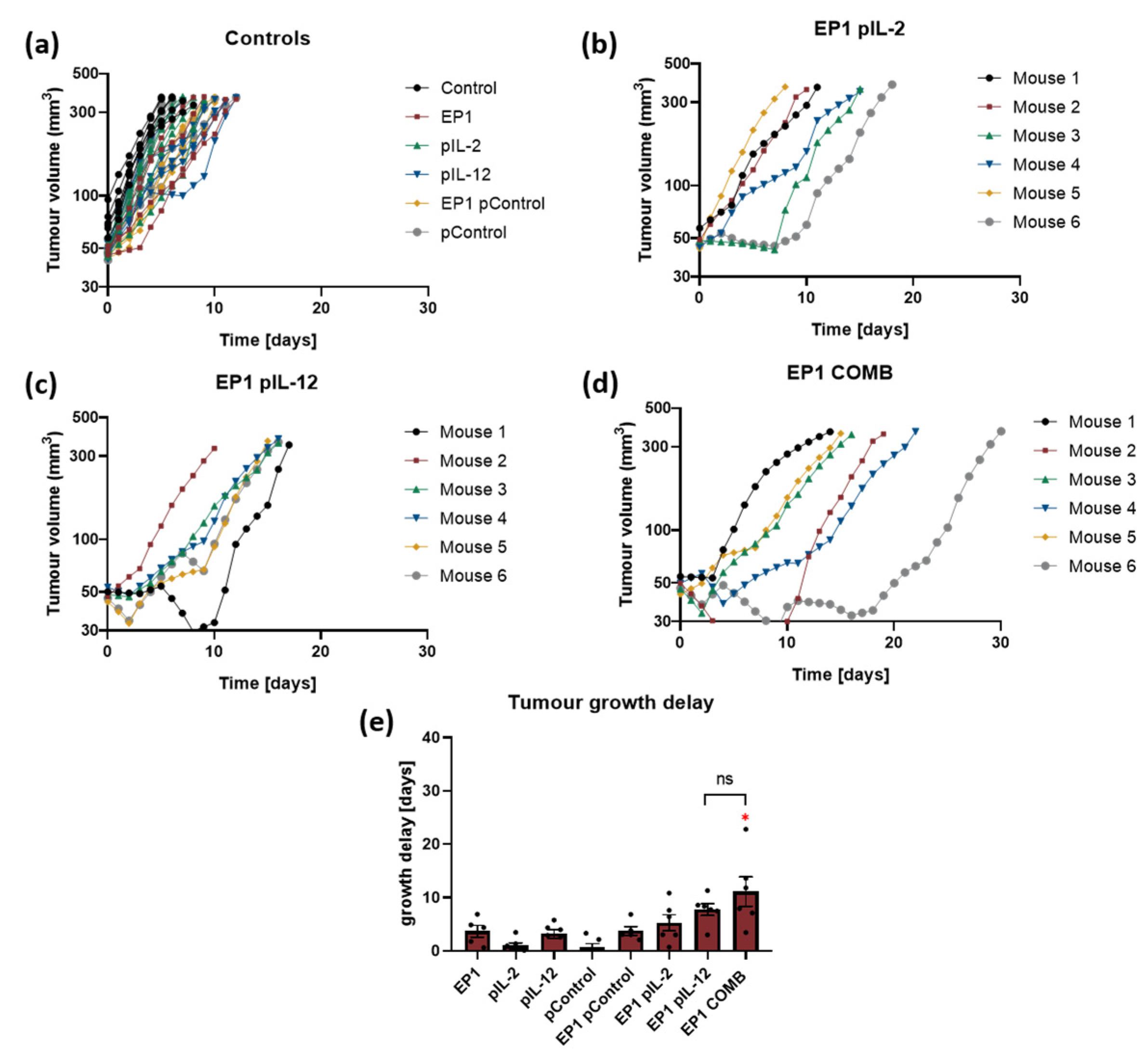
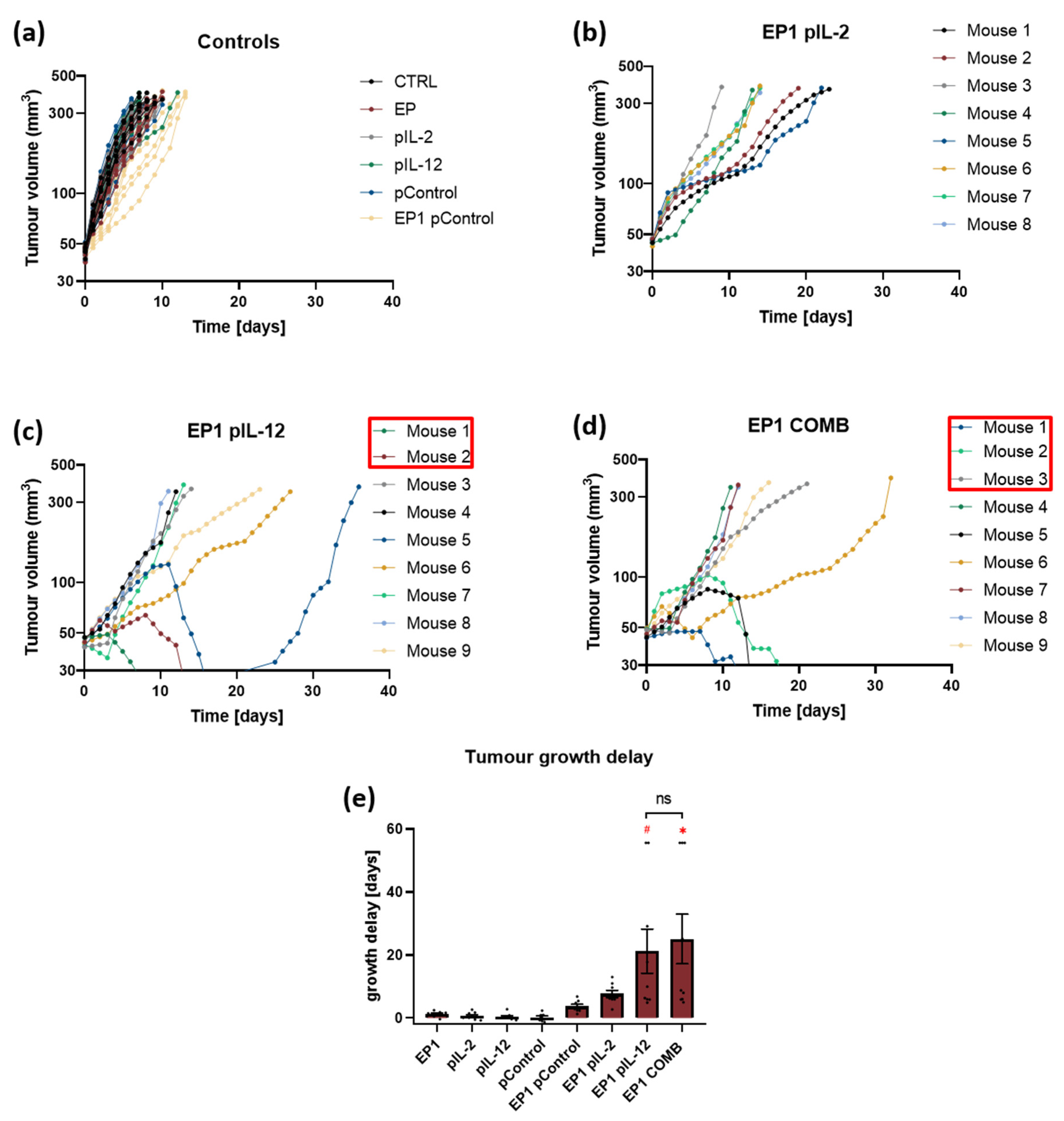
2.4. Therapeutic Effectiveness In Vivo
2.5. Transfection Efficiency In Vivo after Treatment Optimization with Collagenase and Hyaluronidase Pre-Treatment
2.6. Therapeutic Effectiveness In Vivo after Collagenase and Hyaluronidase Pre-Treatment
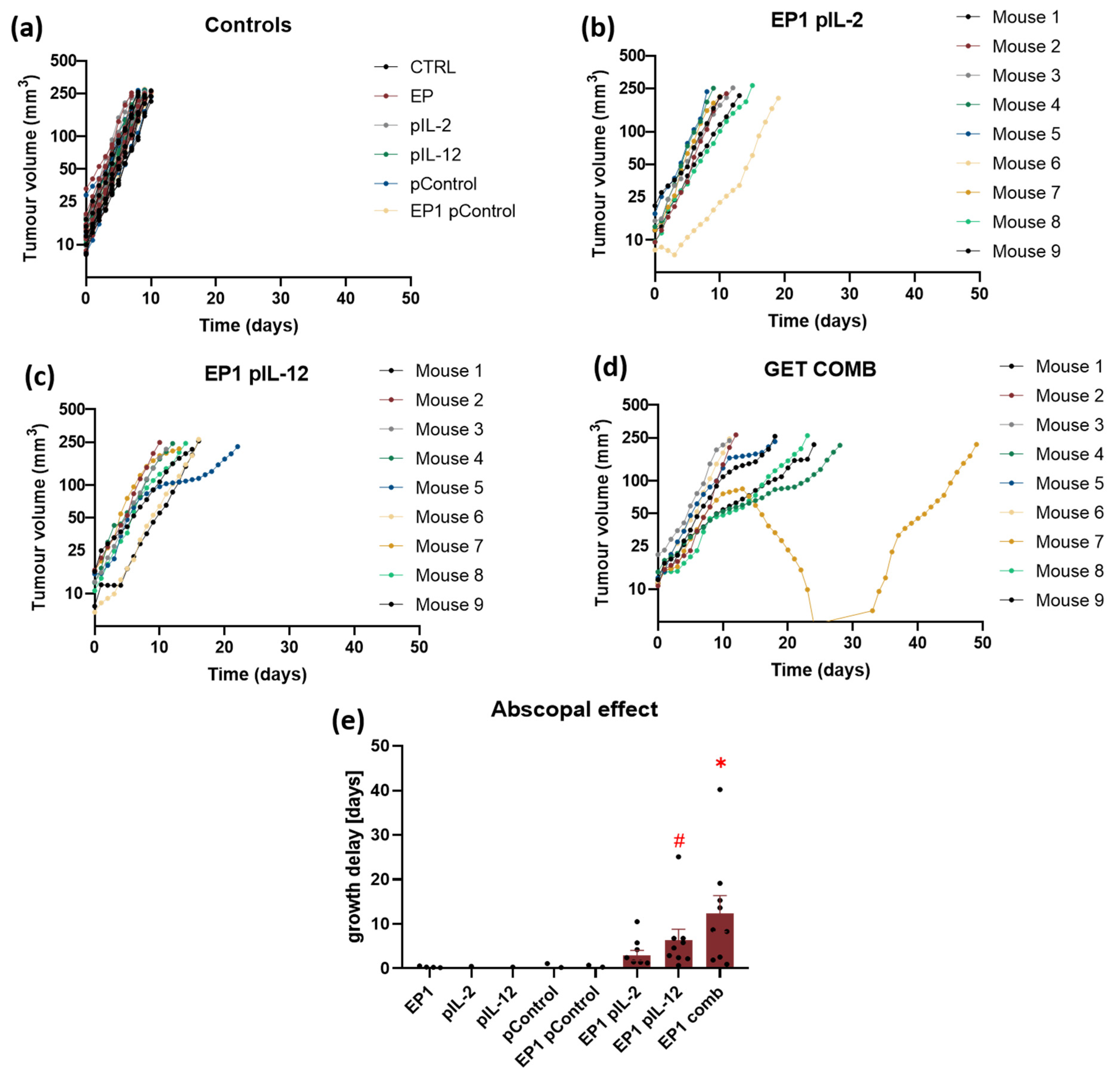
2.7. Abscopal Effect
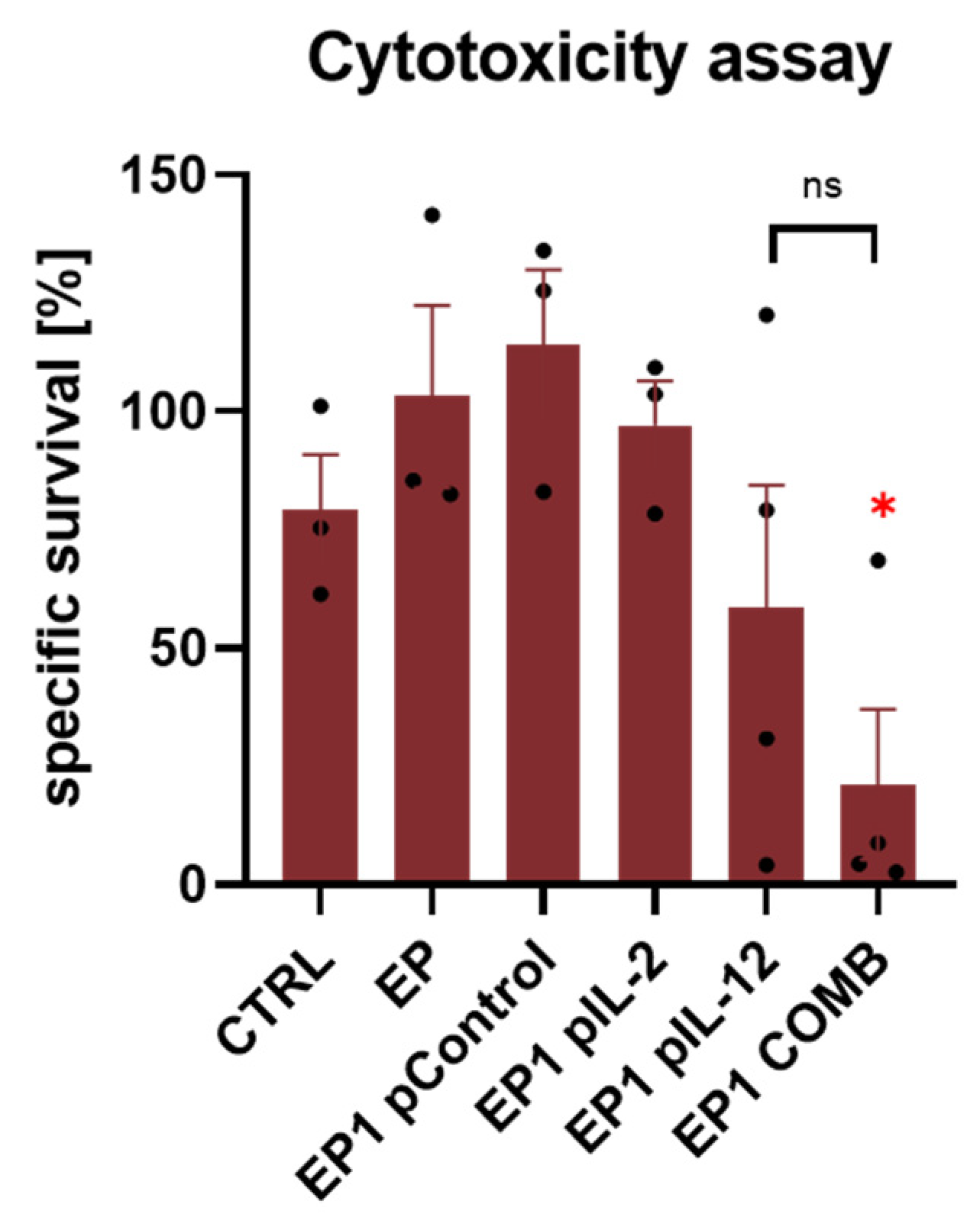
2.8. Ex Vivo Spleen Cytotoxicity Assay
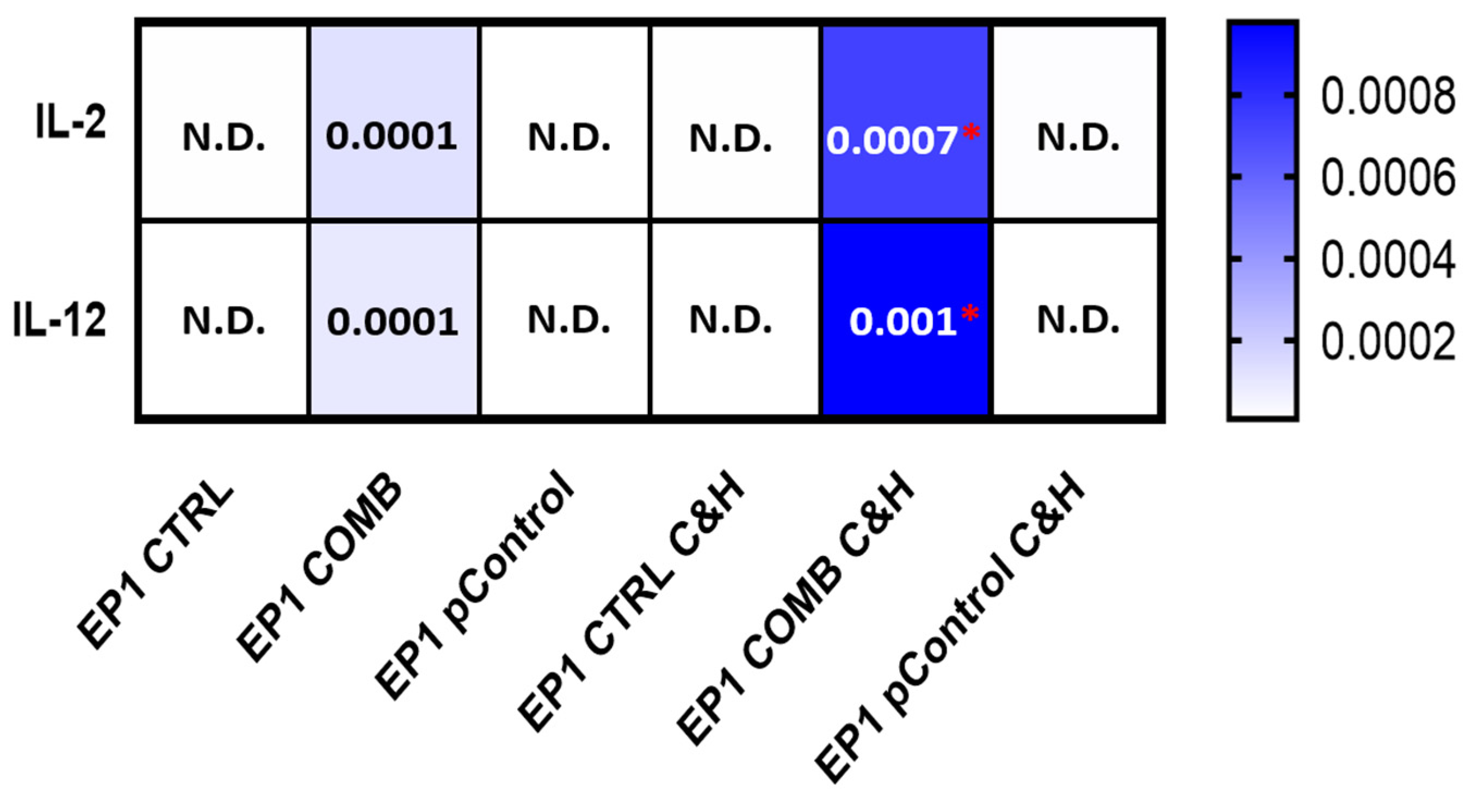
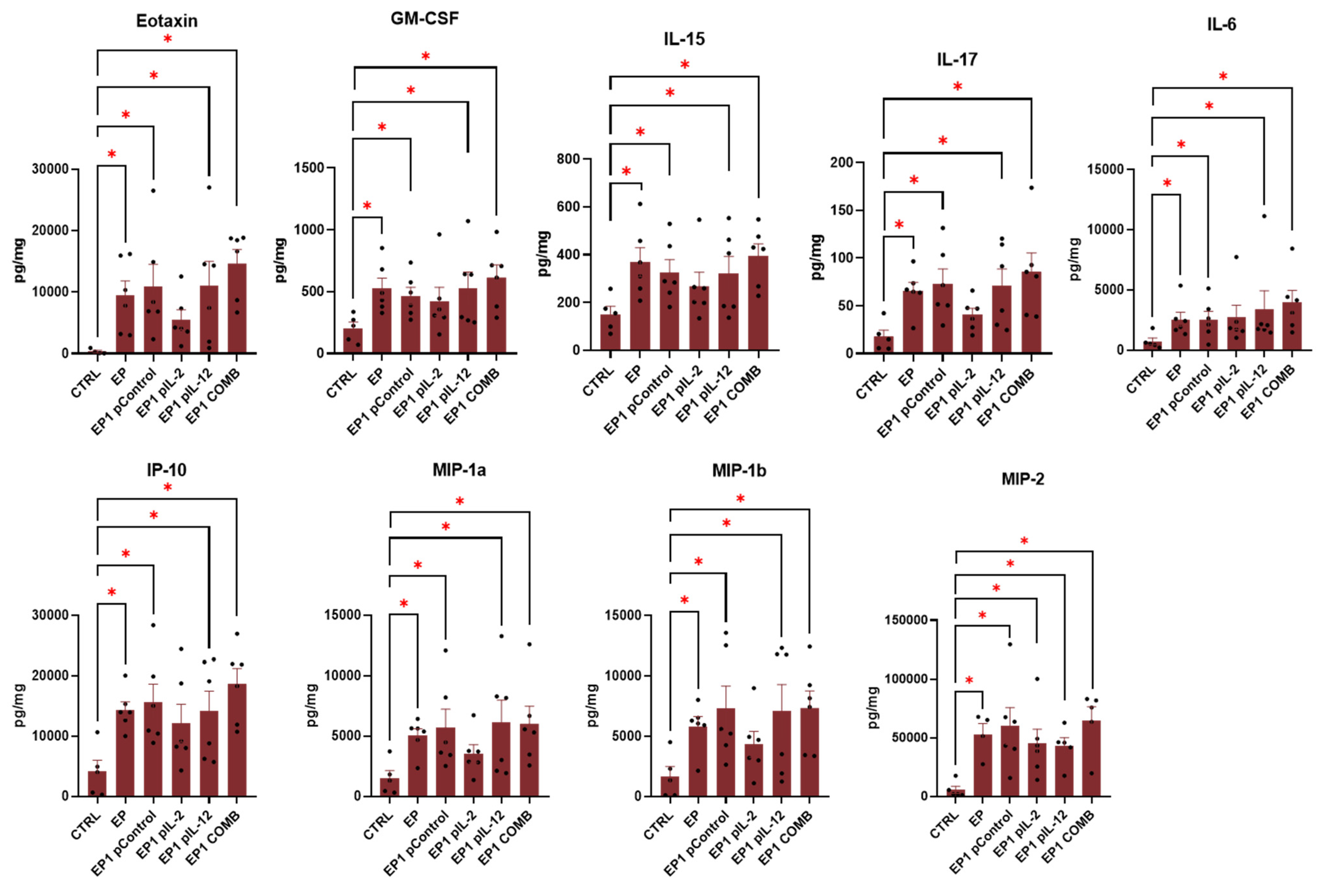
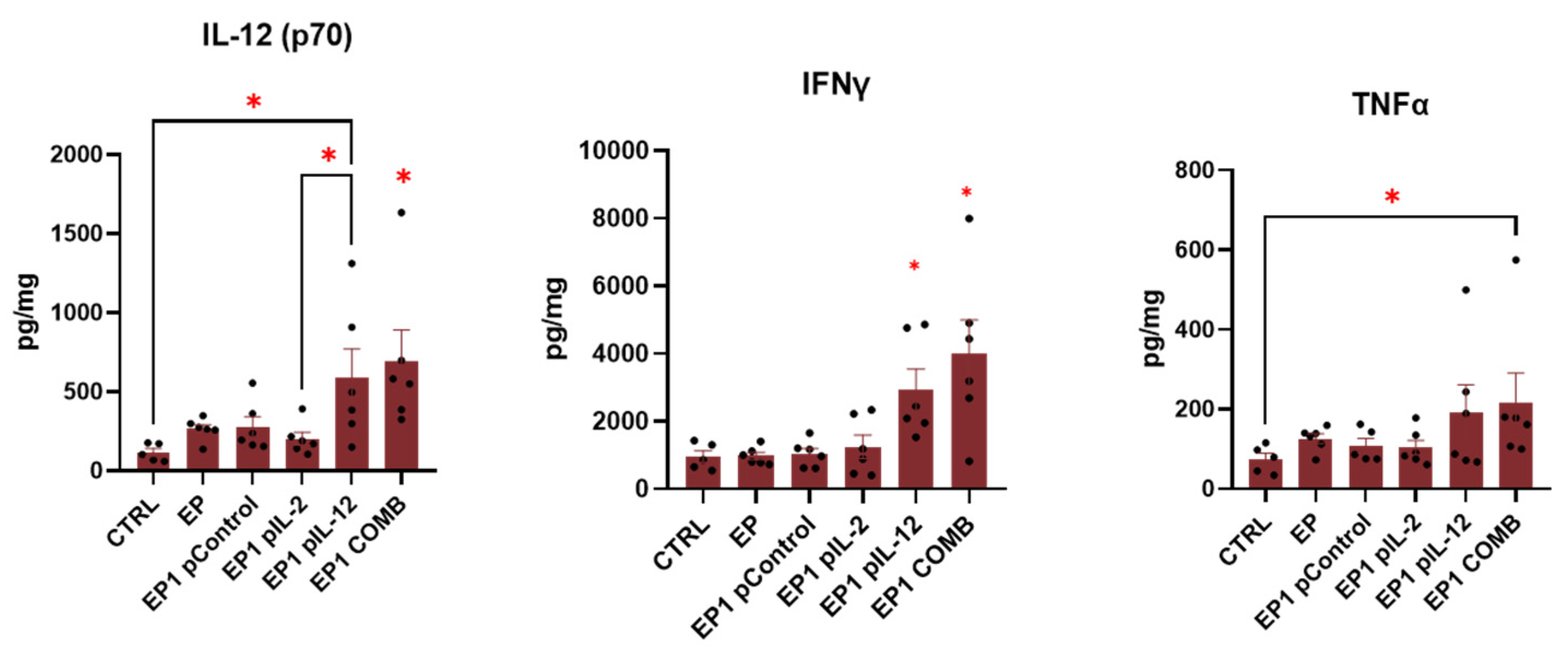
2.9. Expression of Cytokines in Tumour
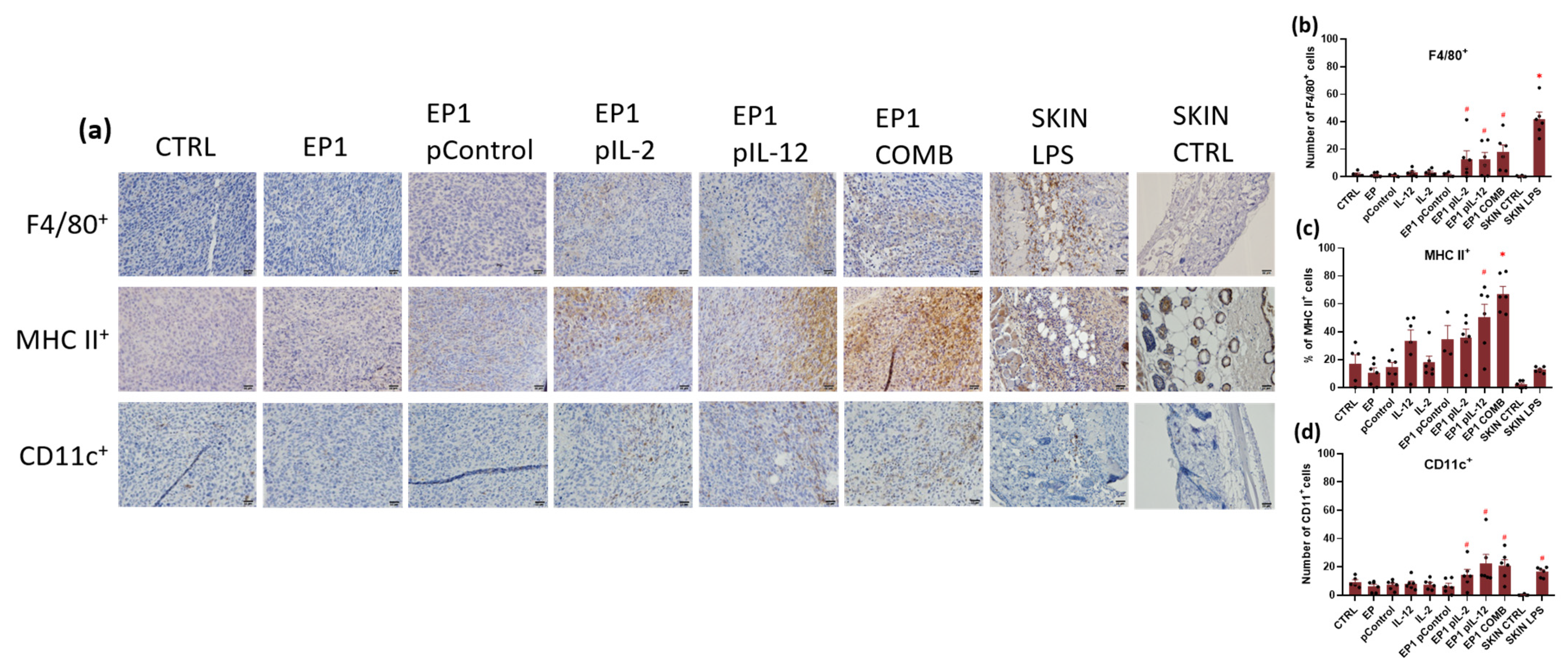
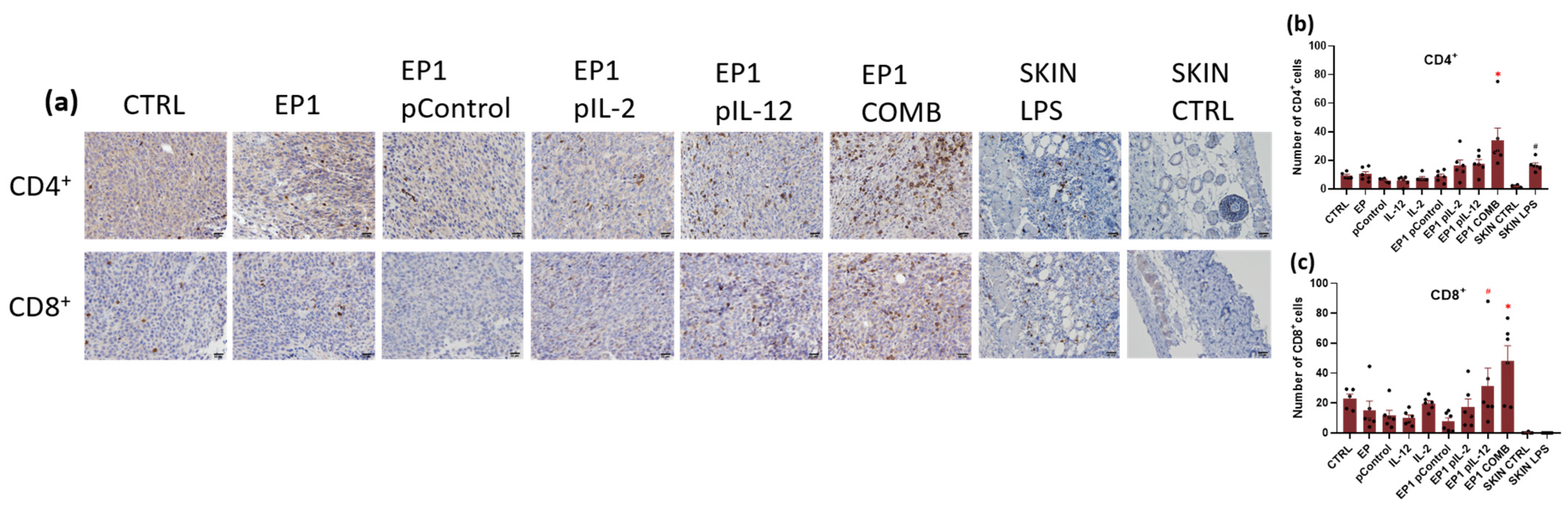
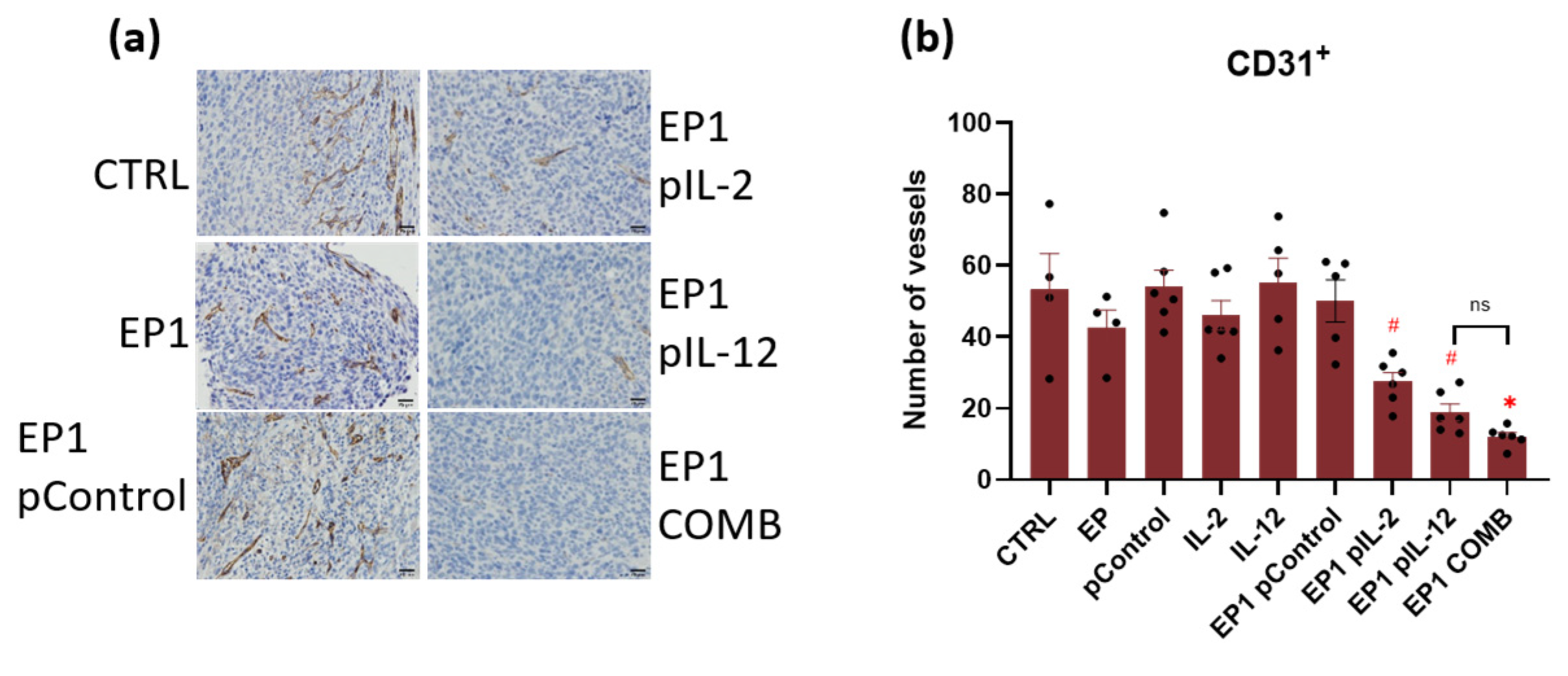
2.10. Immunohistochemical Evaluation of Immune Cell Infiltration in Tumours and Vascularisation after Combined Treatment
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Plasmids
4.3. In Vitro Electroporation Experiments
4.4. Animals and Tumour Induction
4.5. In Vivo Electroporation
4.6. Tumour Growth Measurement
4.7. Ex Vivo Spleen Cytotoxicity Assay
4.8. Multiplex Assay
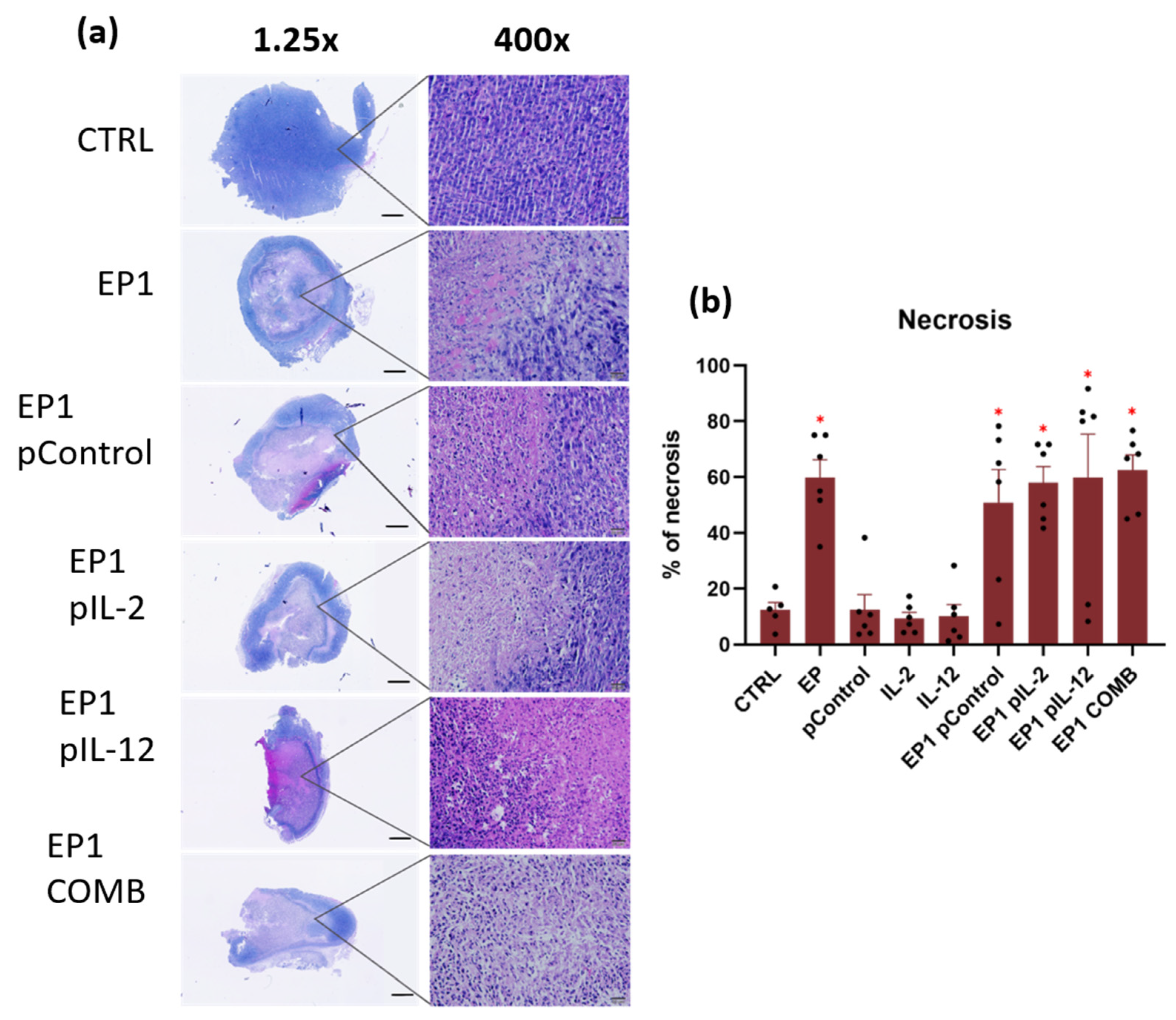
4.9. Histological Analysis
4.10. Immunohistochemistry
4.11. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Littman, D.R. Releasing the Brakes on Cancer Immunotherapy. Cell 2015, 162, 1186–1190. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in Clinical Cancer Immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sokołowska, E.; Błachnio-Zabielska, A.U. A Critical Review of Electroporation as A Plasmid Delivery System in Mouse Skeletal Muscle. Int. J. Mol. Sci. 2019, 20, 2776. [Google Scholar] [CrossRef] [PubMed]
- Rosazza, C.; Haberl Meglic, S.; Zumbusch, A.; Rols, M.-P.; Miklavcic, D. Gene Electrotransfer: A Mechanistic Perspective. Curr. Gene Ther. 2016, 16, 98–129. [Google Scholar] [CrossRef]
- Escoffre, J.M.; Mauroy, C.; Portet, T.; Wasungu, L.; Rosazza, C.; Gilbart, Y.; Mallet, L.; Bellard, E.; Golzio, M.; Rols, M.P.; et al. Gene Electrotransfer: From Biophysical Mechanisms to in Vivo Applications: Part 1—Biophysical Mechanisms. Biophys. Rev. 2009, 1, 177. [Google Scholar] [CrossRef]
- Vivod, G.; Jesenko, T.; Gasljevic, G.; Kovacevic, N.; Bosnjak, M.; Sersa, G.; Merlo, S.; Cemazar, M. Treatment of Vulvar Cancer Recurrences with Electrochemotherapy—A Detailed Analysis of Possible Causes for Unsuccessful Treatment. Radiol. Oncol. 2023, 57, 121–126. [Google Scholar] [CrossRef]
- Tsoneva, I.; Semkova, S.; Bakalova, R.; Zhelev, Z.; Nuss, P.; Staneva, G.; Nikolova, B. Electroporation, Electrochemotherapy and Electro-Assisted Drug Delivery in Cancer. A State-of-the-Art Review. Biophys. Chem. 2022, 286, 106819. [Google Scholar] [CrossRef]
- Kamensek, U.; Rencelj, A.; Jesenko, T.; Remic, T.; Sersa, G.; Cemazar, M. Maintenance and Gene Electrotransfer Efficiency of Antibiotic Resistance Gene-Free Plasmids Encoding Mouse, Canine and Human Interleukin-12 Orthologues. Heliyon 2022, 8, e08879. [Google Scholar] [CrossRef]
- Kamensek, U.; Cemazar, M.; Lampreht Tratar, U.; Ursic, K.; Sersa, G. Antitumor in Situ Vaccination Effect of TNFα and IL-12 Plasmid DNA Electrotransfer in a Murine Melanoma Model. Cancer Immunol. Immunother. 2018, 67, 785–795. [Google Scholar] [CrossRef]
- Nguyen, K.G.; Vrabel, M.R.; Mantooth, S.M.; Hopkins, J.J.; Wagner, E.S.; Gabaldon, T.A.; Zaharoff, D.A. Localized Interleukin-12 for Cancer Immunotherapy. Front. Immunol. 2020, 11, 575597. [Google Scholar] [CrossRef]
- Ursic, K.; Kos, S.; Kamensek, U.; Cemazar, M.; Miceska, S.; Markelc, B.; Bucek, S.; Staresinic, B.; Prevodnik, V.K.; Heller, R.; et al. Potentiation of Electrochemotherapy Effectiveness by Immunostimulation with IL-12 Gene Electrotransfer in Mice Is Dependent on Tumor Immune Status. J. Control. Release 2021, 332, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Malvicini, M.; Rizzo, M.; Alaniz, L.; Piñero, F.; García, M.; Atorrasagasti, C.; Aquino, J.B.; Rozados, V.; Scharovsky, O.G.; Matar, P.; et al. A Novel Synergistic Combination of Cyclophosphamide and Gene Transfer of Interleukin-12 Eradicates Colorectal Carcinoma in Mice. Clin. Cancer Res. 2009, 15, 7256–7265. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Seong, J.; Oh, H.J.; Koom, W.S.; Choi, K.J.; Yun, C.O. A Novel Combination Treatment of Armed Oncolytic Adenovirus Expressing IL-12 and GM-CSF with Radiotherapy in Murine Hepatocarcinoma. J. Radiat. Res. 2011, 52, 646–654. [Google Scholar] [CrossRef]
- Wu, C.J.; Tsai, Y.T.; Lee, I.J.; Wu, P.Y.; Lu, L.S.; Tsao, W.S.; Huang, Y.J.; Chang, C.C.; Ka, S.M.; Tao, M.H. Combination of Radiation and Interleukin 12 Eradicates Large Orthotopic Hepatocellular Carcinoma through Immunomodulation of Tumor Microenvironment. Oncoimmunology 2018, 7, e1477459. [Google Scholar] [CrossRef]
- Sedlar, A.; Dolinsek, T.; Markelc, B.; Prosen, L.; Kranjc, S.; Bosnjak, M.; Blagus, T.; Cemazar, M.; Sersa, G. Potentiation of Electrochemotherapy by Intramuscular IL-12 Gene Electrotransfer in Murine Sarcoma and Carcinoma with Different Immunogenicity. Radiol. Oncol. 2012, 46, 302. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Lundberg, C.; Scott, M.; Edelblute, C.; Heller, R. Enhanced Antitumor Effects by Combining IL-12 Gene Electrotransfer with Anti-PD1 in Preclinical Metastatic Cancer Model. J. Immunol. 2019, 202, 136.2. [Google Scholar] [CrossRef]
- Greaney, S.K.; Algazi, A.P.; Tsai, K.K.; Takamura, K.T.; Chen, L.; Twitty, C.G.; Zhang, L.; Paciorek, A.; Pierce, R.H.; Le, M.H.; et al. Intratumoral Plasmid IL12 Electroporation Therapy in Patients with Advanced Melanoma Induces Systemic and Intratumoral T-Cell Responses. Cancer Immunol. Res. 2020, 8, 246–254. [Google Scholar] [CrossRef]
- Groselj, A.; Bosnjak, M.; Jesenko, T.; Cemazar, M.; Markelc, B.; Strojan, P.; Sersa, G.; Serša, G. Treatment of Skin Tumors with Intratumoral Interleukin 12 Gene Electrotransfer in the Head and Neck Region: A First-in-Human Clinical Trial Protocol. Radiol. Oncol. 2022, 56, 398–408. [Google Scholar] [CrossRef]
- Shirley, S.A.; Lundberg, C.G.; Heller, R. Electrotransfer of IL-15/IL-15Rα Complex Complex for the Treatment of Established Melanoma. Cancers 2020, 12, 3072. [Google Scholar] [CrossRef]
- Horton, H.M.; Lalor, P.A.; Rolland, A.P. IL-2 Plasmid Electroporation: From Preclinical Studies to Phase I Clinical Trial. Methods Mol. Biol. 2008, 423, 361–372. [Google Scholar]
- Komel, T.; Bosnjak, M.; Kranjc Brezar, S.; De Robertis, M.; Mastrodonato, M.; Scillitani, G.; Pesole, G.; Signori, E.; Sersa, G.; Cemazar, M. Gene Electrotransfer of IL-2 and IL-12 Plasmids Effectively Eradicated Murine B16. F10 Melanoma Bioelectrochemistry 2021, 141, 107843. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Neurath, M.F. Interleukin-12: Functional Activities and Implications for Disease. Cytokine Growth Factor Rev. 2015, 26, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Interferon-γ: Teammate or Opponent in the Tumour Microenvironment? Nat. Rev. Immunol. 2021, 22, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Kimura, Y.; Mizuno, H.; Satake, K.; Tahara, H.; Tsukuda, M. Effects of Combined Therapy with Interleukin 2 and Interleukin 12 Gene-Transfected Tumor Vaccine for Head and Neck Carcinoma. Arch. Otolaryngol. Head. Neck Surg. 2003, 129, 1181–1185. [Google Scholar] [CrossRef]
- Zahalka, S.; Starkl, P.; Watzenboeck, M.L.; Farhat, A.; Radhouani, M.; Deckert, F.; Hladik, A.; Lakovits, K.; Oberndorfer, F.; Lassnig, C.; et al. Trained Immunity of Alveolar Macrophages Requires Metabolic Rewiring and Type 1 Interferon Signaling. Mucosal Immunol. 2022, 15, 896–907. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, C.; Sun, Q.; Wang, Y.; Yu, W.; Wei, F.; Ren, X. Trained Immunity of IL-12-, IL-15-, and IL-18-Induced CD3+CD56+ NKT-Like Cells. J. Oncol. 2022, 2022, 8724933. [Google Scholar] [CrossRef]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; van der Meer, J.W.M. Hypothesis: Stimulation of Trained Immunity as Adjunctive Immunotherapy in Cancer. J. Leukoc. Biol. 2017, 102, 1323–1332. [Google Scholar] [CrossRef]
- Morandi, F.; Yazdanifar, M.; Cocco, C.; Bertaina, A.; Airoldi, I. Engineering the Bridge between Innate and Adaptive Immunity for Cancer Immunotherapy: Focus on Γδ T and NK Cells. Cells 2020, 9, 1757. [Google Scholar] [CrossRef]
- Ren, X.; Guo, S.; Guan, X.; Kang, Y.; Liu, J.; Yang, X. Immunological Classification of Tumor Types and Advances in Precision Combination Immunotherapy. Front. Immunol. 2022, 13, 790113. [Google Scholar] [CrossRef]
- Duan, Q.; Zhang, H.; Zheng, J.; Zhang, L. Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 2020, 6, 605–618. [Google Scholar] [CrossRef] [PubMed]
- Floros, T.; Tarhini, A.A. Anticancer Cytokines: Biology and Clinical Effects of IFN-A2, IL-2, IL-15, IL-21, and IL-12. Semin. Oncol. 2015, 42, 539. [Google Scholar] [CrossRef] [PubMed]
- Lasek, W.; Zagożdżon, R.; Jakobisiak, M. Interleukin 12: Still a Promising Candidate for Tumor Immunotherapy? Cancer Immunol. Immunother. 2014, 63, 419. [Google Scholar] [CrossRef] [PubMed]
- Burkart, C.; Mukhopadhyay, A.; Shirley, S.A.; Connolly, R.J.; Wright, J.H.; Bahrami, A.; Campbell, J.S.; Pierce, R.H.; Canton, D.A. Improving Therapeutic Efficacy of IL-12 Intratumoral Gene Electrotransfer through Novel Plasmid Design and Modified Parameters. Gene Ther. 2018, 25, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Heller, L.C.; Cruz, Y.L.; Ferraro, B.; Yang, H.; Heller, R. Plasmid Injection and Application of Electric Pulses Alter Endogenous MRNA and Protein Expression in B16. F10 Mouse Melanomas Cancer Gene Ther. 2010, 17, 864–871. [Google Scholar] [CrossRef]
- Lucas, M.L.; Heller, R. IL-12 Gene Therapy Using an Electrically Mediated Nonviral Approach Reduces Metastatic Growth of Melanoma. DNA Cell Biol. 2003, 22, 755–763. [Google Scholar] [CrossRef]
- Shi, G.; Edelblute, C.; Arpag, S.; Lundberg, C.; Heller, R. IL-12 Gene Electrotransfer Triggers a Change in Immune Response within Mouse Tumors. Cancers 2018, 10, 498. [Google Scholar] [CrossRef]
- Tugues, S.; Burkhard, S.H.; Ohs, I.; Vrohlings, M.; Nussbaum, K.; Vom Berg, J.; Kulig, P.; Becher, B. New Insights into IL-12-Mediated Tumor Suppression. Cell Death Differ. 2015, 22, 237–246. [Google Scholar] [CrossRef]
- Vannini, A.; Leoni, V.; Campadelli-Fiume, G. Targeted Delivery of IL-12 Adjuvants Immunotherapy by Oncolytic Viruses. Adv. Exp. Med. Biol. 2021, 1290, 67–80. [Google Scholar]
- Do, H.D.; Marie, C.; Bessoles, S.; Dhotel, H.; Seguin, J.; Larrat, B.; Doan, B.T.; Scherman, D.; Escriou, V.; Hacein-Bey-Abina, S.; et al. Combination of Thermal Ablation by Focused Ultrasound, PFAR4-IL-12 Transfection and Lipidic Adjuvant Provide a Distal Immune Response. Explor. Target. Anti-Tumor Ther. 2022, 3, 398. [Google Scholar] [CrossRef]
- Cemazar, M.; Golzio, M.; Sersa, G.; Hojman, P.; Kranjc, S.; Mesojednik, S.; Rols, M.P.; Teissie, J. Control by Pulse Parameters of DNA Electrotransfer into Solid Tumors in Mice. Gene Ther. 2009, 16, 635–644. [Google Scholar] [CrossRef]
- Cemazar, M.; Golzio, M.; Sersa, G.; Escoffre, J.M.; Coer, A.; Vidic, S.; Teissie, J. Hyaluronidase and Collagenase Increase the Transfection Efficiency of Gene Electrotransfer in Various Murine Tumors. Hum. Gene Ther. 2012, 23, 128. [Google Scholar] [CrossRef]
- Justesen, T.F.; Orhan, A.; Raskov, H.; Nolsoe, C.; Gögenur, I. Electroporation and Immunotherapy—Unleashing the Abscopal Effect. Cancers 2022, 14, 2876. [Google Scholar] [CrossRef]
- Raskov, H.; Orhan, A.; Christensen, J.P.; Gögenur, I. Cytotoxic CD8+ T Cells in Cancer and Cancer Immunotherapy. Br. J. Cancer 2020, 124, 359–367. [Google Scholar] [CrossRef]
- Zhang, N.; Li, Z.; Han, X.; Zhu, Z.; Li, Z.; Zhao, Y.; Liu, Z.; Lv, Y. Irreversible Electroporation: An Emerging Immunomodulatory Therapy on Solid Tumors. Front. Immunol. 2022, 12, 5674. [Google Scholar] [CrossRef]
- Chiarella, P.; Michele Fazio, V.; Signori, E. Electroporation in DNA Vaccination Protocols Against Cancer. Curr. Drug Metab. 2013, 14, 291–299. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Saunders, M.E. Cytokines and Cytokine Receptors. Immune Response 2006, 463–516. [Google Scholar] [CrossRef]
- Zhu, F.; Liu, P.; Li, J.; Zhang, Y. Eotaxin-1 Promotes Prostate Cancer Cell Invasion via Activation of the CCR3-ERK Pathway and Upregulation of MMP-3 Expression. Oncol. Rep. 2014, 31, 2049–2054. [Google Scholar] [CrossRef] [PubMed]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Lucarini, V.; Marone, G.; Mattei, F.; Marone, G.; Schiavoni, G. Eosinophils: The Unsung Heroes in Cancer? Oncoimmunology 2018, 7, e1393134. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Su, H.; Zhong, W.; Yuan, Y.; Yu, Z.; Fang, Y.; Zhou, H.; Li, C.; Huang, K. Interleukin-17 Is a Favorable Prognostic Marker for Colorectal Cancer. Clin. Transl. Oncol. 2015, 17, 50–56. [Google Scholar] [CrossRef]
- Lotfi, N.; Thome, R.; Rezaei, N.; Zhang, G.X.; Rezaei, A.; Rostami, A.; Esmaeil, N. Roles of GM-CSF in the Pathogenesis of Autoimmune Diseases: An Update. Front. Immunol. 2019, 10, 1265. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.; Chen, L.; Wei, X. Inflammatory Cytokines in Cancer: Comprehensive Understanding and Clinical Progress in Gene Therapy. Cells 2021, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Narazaki, M.; Kishimoto, T. IL-6 in Inflammation, Immunity, and Disease. Cold Spring Harb. Perspect. Biol. 2014, 6, 16295–16296. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.H.; Dziejman, M.; Liu, M.T.; Leung, J.H.; Lane, T.E.; Luster, A.D. IFN-Gamma-Inducible Protein 10 (IP-10; CXCL10)-Deficient Mice Reveal a Role for IP-10 in Effector T Cell Generation and Trafficking. J. Immunol. 2002, 168, 3195–3204. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Stiles, J.K. The Emerging Role of CXCL10 in Cancer (Review). Oncol. Lett. 2011, 2, 583. [Google Scholar] [CrossRef]
- Mlecnik, B.; Tosolini, M.; Charoentong, P.; Kirilovsky, A.; Bindea, G.; Berger, A.; Camus, M.; Gillard, M.; Bruneval, P.; Fridman, W.H.; et al. Biomolecular Network Reconstruction Identifies T-Cell Homing Factors Associated with Survival in Colorectal Cancer. Gastroenterology 2010, 138, 1429–1440. [Google Scholar] [CrossRef]
- Chonov, D.C.; Ignatova, M.M.K.; Ananiev, J.R.; Gulubova, M.V. IL-6 Activities in the Tumour Microenvironment. Part 1. Open Access Maced. J. Med. Sci. 2019, 7, 2391. [Google Scholar] [CrossRef]
- Li, S.; Xia, X.; Mellieon, F.M.; Liu, J.; Steele, S. Candidate Genes Associated with Tumor Regression Mediated by Intratumoral Il-12 Electroporation Gene Therapy. Mol. Ther. 2004, 9, 347–354. [Google Scholar] [CrossRef]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 Polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef]
- Josephs, S.F.; Ichim, T.E.; Prince, S.M.; Kesari, S.; Marincola, F.M.; Escobedo, A.R.; Jafri, A. Unleashing Endogenous TNF-Alpha as a Cancer Immunotherapeutic. J. Transl. Med. 2018, 16, 242. [Google Scholar] [CrossRef]
- Lipski, D.A.; Dewispelaere, R.; Foucart, V.; Caspers, L.E.; Defrance, M.; Bruyns, C.; Willermain, F. MHC Class II Expression and Potential Antigen-Presenting Cells in the Retina during Experimental Autoimmune Uveitis. J. Neuroinflammation 2017, 14, 136. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; He, Y.; Huang, J.; Tao, Y.; Liu, S. Metabolism of Dendritic Cells in Tumor Microenvironment: For Immunotherapy. Front. Immunol. 2021, 12, 317. [Google Scholar] [CrossRef]
- Yang, Y.; Cao, Y. The Impact of VEGF on Cancer Metastasis and Systemic Disease. Semin. Cancer Biol. 2022, 86, 251–261. [Google Scholar] [CrossRef]
- Kieran, M.W.; Kalluri, R.; Cho, Y.J. The VEGF Pathway in Cancer and Disease: Responses, Resistance, and the Path Forward. Cold Spring Harb. Perspect. Med. 2012, 2, a006593. [Google Scholar] [CrossRef] [PubMed]
- Gomulka, K.; Liebhart, J.; Lange, A.; Medrala, W. Vascular Endothelial Growth Factor-Activated Basophils in Asthmatics. Adv. Dermatology Allergol. Dermatologii i Alergol. 2020, 37, 584. [Google Scholar] [CrossRef]
- Lérias, J.R.; de Sousa, E.; Paraschoudi, G.; Martins, J.; Condeço, C.; Figueiredo, N.; Carvalho, C.; Dodoo, E.; Maia, A.; Castillo-Martin, M.; et al. Trained Immunity for Personalized Cancer Immunotherapy: Current Knowledge and Future Opportunities. Front. Microbiol. 2020, 10, 2924. [Google Scholar] [CrossRef] [PubMed]
- Bosnjak, M.; Dolinsek, T.; Cemazar, M.; Kranjc, S.; Blagus, T.; Markelc, B.; Stimac, M.; Zavrsnik, J.; Kamensek, U.; Heller, L.; et al. Gene Electrotransfer of Plasmid AMEP, an Integrin-Targeted Therapy, Has Antitumor and Antiangiogenic Action in Murine B16 Melanoma. Gene Ther. 2015, 22, 578–590. [Google Scholar] [CrossRef]
- Langford, D.J.; Bailey, A.L.; Chanda, M.L.; Clarke, S.E.; Drummond, T.E.; Echols, S.; Glick, S.; Ingrao, J.; Klassen-Ross, T.; Lacroix-Fralish, M.L.; et al. Coding of Facial Expressions of Pain in the Laboratory Mouse. Nat. Methods 2010, 7, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Dolor, A.; Szoka, F.C. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Mol. Pharm. 2018, 15, 2069–2083. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komel, T.; Omerzel, M.; Kamensek, U.; Znidar, K.; Lampreht Tratar, U.; Kranjc Brezar, S.; Dolinar, K.; Pirkmajer, S.; Sersa, G.; Cemazar, M. Gene Immunotherapy of Colon Carcinoma with IL-2 and IL-12 Using Gene Electrotransfer. Int. J. Mol. Sci. 2023, 24, 12900. https://doi.org/10.3390/ijms241612900
Komel T, Omerzel M, Kamensek U, Znidar K, Lampreht Tratar U, Kranjc Brezar S, Dolinar K, Pirkmajer S, Sersa G, Cemazar M. Gene Immunotherapy of Colon Carcinoma with IL-2 and IL-12 Using Gene Electrotransfer. International Journal of Molecular Sciences. 2023; 24(16):12900. https://doi.org/10.3390/ijms241612900
Chicago/Turabian StyleKomel, Tilen, Masa Omerzel, Urska Kamensek, Katarina Znidar, Ursa Lampreht Tratar, Simona Kranjc Brezar, Klemen Dolinar, Sergej Pirkmajer, Gregor Sersa, and Maja Cemazar. 2023. "Gene Immunotherapy of Colon Carcinoma with IL-2 and IL-12 Using Gene Electrotransfer" International Journal of Molecular Sciences 24, no. 16: 12900. https://doi.org/10.3390/ijms241612900
APA StyleKomel, T., Omerzel, M., Kamensek, U., Znidar, K., Lampreht Tratar, U., Kranjc Brezar, S., Dolinar, K., Pirkmajer, S., Sersa, G., & Cemazar, M. (2023). Gene Immunotherapy of Colon Carcinoma with IL-2 and IL-12 Using Gene Electrotransfer. International Journal of Molecular Sciences, 24(16), 12900. https://doi.org/10.3390/ijms241612900