
Apoptosis Regulation in Osteoarthritis and the Influence of Lipid Interactions
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
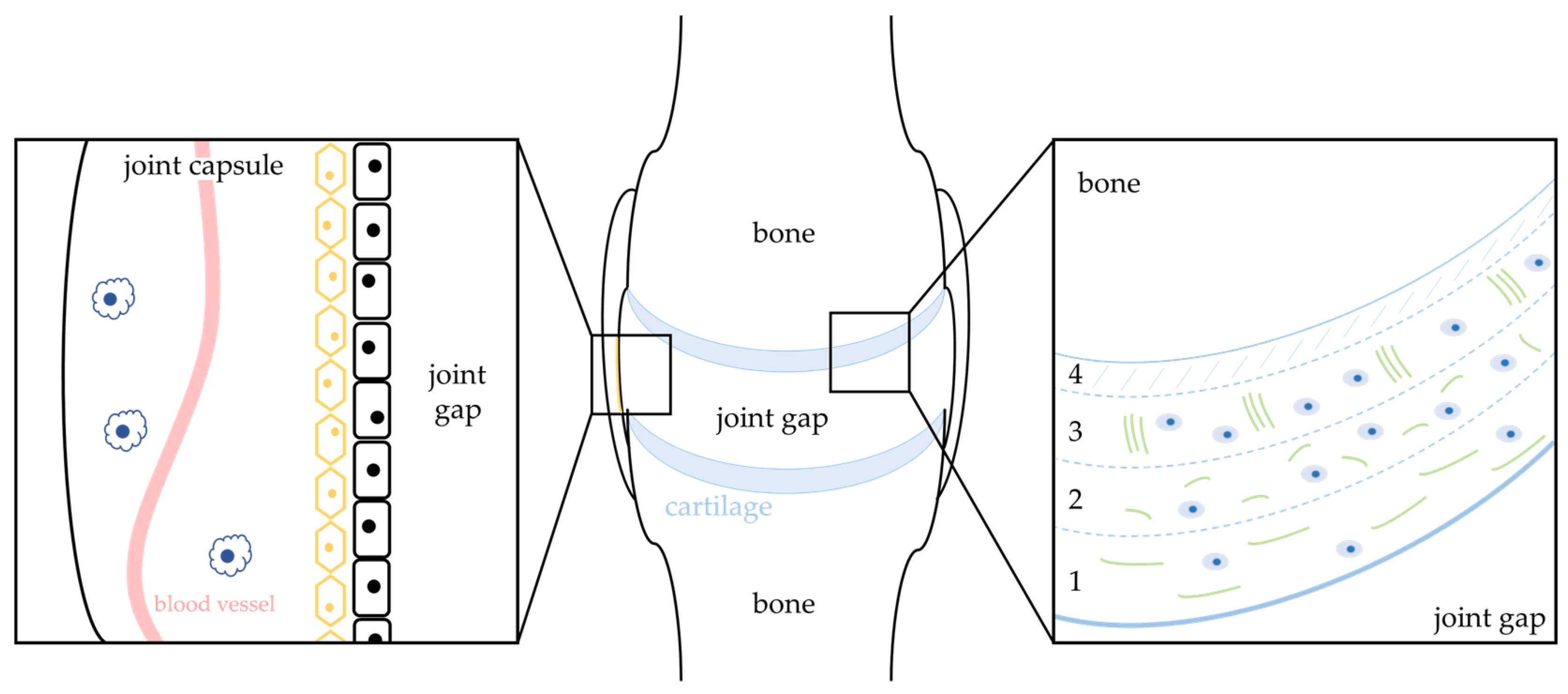
:1. Osteoarthritis
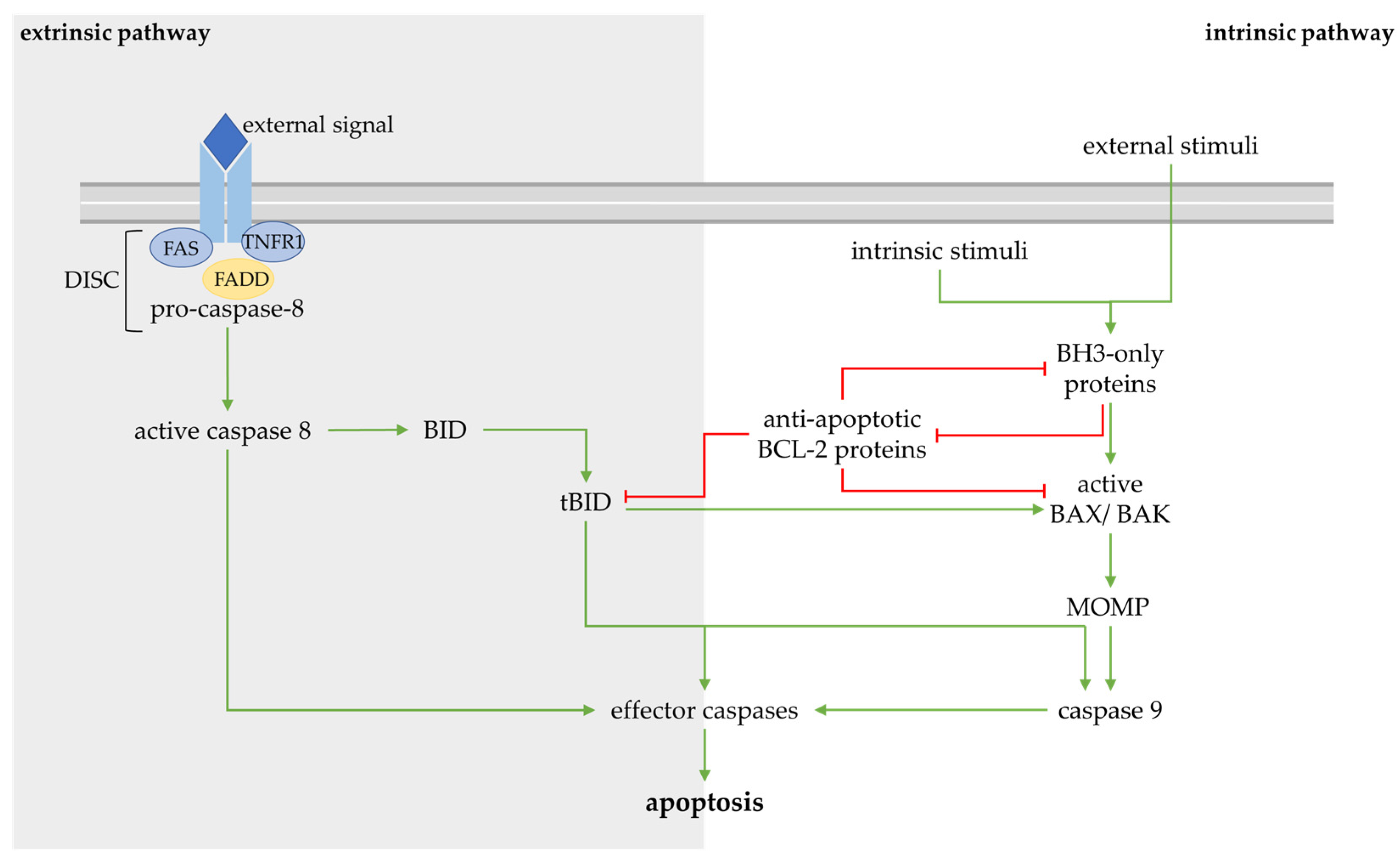
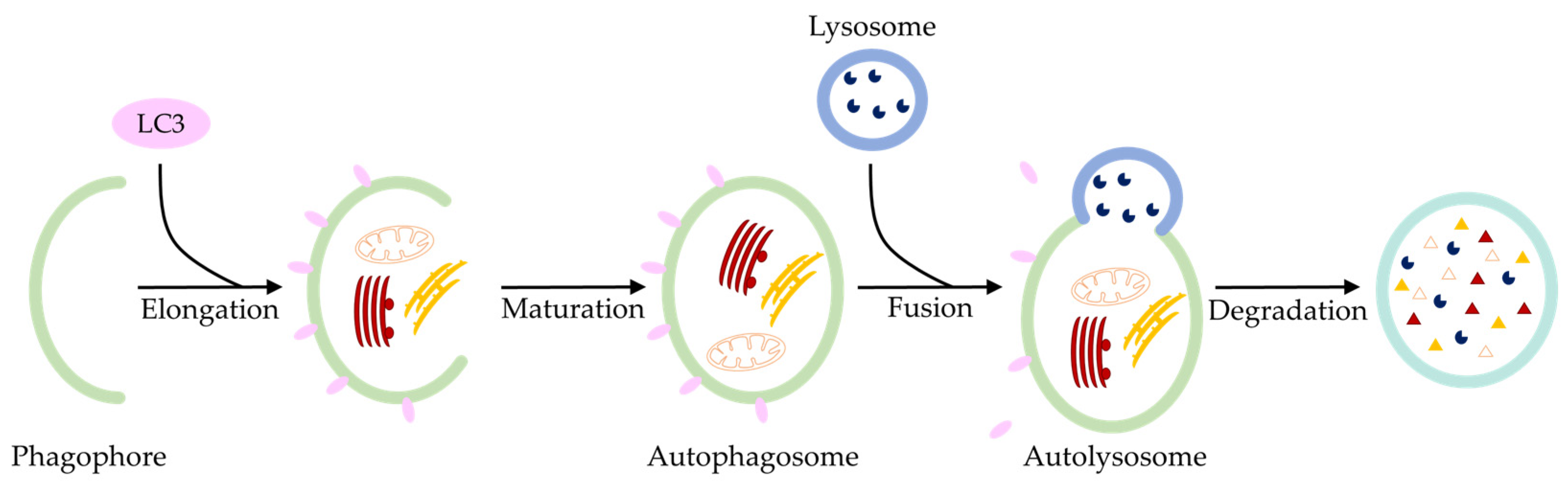
2. Regulated Cell Death in the Pathogenesis of Osteoarthritis
3. Apoptosis and Inflammation in Osteoarthritis
4. Lipids Potentially Modulate Cell Degeneration in Osteoarthritis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Im, G.-I.; Moon, J.-Y. Emerging Concepts of Endotypes/Phenotypes in Regenerative Medicine for Osteoarthritis. Tissue Eng. Regen. Med. 2022, 19, 321–324. [Google Scholar] [CrossRef]
- Angelini, F.; Widera, P.; Mobasheri, A.; Blair, J.; Struglics, A.; Uebelhoer, M.; Henrotin, Y.; Marijnissen, A.C.; Kloppenburg, M.; Blanco, F.J.; et al. Osteoarthritis endotype discovery via clustering of biochemical marker data. Ann. Rheum. Dis. 2022, 81, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Chen, X.; Lin, Z.; Alahdal, M.; Wang, D.; Liu, J.; Li, W. The Homeostasis of Cartilage Matrix Remodeling and the Regulation of Volume-Sensitive Ion Channel. Aging Dis. 2022, 13, 787. [Google Scholar] [CrossRef] [PubMed]
- Caramés, B.; Olmer, M.; Kiosses, W.B.; Lotz, M.K. The Relationship of Autophagy Defects to Cartilage Damage During Joint Aging in a Mouse Model. Arthritis Rheumatol. 2015, 67, 1568–1576. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Im, G.-I. Autophagy in osteoarthritis. Connect. Tissue Res. 2017, 58, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, J.; Lu, L.; Qiu, Z.-Y.; Zhang, X.; Yu, S.-B.; Wu, Y.-P.; Wang, M.-Q. Enhancement of chondrocyte autophagy is an early response in the degenerative cartilage of the temporomandibular joint to biomechanical dental stimulation. Apoptosis 2013, 18, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Hwang, H.; Kim, H. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef]
- Speziali, A.; Delcogliano, M.; Tei, M.; Placella, G.; Chillemi, M.; Tiribuzi, R.; Cerulli, G. Chondropenia: Current concept review. Musculoskelet. Surg. 2015, 99, 189–200. [Google Scholar] [CrossRef]
- Im, G.-I. The Concept of Early Osteoarthritis and Its Significance in Regenerative Medicine. Tissue Eng. Regen. Med. 2022, 19, 431–436. [Google Scholar] [CrossRef]
- Aigner, T.; Söder, S.; Gebhard, P.M.; McAlinden, A.; Haag, J. Mechanisms of Disease: Role of chondrocytes in the pathogenesis of osteoarthritis—Structure, chaos and senescence. Nat. Rev. Rheumatol. 2007, 3, 391–399. [Google Scholar] [CrossRef]
- Chimenti, M.S.; Triggianese, P.; Conigliaro, P.; Candi, E.; Melino, G.; Perricone, R. The interplay between inflammation and metabolism in rheumatoid arthritis. Cell Death Dis. 2015, 6, e1887. [Google Scholar] [CrossRef] [PubMed]
- Maeda, K.; Yoshida, K.; Nishizawa, T.; Otani, K.; Yamashita, Y.; Okabe, H.; Hadano, Y.; Kayama, T.; Kurosaka, D.; Saito, M. Inflammation and Bone Metabolism in Rheumatoid Arthritis: Molecular Mechanisms of Joint Destruction and Pharmacological Treatments. Int. J. Mol. Sci. 2022, 23, 2871. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, S.E.; Firestein, G.S. Rheumatoid arthritis: Regulation of synovial inflammation. Int. J. Biochem. Cell Biol. 2004, 36, 372–378. [Google Scholar] [CrossRef]
- Matsuda, K.; Shiba, N.; Hiraoka, K. New Insights into the Role of Synovial Fibroblasts Leading to Joint Destruction in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 5173. [Google Scholar] [CrossRef]
- Aigner, T.; Hemmel, M.; Neureiter, D.; Gebhard, P.M.; Zeiler, G.; Kirchner, T.; McKenna, L. Apoptotic cell death is not a widespread phenomenon in normal aging and osteoarthritis human articular knee cartilage: A study of proliferation, programmed cell death (apoptosis), and viability of chondrocytes in normal and osteoarthritic human knee cartilage. Arthritis Rheum. 2001, 44, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Bullough, P.G. Orthopaedic Pathology, 5th ed.; Expert consult; Online and print; Mosby/Elsevier: Maryland Heights, MO, USA, 2010; ISBN 978-0-323-05471-3. [Google Scholar]
- Meachim, G.; Ghadially, F.N.; Collins, D.H. Regressive Changes in the Superficial Layer of Human Articular Cartilage. Ann. Rheum. Dis. 1965, 24, 23–30. [Google Scholar] [CrossRef]
- Blanco, F.J.; Guitian, R.; Vázquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Heraud, F. Apoptosis in normal and osteoarthritic human articular cartilage. Ann. Rheum. Dis. 2000, 59, 959–965. [Google Scholar] [CrossRef]
- Kerr, J.F.R. History of the events leading to the formulation of the apoptosis concept. Toxicology 2002, 181–182, 471–474. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Fadok, V.A.; Voelker, D.R.; Campbell, P.A.; Cohen, J.J.; Bratton, D.L.; Henson, P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992, 148, 2207–2216. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Gores, G.J. Life and death by death receptors. FASEB J. 2009, 23, 1625–1637. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Renatus, M.; Scott, F.L.; Sperandio, S.; Shin, H.; Pedersen, I.M.; Ricci, J.-E.; Edris, W.A.; Sutherlin, D.P.; Green, D.R.; et al. A Unified Model for Apical Caspase Activation. Mol. Cell 2003, 11, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, C. Two CD95 (APO-1/Fas) signaling pathways. EMBO J. 1998, 17, 1675–1687. [Google Scholar] [CrossRef]
- Vaux, D.L.; Cory, S.; Adams, J.M. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-x L -Bak Peptide Complex: Recognition Between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef] [PubMed]
- Oltval, Z.N.; Milliman, C.L.; Korsmeyer, S.J. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programed cell death. Cell 1993, 74, 609–619. [Google Scholar] [CrossRef]
- Zha, H.; Aimé-Sempé, C.; Sato, T.; Reed, J.C. Proapoptotic Protein Bax Heterodimerizes with Bcl-2 and Homodimerizes with Bax via a Novel Domain (BH3) Distinct from BH1 and BH2. J. Biol. Chem. 1996, 271, 7440–7444. [Google Scholar] [CrossRef]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial Membrane Permeabilization in Cell Death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef]
- Edlich, F.; Banerjee, S.; Suzuki, M.; Cleland, M.M.; Arnoult, D.; Wang, C.; Neutzner, A.; Tjandra, N.; Youle, R.J. Bcl-xL Retrotranslocates Bax from the Mitochondria into the Cytosol. Cell 2011, 145, 104–116. [Google Scholar] [CrossRef]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Youle, R.J.; Edlich, F. The C-terminal helix of Bcl-xL mediates Bax retrotranslocation from the mitochondria. Cell Death Differ. 2013, 20, 333–342. [Google Scholar] [CrossRef] [PubMed]
- Todt, F.; Cakir, Z.; Reichenbach, F.; Emschermann, F.; Lauterwasser, J.; Kaiser, A.; Ichim, G.; Tait, S.W.; Frank, S.; Langer, H.F.; et al. Differential retrotranslocation of mitochondrial Bax and Bak. EMBO J. 2015, 34, 67–80. [Google Scholar] [CrossRef] [PubMed]
- Eskes, R.; Antonsson, B.; Osen-Sand, A.; Montessuit, S.; Richter, C.; Sadoul, R.; Mazzei, G.; Nichols, A.; Martinou, J.-C. Bax-induced Cytochrome C Release from Mitochondria Is Independent of the Permeability Transition Pore but Highly Dependent on Mg2+ Ions. J. Cell Biol. 1998, 143, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Slee, E.; Adrain, C.; Martin, S. Serial killers: Ordering caspase activation events in apoptosis. Cell Death Differ. 1999, 6, 1067–1074. [Google Scholar] [CrossRef]
- Kuribayashi, K.; Mayes, P.A.; El-Deiry, W.S. What are caspases 3 and 7 doing upstream of the mitochondria? Cancer Biol. Ther. 2006, 5, 763–765. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.-M.; Wang, K.; Gross, A.; Zhao, Y.; Zinkel, S.; Klocke, B.; Roth, K.A.; Korsmeyer, S.J. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999, 400, 886–891. [Google Scholar] [CrossRef]
- Lauterwasser, J.; Fimm-Todt, F.; Oelgeklaus, A.; Schreiner, A.; Funk, K.; Falquez-Medina, H.; Klesse, R.; Jahreis, G.; Zerbes, R.M.; O’Neill, K.; et al. Hexokinases inhibit death receptor–dependent apoptosis on the mitochondria. Proc. Natl. Acad. Sci. USA 2021, 118, e2021175118. [Google Scholar] [CrossRef]
- Grinberg, M.; Sarig, R.; Zaltsman, Y.; Frumkin, D.; Grammatikakis, N.; Reuveny, E.; Gross, A. tBID Homooligomerizes in the Mitochondrial Membrane to Induce Apoptosis. J. Biol. Chem. 2002, 277, 12237–12245. [Google Scholar] [CrossRef]
- Schendel, S.L.; Azimov, R.; Pawłowski, K.; Godzik, A.; Kagan, B.L.; Reed, J.C. Ion Channel Activity of the BH3 Only Bcl-2 Family Member, BID. J. Biol. Chem. 1999, 274, 21932–21936. [Google Scholar] [CrossRef]
- Roach, H.I.; Aigner, T.; Kouri, J.B. Chondroptosis: A variant of apoptotic cell death in chondrocytes? Apoptosis 2004, 9, 265–277. [Google Scholar] [CrossRef]
- Sperandio, S.; De Belle, I.; Bredesen, D.E. An alternative, nonapoptotic form of programmed cell death. Proc. Natl. Acad. Sci. USA 2000, 97, 14376–14381. [Google Scholar] [CrossRef] [PubMed]
- Aigner, T.; Kim, H.A.; Roach, H.I. Apoptosis in osteoarthritis. Rheum. Dis. Clin. N. Am. 2004, 30, 639–653. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochs, R.L.; Rosen, F.; Quach, J.; McCabe, G.; Solan, J.; Seegmiller, J.E.; Terkeltaub, R.; Lotz, M. Chondrocyte-derived apoptotic bodies and calcification of articular cartilage. Proc. Natl. Acad. Sci. USA 1998, 95, 3094–3099. [Google Scholar] [CrossRef]
- Greenwald, R.A.; Moy, W.W. Inhibition of collagen gelation by action of the superoxide radical. Arthritis Rheum. 1979, 22, 251–259. [Google Scholar] [CrossRef]
- Bates, E.J.; Johnson, C.C.; Lowther, D.A. Inhibition of proteoglycan synthesis by hydrogen peroxide in cultured bovine articular cartilage. Biochim. Biophys. Acta (BBA) Gen. Subj. 1985, 838, 221–228. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.; Stryer, L. Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2018; ISBN 978-3-662-54619-2. [Google Scholar]
- Grishko, V.I.; Ho, R.; Wilson, G.L.; Pearsall, A.W. Diminished mitochondrial DNA integrity and repair capacity in OA chondrocytes. Osteoarthr. Cartil. 2009, 17, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Terkeltaub, R. Invited review: The mitochondrion in osteoarthritis. Mitochondrion 2002, 1, 301–319. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Battista, J.D.; Lajeunesse, D. Biochemical Factors in Joint Articular Tissue Degradation in Osteoarthritis. In Osteoarthritis; Springer: Berlin/Heidelberg, Germany, 1999; pp. 156–187. ISBN 978-3-540-65127-7. [Google Scholar]
- López-Armada, M.J.; Caramés, B.; Lires-Deán, M.; Cillero-Pastor, B.; Ruiz-Romero, C.; Galdo, F.; Blanco, F.J. Cytokines, tumor necrosis factor-α and interleukin-1β, differentially regulate apoptosis in osteoarthritis cultured human chondrocytes. Osteoarthr. Cartil. 2006, 14, 660–669. [Google Scholar] [CrossRef]
- Kubota, E.; Imamura, H.; Kubota, T.; Shibata, T.; Murakami, K.-I. Interleukin 1β and stromelysin (MMP3) activity of synovial fluid as possible markers of osteoarthritis in the temporomandibular joint. J. Oral Maxillofac. Surg. 1997, 55, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.D.; Triantafillou, S.; Parker, A.; Youssef, P.P.; Coleman, M. Synovial membrane inflammation and cytokine production in patients with early osteoarthritis. J. Rheumatol. 1997, 24, 365–371. [Google Scholar]
- Palmer, R.M.J.; Hickery, M.S.; Charles, I.G.; Moncada, S.; Bayliss, M.T. Induction of Nitric Oxide Synthase in Human Chondrocytes. Biochem. Biophys. Res. Commun. 1993, 193, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Old, L.J. Tumor Necrosis Factor (TNF). Science 1985, 230, 630–632. [Google Scholar] [CrossRef]
- Dostert, C.; Grusdat, M.; Letellier, E.; Brenner, D. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev. 2019, 99, 115–160. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Banerjee, P.; Nikolakaki, E.; Dai, T.; Rubie, E.A.; Ahmad, M.F.; Avruch, J.; Woodgett, J.R. The stress-activated protein kinase subfamily of c-Jun kinases. Nature 1994, 369, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef]
- Watson, A.; Eilers, A.; Lallemand, D.; Kyriakis, J.; Rubin, L.L.; Ham, J. Phosphorylation of c-Jun Is Necessary for Apoptosis Induced by Survival Signal Withdrawal in Cerebellar Granule Neurons. J. Neurosci. 1998, 18, 751–762. [Google Scholar] [CrossRef] [PubMed]
- Kasibhatla, S.; Brunner, T.; Genestier, L.; Echeverri, F.; Mahboubi, A.; Green, D.R. DNA Damaging Agents Induce Expression of Fas Ligand and Subsequent Apoptosis in T Lymphocytes via the Activation of NF-κB and AP-1. Mol. Cell 1998, 1, 543–551. [Google Scholar] [CrossRef]
- Nakano, K.; Vousden, K.H. PUMA, a Novel Proapoptotic Gene, Is Induced by p53. Mol. Cell 2001, 7, 683–694. [Google Scholar] [CrossRef]
- Fan, Z.; Söder, S.; Oehler, S.; Fundel, K.; Aigner, T. Activation of Interleukin-1 Signaling Cascades in Normal and Osteoarthritic Articular Cartilage. Am. J. Pathol. 2007, 171, 938–946. [Google Scholar] [CrossRef]
- Kumar, S.; Votta, B.J.; Rieman, D.J.; Badger, A.M.; Gowen, M.; Lee, J.C. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J. Cell. Physiol. 2001, 187, 294–303. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Heitmeyer, S.A.; Hookfin, E.B.; Hsieh, L.; Buchalova, M.; Taiwo, Y.O.; Janusz, M.J. P38 MAP kinase inhibitors as potential therapeutics for the treatment of joint degeneration and pain associated with osteoarthritis. J. Inflamm. 2008, 5, 22. [Google Scholar] [CrossRef]
- Wu, G.-J.; Chen, T.-G.; Chang, H.-C.; Chiu, W.-T.; Chang, C.-C.; Chen, R.-M. Nitric oxide from both exogenous and endogenous sources activates mitochondria-dependent events and induces insults to human chondrocytes. J. Cell. Biochem. 2007, 101, 1520–1531. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.-P.; Jovanovic, D.; Fernandes, J.C.; Manning, P.; Connor, J.R.; Currie, M.G.; Di Battista, J.A.; Martel-Pelletier, J. Reduced progression of experimental osteoarthritis in vivo by selective inhibition of inducible nitric oxide synthase. Arthritis Rheum. 1998, 41, 1275–1286. [Google Scholar] [CrossRef]
- Kühn, K.; Hashimoto, S.; Lotz, M. IL-1β Protects Human Chondrocytes from CD95-Induced Apoptosis. J. Immunol. 2000, 164, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Nugent, A.E.; Speicher, D.M.; Gradisar, I.; McBurney, D.L.; Baraga, A.; Doane, K.J.; Horton, W.E. Advanced Osteoarthritis in Humans Is Associated With Altered Collagen VI Expression and Upregulation of ER-stress Markers Grp78 and Bag-1. J. Histochem. Cytochem. 2009, 57, 923–931. [Google Scholar] [CrossRef]
- Lin, J.H.; Walter, P.; Yen, T.S.B. Endoplasmic Reticulum Stress in Disease Pathogenesis. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 399–425. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef]
- Takada, K.; Hirose, J.; Senba, K.; Yamabe, S.; Oike, Y.; Gotoh, T.; Mizuta, H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int. J. Exp. Pathol. 2011, 92, 232–242. [Google Scholar] [CrossRef]
- Yudoh, K.; Nakamura, H.; Masuko-Hongo, K.; Kato, T.; Nishioka, K. Catabolic stress induces expression of hypoxia-inducible factor (HIF)-1α in articular chondrocytes: Involvement of HIF-1α in the pathogenesis of osteoarthritis. Arthritis Res. Ther. 2005, 7, R904. [Google Scholar] [CrossRef]
- Almonte-Becerril, M.; Navarro-Garcia, F.; Gonzalez-Robles, A.; Vega-Lopez, M.A.; Lavalle, C.; Kouri, J.B. Cell death of chondrocytes is a combination between apoptosis and autophagy during the pathogenesis of Osteoarthritis within an experimental model. Apoptosis 2010, 15, 631–638. [Google Scholar] [CrossRef]
- Vancompernolle, K.; Van Herreweghe, F.; Pynaert, G.; Van De Craen, M.; De Vos, K.; Totty, N.; Sterling, A.; Fiers, W.; Vandenabeele, P.; Grooten, J. Atractyloside-induced release of cathepsin B, a protease with caspase-processing activity. FEBS Lett. 1998, 438, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Deussing, J.; Miyoshi, H.; Bronk, S.F.; Svingen, P.A.; Peters, C.; Kaufmann, S.H.; Gores, G.J. Cathepsin B contributes to TNF-α–mediated hepatocyte apoptosis by promoting mitochondrial release of cytochrome c. J. Clin. Investig. 2000, 106, 1127–1137. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Bhalla, K.; Kim, C.N.; Ibrado, A.M.; Cai, J.; Peng, T.-I.; Jones, D.P.; Wang, X. Prevention of Apoptosis by Bcl-2: Release of Cytochrome c from Mitochondria Blocked. Science 1997, 275, 1129–1132. [Google Scholar] [CrossRef]
- Werneburg, N.; Guicciardi, M.E.; Yin, X.-M.; Gores, G.J. TNF-α-mediated lysosomal permeabilization is FAN and caspase 8/Bid dependent. Am. J. Physiol.-Gastrointest. Liver Physiol. 2004, 287, G436–G443. [Google Scholar] [CrossRef]
- Kågedal, K.; Johansson, A.-C.; Johansson, U.; Heimlich, G.; Roberg, K.; Wang, N.S.; Jürgensmeier, J.M.; Öllinger, K. Lysosomal membrane permeabilization during apoptosis-involvement of Bax?: Bax-mediated lysosomal membrane permeabilization. Int. J. Exp. Pathol. 2005, 86, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Van Den Berg, W.B. Osteoarthritis year 2010 in review: Pathomechanisms. Osteoarthr. Cartil. 2011, 19, 338–341. [Google Scholar] [CrossRef]
- Nuttall, M.E.; Nadeau, D.P.; Fisher, P.W.; Wang, F.; Keller, P.M.; Dewolf, W.E., Jr.; Goldring, M.B.; Badger, A.M.; Lee, D.; Levy, M.A.; et al. Inhibition of caspase-3-like activity prevents apoptosis while retaining functionality of human chondrocytes in vitro. J. Orthop. Res. 2000, 18, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, J.P.; Fernandes, J.C.; Jovanovic, D.V.; Reboul, P.; Martel-Pelletier, J. Chondrocyte death in experimental osteoarthritis is mediated by MEK 1/2 and p38 pathways: Role of cyclooxygenase-2 and inducible nitric oxide synthase. J. Rheumatol. 2001, 28, 2509–2519. [Google Scholar]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006, 54, 1814–1821. [Google Scholar] [CrossRef]
- Sugiyama, M.; Tsukazaki, T.; Yonekura, A.; Matsuzaki, S.; Yamashita, S.; Iwasaki, K. Localisation of apoptosis and expression of apoptosis related proteins in the synovium of patients with rheumatoid arthritis. Ann. Rheum. Dis. 1996, 55, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Perlman, H.; Pagliari, L.J.; Liu, H.; Koch, A.E.; Haines, G.K.; Pope, R.M. Rheumatoid arthritis synovial macrophages express the Fas-associated death domain-like interleukin-1?-converting enzyme-inhibitory protein and are refractory to Fas-mediated apoptosis. Arthritis Rheum. 2001, 44, 21–30. [Google Scholar] [CrossRef]
- Perlman, H.; Pagliari, L.J.; Georganas, C.; Mano, T.; Walsh, K.; Pope, R.M. Flice-Inhibitory Protein Expression during Macrophage Differentiation Confers Resistance to FAS-Mediated Apoptosis. J. Exp. Med. 1999, 190, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Pagliari, L.J.; Perlman, H.; Liu, H.; Pope, R.M. Macrophages Require Constitutive NF-κB Activation To Maintain A1 Expression and Mitochondrial Homeostasis. Mol. Cell. Biol. 2000, 20, 8855–8865. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; Echeverri, F.; Yeo, M.; Zvaifler, N.J.; Green, D.R. Somatic mutations in the p53 tumor suppressor gene in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 1997, 94, 10895–10900. [Google Scholar] [CrossRef]
- Aupperle, K.R.; Boyle, D.L.; Hendrix, M.; Seftor, E.A.; Zvaifler, N.J.; Barbosa, M.; Firestein, G.S. Regulation of synoviocyte proliferation, apoptosis, and invasion by the p53 tumor suppressor gene. Am. J. Pathol. 1998, 152, 1091–1098. [Google Scholar]
- Han, Z.; Boyle, D.L.; Shi, Y.; Green, D.R.; Firestein, G.S. Dominant-negative p53 mutations in rheumatoid arthritis. Arthritis Rheum. 1999, 42, 1088–1092. [Google Scholar] [CrossRef]
- Chen, X.; Gong, W.; Shao, X.; Shi, T.; Zhang, L.; Dong, J.; Shi, Y.; Shen, S.; Qin, J.; Jiang, Q.; et al. METTL3-mediated m6A modification of ATG7 regulates autophagy-GATA4 axis to promote cellular senescence and osteoarthritis progression. Ann. Rheum. Dis. 2022, 81, 85–97. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent cells and osteoarthritis: A painful connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Martin, J.A.; Brown, T.D.; Heiner, A.D.; Buckwalter, J.A. Chondrocyte Senescence, Joint Loading and Osteoarthritis. Clin. Orthop. Relat. Res. 2004, 427, S96–S103. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Patil, C.K.; Rodier, F.; Sun, Y.; Muñoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.-Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef] [PubMed]
- Lucken-Ardjomande, S.; Montessuit, S.; Martinou, J.-C. Contributions to Bax insertion and oligomerization of lipids of the mitochondrial outer membrane. Cell Death Differ. 2008, 15, 929–937. [Google Scholar] [CrossRef] [PubMed]
- Raemy, E.; Montessuit, S.; Pierredon, S.; Van Kampen, A.H.; Vaz, F.M.; Martinou, J.-C. Cardiolipin or MTCH2 can serve as tBID receptors during apoptosis. Cell Death Differ. 2016, 23, 1165–1174. [Google Scholar] [CrossRef]
- Landeta, O.; Landajuela, A.; Gil, D.; Taneva, S.; DiPrimo, C.; Sot, B.; Valle, M.; Frolov, V.A.; Basañez, G. Reconstitution of Proapoptotic BAK Function in Liposomes Reveals a Dual Role for Mitochondrial Lipids in the BAK-driven Membrane Permeabilization Process. J. Biol. Chem. 2011, 286, 8213–8230. [Google Scholar] [CrossRef] [PubMed]
- De Kroon, A.I.P.M.; Dolis, D.; Mayer, A.; Lill, R.; De Kruijff, B. Phospholipid composition of highly purified mitochondrial outer membranes of rat liver and Neurospora crassa. Is cardiolipin present in the mitochondrial outer membrane? Biochim. Biophys. Acta (BBA) Biomembr. 1997, 1325, 108–116. [Google Scholar] [CrossRef]
- Lutter, M.; Fang, M.; Luo, X.; Nishijima, M.; Xie, X.; Wang, X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat. Cell Biol. 2000, 2, 754–756. [Google Scholar] [CrossRef]
- Liu, J.; Dai, Q.; Chen, J.; Durrant, D.; Freeman, A.; Liu, T.; Grossman, D.; Lee, R.M. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol. Cancer Res. 2003, 1, 892–902. [Google Scholar]
- Martínez-Abundis, E.; García, N.; Correa, F.; Franco, M.; Zazueta, C. Changes in specific lipids regulate BAX-induced mitochondrial permeability transition: Microdomain components-effect on BAX-induced mPT. FEBS J. 2007, 274, 6500–6510. [Google Scholar] [CrossRef]
- Martínez-Abundis, E.; Correa, F.; Pavón, N.; Zazueta, C. Bax distribution into mitochondrial detergent-resistant microdomains is related to ceramide and cholesterol content in postischemic hearts: Bax translocation into mitochondrial microdomains. FEBS J. 2009, 276, 5579–5588. [Google Scholar] [CrossRef]
- Ganesan, V.; Perera, M.N.; Colombini, D.; Datskovskiy, D.; Chadha, K.; Colombini, M. Ceramide and activated Bax act synergistically to permeabilize the mitochondrial outer membrane. Apoptosis 2010, 15, 553–562. [Google Scholar] [CrossRef]
- Perera, M.N.; Lin, S.H.; Peterson, Y.K.; Bielawska, A.; Szulc, Z.M.; Bittman, R.; Colombini, M. Bax and Bcl-xL exert their regulation on different sites of the ceramide channel. Biochem. J. 2012, 445, 81–91. [Google Scholar] [CrossRef] [PubMed]
- Cowan, A.D.; Smith, N.A.; Sandow, J.J.; Kapp, E.A.; Rustam, Y.H.; Murphy, J.M.; Brouwer, J.M.; Bernardini, J.P.; Roy, M.J.; Wardak, A.Z.; et al. BAK core dimers bind lipids and can be bridged by them. Nat. Struct. Mol. Biol. 2020, 27, 1024–1031. [Google Scholar] [CrossRef]
- Portanova, J.P.; Zhang, Y.; Anderson, G.D.; Hauser, S.D.; Masferrer, J.L.; Seibert, K.; Gregory, S.A.; Isakson, P.C. Selective neutralization of prostaglandin E2 blocks inflammation, hyperalgesia, and interleukin 6 production in vivo. J. Exp. Med. 1996, 184, 883–891. [Google Scholar] [CrossRef]
- Lalier, L.; Cartron, P.-F.; Pedelaborde, F.; Olivier, C.; Loussouarn, D.; Martin, S.A.; Meflah, K.; Menanteau, J.; Vallette, F.M. Increase in PGE2 biosynthesis induces a Bax dependent apoptosis correlated to patients’ survival in glioblastoma multiforme. Oncogene 2007, 26, 4999–5009. [Google Scholar] [CrossRef] [PubMed]
- Lalier, L.; Cartron, P.-F.; Olivier, C.; Logé, C.; Bougras, G.; Robert, J.-M.; Oliver, L.; Vallette, F.M. Prostaglandins antagonistically control Bax activation during apoptosis. Cell Death Differ. 2011, 18, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-S.; Rhim, H.; Jeong, S.-W.; Kim, J.W.; Kim, I.-K. Induction of apoptosis dependent on caspase activities and growth arrest in HL-60 cells by PGA2. Prostaglandins Other Lipid Mediat. 2002, 70, 169–183. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werry, F.; Mazur, E.; Theyse, L.F.H.; Edlich, F. Apoptosis Regulation in Osteoarthritis and the Influence of Lipid Interactions. Int. J. Mol. Sci. 2023, 24, 13028. https://doi.org/10.3390/ijms241713028
Werry F, Mazur E, Theyse LFH, Edlich F. Apoptosis Regulation in Osteoarthritis and the Influence of Lipid Interactions. International Journal of Molecular Sciences. 2023; 24(17):13028. https://doi.org/10.3390/ijms241713028
Chicago/Turabian StyleWerry, Frederike, Emilia Mazur, Lars F. H. Theyse, and Frank Edlich. 2023. "Apoptosis Regulation in Osteoarthritis and the Influence of Lipid Interactions" International Journal of Molecular Sciences 24, no. 17: 13028. https://doi.org/10.3390/ijms241713028