
Repeated Methamphetamine Administration Results in Axon Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta and Locus Coeruleus Neurons in Male but Not Female Mice

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
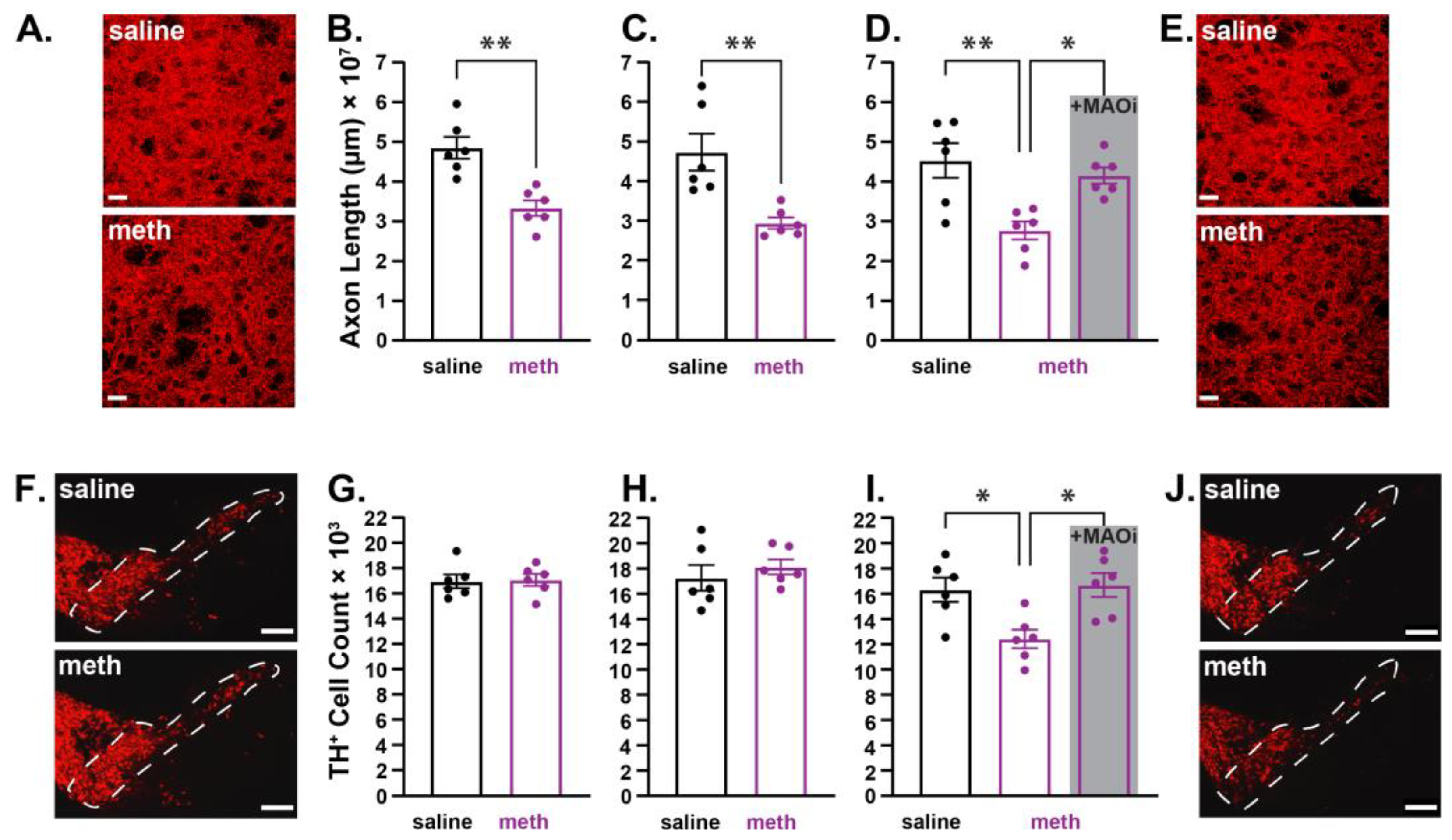
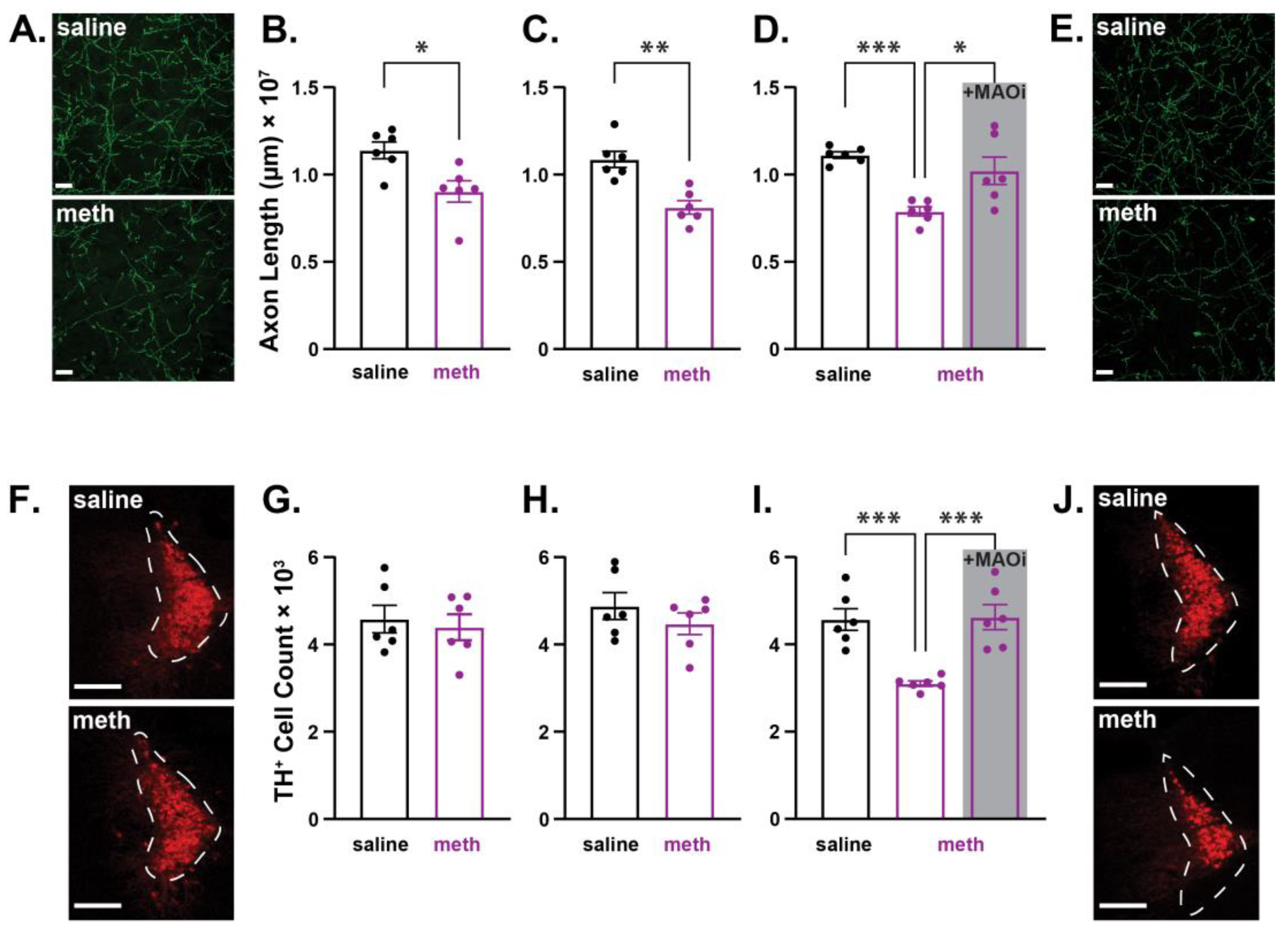
2.1. Chronic Methamphetamine Administration Resulted in Axonal Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta Dopamine Neurons in Male Mice
2.2. Chronic Administration of Methamphetamine Resulted in Axonal Loss Prior to Somatic Loss of Locus Coeruleus Norepinephrine Neurons in Male Mice
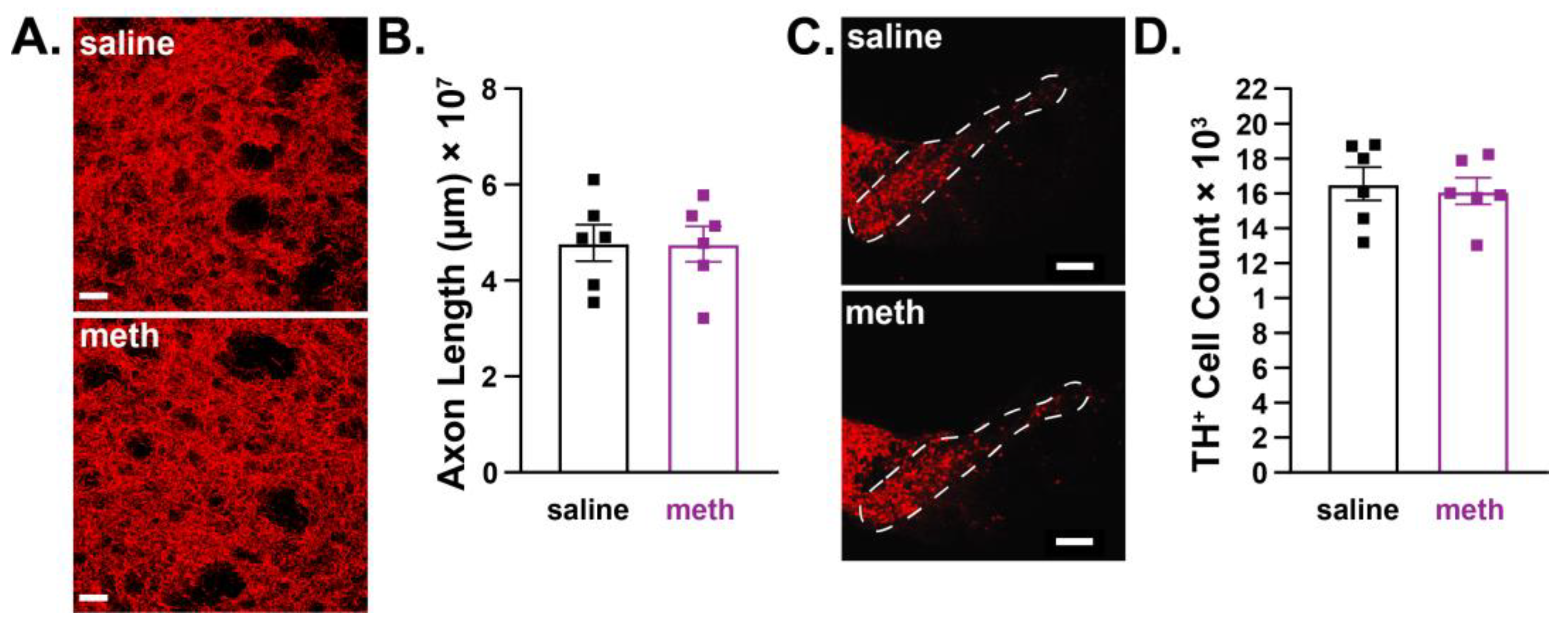
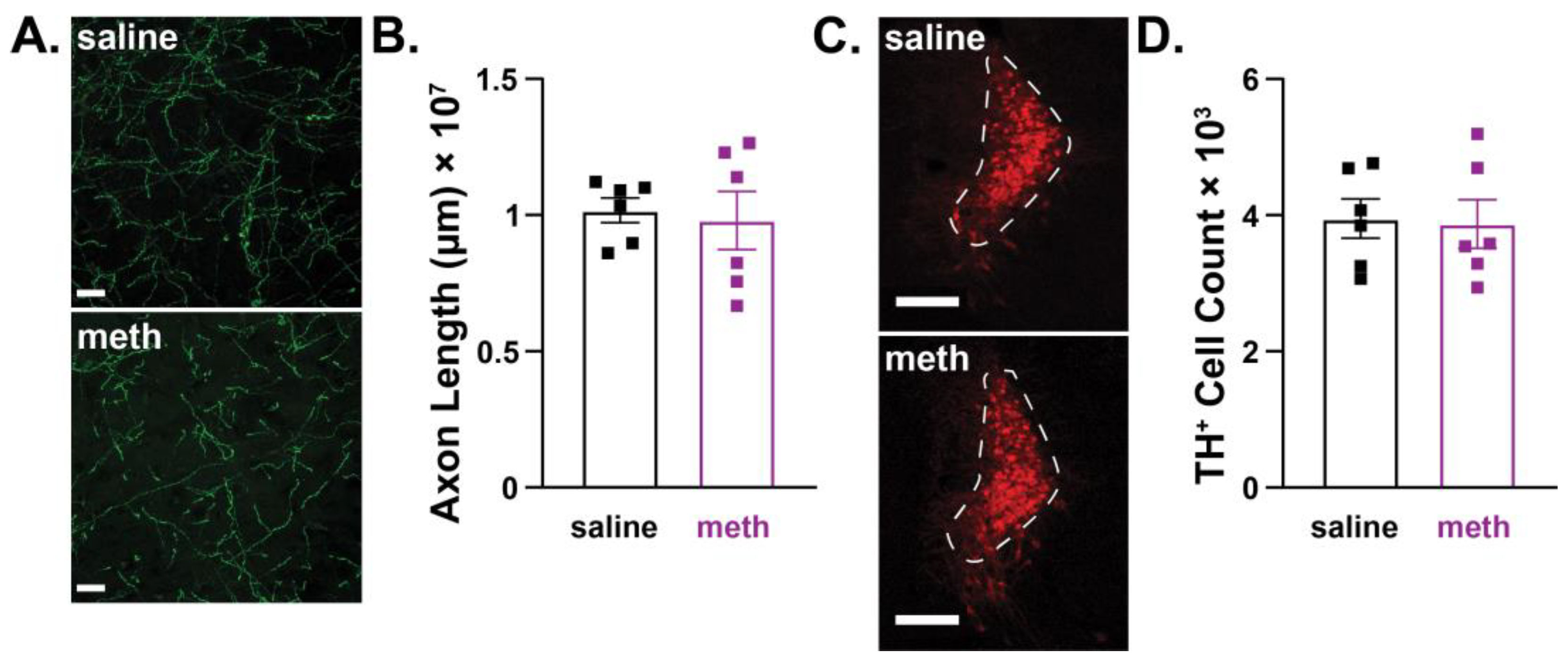
2.3. Female Mice were Resistant to Chronic Methamphetamine-Induced Degeneration of Substantia Nigra Pars Compacta Dopamine Neurons
2.4. Female Mice were Resistant to Chronic Methamphetamine-Induced Degeneration of Locus Coeruleus Norepinephrine Neurons
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Experimental Subjects
5.2. In Vivo Drug Administration
5.3. Immunohistochemistry
5.4. Stereological Quantification
5.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2016 National Survey on Drug Use and Health; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2017. [Google Scholar]
- Substance Abuse and Mental Health Services Administration. Key Substance Use and Mental Health Indicators in the United States: Results from the 2021 National Survey on Drug Use and Health; Substance Abuse and Mental Health Services Administration: Rockville, MD, USA, 2022. [Google Scholar]
- Jones, C.M.; Houry, D.; Han, B.; Baldwin, G.; Vivolo-Kantor, A.; Compton, W.M. Methamphetamine use in the United States: Epidemiological update and implications for prevention, treatment, and harm reduction. Ann. N. Y. Acad. Sci. 2022, 1508, 3–22. [Google Scholar] [CrossRef] [PubMed]
- Jayanthi, S.; Daiwile, A.P.; Cadet, J.L. Neurotoxicity of methamphetamine: Main effects and mechanisms. Exp. Neurol. 2021, 344, 113795. [Google Scholar] [CrossRef] [PubMed]
- Shaerzadeh, F.; Streit, W.J.; Heysieattalab, S.; Khoshbouei, H. Methamphetamine neurotoxicity, microglia, and neuroinflammation. J. Neuroinflam. 2018, 15, 341. [Google Scholar] [CrossRef] [PubMed]
- McCann, U.D.; Wong, D.F.; Yokoi, F.; Villemagne, V.; Dannals, R.F.; Ricaurte, G.A. Reduced striatal dopamine transporter density in abstinent methamphetamine and methcathinone users: Evidence from positron emission tomography studies with [11C]WIN-35,428. J. Neurosci. 1998, 18, 8417–8422. [Google Scholar] [CrossRef]
- Volkow, N.D.; Chang, L.; Wang, G.J.; Fowler, J.S.; Leonido-Yee, M.; Franceschi, D.; Sedler, M.J.; Gatley, S.J.; Hitzemann, R.; Ding, Y.S.; et al. Association of dopamine transporter reduction with psychomotor impairment in methamphetamine abusers. Am. J. Psychiatry 2001, 158, 377–382. [Google Scholar] [CrossRef]
- McCann, U.D.; Kuwabara, H.; Kumar, A.; Palermo, M.; Abbey, R.; Brasic, J.; Ye, W.; Alexander, M.; Dannals, R.F.; Wong, D.F.; et al. Persistent cognitive and dopamine transporter deficits in abstinent methamphetamine users. Synapse 2008, 62, 91–100. [Google Scholar] [CrossRef]
- Johanson, C.E.; Frey, K.A.; Lundahl, L.H.; Keenan, P.; Lockhart, N.; Roll, J.; Galloway, G.P.; Koeppe, R.A.; Kilbourn, M.R.; Robbins, T.; et al. Cognitive function and nigrostriatal markers in abstinent methamphetamine abusers. Psychopharmacology 2006, 185, 327–338. [Google Scholar] [CrossRef]
- Sekine, Y.; Iyo, M.; Ouchi, Y.; Matsunaga, T.; Tsukada, H.; Okada, H.; Yoshikawa, E.; Futatsubashi, M.; Takei, N.; Mori, N. Methamphetamine-related psychiatric symptoms and reduced brain dopamine transporters studied with PET. Am. J. Psychiatry 2001, 158, 1206–1214. [Google Scholar] [CrossRef]
- Moszczynska, A.; Callan, S.P. Molecular, Behavioral, and Physiological Consequences of Methamphetamine Neurotoxicity: Implications for Treatment. J. Pharmacol. Exp. Ther. 2017, 362, 474. [Google Scholar] [CrossRef]
- Wilson, J.M.; Kalasinsky, K.S.; Levey, A.I.; Bergeron, C.; Reiber, G.; Anthony, R.M.; Schmunk, G.A.; Shannak, K.; Haycock, J.W.; Kish, S.J. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat. Med. 1996, 2, 699–703. [Google Scholar] [CrossRef]
- Kitamura, O.; Tokunaga, I.; Gotohda, T.; Kubo, S. Immunohistochemical investigation of dopaminergic terminal markers and caspase-3 activation in the striatum of human methamphetamine users. Int. J. Leg. Med. 2007, 121, 163–168. [Google Scholar] [CrossRef]
- Ares-Santos, S.; Granado, N.; Espadas, I.; Martinez-Murillo, R.; Moratalla, R. Methamphetamine causes degeneration of dopamine cell bodies and terminals of the nigrostriatal pathway evidenced by silver staining. Neuropsychopharmacology 2014, 39, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Blaker, A.L.; Rodriguez, E.A.; Yamamoto, B.K. Neurotoxicity to dopamine neurons after the serial exposure to alcohol and methamphetamine: Protection by COX-2 antagonism. Brain Behav. Immun. 2019, 81, 317–328. [Google Scholar] [CrossRef]
- Dang, D.K.; Shin, E.J.; Nam, Y.; Ryoo, S.; Jeong, J.H.; Jang, C.G.; Nabeshima, T.; Hong, J.S.; Kim, H.C. Apocynin prevents mitochondrial burdens, microglial activation, and pro-apoptosis induced by a toxic dose of methamphetamine in the striatum of mice via inhibition of p47phox activation by ERK. J. Neuroinflam. 2016, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Eyerman, D.J.; Yamamoto, B.K. Lobeline attenuates methamphetamine-induced changes in vesicular monoamine transporter 2 immunoreactivity and monoamine depletions in the striatum. J. Pharmacol. Exp. Ther. 2005, 312, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Eyerman, D.J.; Yamamoto, B.K. A rapid oxidation and persistent decrease in the vesicular monoamine transporter 2 after methamphetamine. J. Neurochem. 2007, 103, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Frey, K.; Kilbourn, M.; Robinson, T. Reduced striatal vesicular monoamine transporters after neurotoxic but not after behaviorally-sensitizing doses of methamphetamine. Eur. J. Pharmacol. 1997, 334, 273–279. [Google Scholar] [CrossRef]
- Fumagalli, F.; Gainetdinov, R.R.; Valenzano, K.J.; Caron, M.G. Role of dopamine transporter in methamphetamine-induced neurotoxicity: Evidence from mice lacking the transporter. J. Neurosci. 1998, 18, 4861–4869. [Google Scholar] [CrossRef]
- Granado, N.; Ares-Santos, S.; O’Shea, E.; Vicario-Abejón, C.; Colado, M.I.; Moratalla, R. Selective vulnerability in striosomes and in the nigrostriatal dopaminergic pathway after methamphetamine administration: Early loss of TH in striosomes after methamphetamine. Neurotox. Res. 2010, 18, 48–58. [Google Scholar] [CrossRef]
- Granado, N.; Ares-Santos, S.; Tizabi, Y.; Moratalla, R. Striatal Reinnervation Process after Acute Methamphetamine-Induced Dopaminergic Degeneration in Mice. Neurotox. Res. 2018, 34, 627–639. [Google Scholar] [CrossRef]
- Guillot, T.S.; Shepherd, K.R.; Richardson, J.R.; Wang, M.Z.; Li, Y.; Emson, P.C.; Miller, G.W. Reduced vesicular storage of dopamine exacerbates methamphetamine-induced neurodegeneration and astrogliosis. J. Neurochem. 2008, 106, 2205–2217. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Staal, R.G.; Sonsalla, P.K. Analysis of VMAT2 binding after methamphetamine or MPTP treatment: Disparity between homogenates and vesicle preparations. J. Neurochem. 2000, 74, 2217–2220. [Google Scholar] [CrossRef] [PubMed]
- Lohr, K.M.; Stout, K.A.; Dunn, A.R.; Wang, M.; Salahpour, A.; Guillot, T.S.; Miller, G.W. Increased Vesicular Monoamine Transporter 2 (VMAT2; Slc18a2) Protects against Methamphetamine Toxicity. ACS Chem. Neurosci. 2015, 6, 790–799. [Google Scholar] [CrossRef]
- Mark, K.A.; Soghomonian, J.J.; Yamamoto, B.K. High-dose methamphetamine acutely activates the striatonigral pathway to increase striatal glutamate and mediate long-term dopamine toxicity. J. Neurosci. 2004, 24, 11449–11456. [Google Scholar] [CrossRef] [PubMed]
- McConnell, S.E.; O’Banion, M.K.; Cory-Slechta, D.A.; Olschowka, J.A.; Opanashuk, L.A. Characterization of binge-dosed methamphetamine-induced neurotoxicity and neuroinflammation. Neurotoxicology 2015, 50, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Moszczynska, A.; Yamamoto, B.K. Methamphetamine oxidatively damages parkin and decreases the activity of 26S proteasome in vivo. J. Neurochem. 2011, 116, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Northrop, N.A.; Yamamoto, B.K. Cyclooxygenase activity contributes to the monoaminergic damage caused by serial exposure to stress and methamphetamine. Neuropharmacology 2013, 72, 96–105. [Google Scholar] [CrossRef]
- O’Callaghan, J.P.; Miller, D.B. Neurotoxicity profiles of substituted amphetamines in the C57BL/6J mouse. J. Pharmacol. Exp. Ther. 1994, 270, 741–751. [Google Scholar]
- Reichel, C.M.; Ramsey, L.A.; Schwendt, M.; McGinty, J.F.; See, R.E. Methamphetamine-induced changes in the object recognition memory circuit. Neuropharmacology 2012, 62, 1119–1126. [Google Scholar] [CrossRef]
- Salvatore, M.F.; Nejtek, V.A.; Khoshbouei, H. Prolonged increase in ser31 tyrosine hydroxylase phosphorylation in substantia nigra following cessation of chronic methamphetamine. Neurotoxicology 2018, 67, 121–128. [Google Scholar] [CrossRef]
- Seiden, L.S.; Commins, D.L.; Vosmer, G.; Axt, K.; Marek, G. Neurotoxicity in dopamine and 5-hydroxytryptamine terminal fields: A regional analysis in nigrostriatal and mesolimbic projections. Ann. N. Y. Acad. Sci. 1988, 537, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Sonsalla, P.K.; Jochnowitz, N.D.; Zeevalk, G.D.; Oostveen, J.A.; Hall, E.D. Treatment of mice with methamphetamine produces cell loss in the substantia nigra. Brain Res. 1996, 738, 172–175. [Google Scholar] [CrossRef] [PubMed]
- Truong, J.G.; Wilkins, D.G.; Baudys, J.; Crouch, D.J.; Johnson-Davis, K.L.; Gibb, J.W.; Hanson, G.R.; Fleckenstein, A.E. Age-dependent methamphetamine-induced alterations in vesicular monoamine transporter-2 function: Implications for neurotoxicity. J. Pharmacol. Exp. Ther. 2005, 314, 1087–1092. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Lee, Y.B.; Graves, S.M. Chronic methamphetamine-induced neurodegeneration: Differential vulnerability of ventral tegmental area and substantia nigra pars compacta dopamine neurons. Neuropharmacology 2021, 200, 108817. [Google Scholar] [CrossRef]
- Graves, S.M.; Schwarzschild, S.E.; Tai, R.A.; Chen, Y.; Surmeier, D.J. Mitochondrial oxidant stress mediates methamphetamine neurotoxicity in substantia nigra dopaminergic neurons. Neurobiol. Dis. 2021, 156, 105409. [Google Scholar] [CrossRef]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20. [Google Scholar] [CrossRef]
- Sulzer, D.; Sonders, M.S.; Poulsen, N.W.; Galli, A. Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 2005, 75, 406–433. [Google Scholar] [CrossRef]
- Freyberg, Z.; Sonders, M.S.; Aguilar, J.I.; Hiranita, T.; Karam, C.S.; Flores, J.; Pizzo, A.B.; Zhang, Y.; Farino, Z.J.; Chen, A.; et al. Mechanisms of amphetamine action illuminated through optical monitoring of dopamine synaptic vesicles in Drosophila brain. Nat. Commun. 2016, 7, 10652. [Google Scholar] [CrossRef]
- Rothman, R.B.; Baumann, M.H.; Dersch, C.M.; Romero, D.V.; Rice, K.C.; Carroll, F.I.; Partilla, J.S. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse 2001, 39, 32–41. [Google Scholar] [CrossRef]
- Du, Y.; Choi, S.; Pilski, A.; Graves, S.M. Differential vulnerability of locus coeruleus and dorsal raphe neurons to chronic methamphetamine-induced degeneration. Front. Cell Neurosci. 2022, 16, 949923. [Google Scholar] [CrossRef]
- Bourque, M.; Dluzen, D.E.; Di Paolo, T. Male/Female differences in neuroprotection and neuromodulation of brain dopamine. Front. Endocrinol. 2011, 2, 35. [Google Scholar] [CrossRef] [PubMed]
- Bourque, M.; Liu, B.; Dluzen, D.E.; Di Paolo, T. Sex differences in methamphetamine toxicity in mice: Effect on brain dopamine signaling pathways. Psychoneuroendocrinology 2011, 36, 955–969. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Dluzen, D.E. Effect of estrogen upon methamphetamine-induced neurotoxicity within the impaired nigrostriatal dopaminergic system. Synapse 2006, 60, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Dluzen, D.E.; Mcdermott, J.L. Estrogen, Testosterone, and Methamphetamine Toxicity. Ann. N. Y. Acad. Sci. 2006, 1074, 282–294. [Google Scholar] [CrossRef]
- Dluzen, D.E.; McDermott, J.L. Neuroprotective role of estrogen upon methamphetamine and related neurotoxins within the nigrostriatal dopaminergic system. Ann. N. Y. Acad. Sci. 2000, 914, 112–126. [Google Scholar] [CrossRef]
- Gajjar, T.M.; Anderson, L.I.; Dluzen, D.E. Acute effects of estrogen upon methamphetamine induced neurotoxicity of the nigrostriatal dopaminergic system. J. Neural Transm. 2003, 110, 1215–1224. [Google Scholar] [CrossRef]
- Dluzen, D.E.; Anderson, L.I.; Pilati, C.F. Methamphetamine–gonadal steroid hormonal interactions:: Effects upon acute toxicity and striatal dopamine concentrations. Neurotoxicol. Teratol. 2002, 24, 267–273. [Google Scholar] [CrossRef]
- Liu, B.; Dluzen, D.E. Effects of Estrogen and Related Agents upon Methamphetamine-Induced Neurotoxicity within an Impaired Nigrostriatal Dopaminergic System of Ovariectomized Mice. Neuroendocrinology 2006, 83, 295–302. [Google Scholar] [CrossRef]
- Kim, K.K.; Adelstein, R.S.; Kawamoto, S. Identification of neuronal nuclei (NeuN) as Fox-3, a new member of the Fox-1 gene family of splicing factors. J. Biol. Chem. 2009, 284, 31052–31061. [Google Scholar] [CrossRef]
- González-Rodríguez, P.; Zampese, E.; Stout, K.A.; Guzman, J.N.; Ilijic, E.; Yang, B.; Tkatch, T.; Stavarache, M.A.; Wokosin, D.L.; Gao, L.; et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature 2021, 599, 650–656. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sánchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef]
- Matschke, L.A.; Bertoune, M.; Roeper, J.; Snutch, T.P.; Oertel, W.H.; Rinné, S.; Decher, N. A concerted action of L- and T-type Ca2+ channels regulates locus coeruleus pacemaking. Mol. Cell Neurosci. 2015, 68, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Padilla, J.; Guzman, J.N.; Ilijic, E.; Kondapalli, J.; Galtieri, D.J.; Yang, B.; Schieber, S.; Oertel, W.; Wokosin, D.; Schumacker, P.T.; et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 2014, 17, 832–840. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045. [Google Scholar] [CrossRef]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef]
- Ilijic, E.; Guzman, J.N.; Surmeier, D.J. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 43, 364–371. [Google Scholar] [CrossRef]
- Mermelstein, P.; Becker, J.; Surmeier, D. Estradiol reduces calcium currents in rat neostriatal neurons via a membrane receptor. J. Neurosci. 1996, 16, 595–604. [Google Scholar] [CrossRef]
- Kousik, S.M.; Carvey, P.M.; Napier, T.C. Methamphetamine self-administration results in persistent dopaminergic pathology: Implications for Parkinson’s disease risk and reward-seeking. Eur. J. Neurosci. 2014, 40, 2707–2714. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013, 28, 41–50. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- O’Keeffe, G.W.; Sullivan, A.M. Evidence for dopaminergic axonal degeneration as an early pathological process in Parkinson’s disease. Park. Relat. Disord. 2018, 56, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferro, P.; Burke, R.E. Retrograde Axonal Degeneration in Parkinson Disease. J. Park. Dis. 2016, 6, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kurowska, Z.; Kordower, J.H.; Stoessl, A.J.; Burke, R.E.; Brundin, P.; Yue, Z.; Brady, S.T.; Milbrandt, J.; Trapp, B.D.; Sherer, T.B.; et al. Is Axonal Degeneration a Key Early Event in Parkinson’s Disease? J. Park. Dis. 2016, 6, 703–707. [Google Scholar] [CrossRef]
- Cheng, H.C.; Ulane, C.M.; Burke, R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010, 67, 715–725. [Google Scholar] [CrossRef]
- Baloyannis, S.J.; Costa, V.; Baloyannis, I.S. Morphological alterations of the synapses in the locus coeruleus in Parkinson’s disease. J. Neurol. Sci. 2006, 248, 35–41. [Google Scholar] [CrossRef]
- Doppler, C.E.J.; Kinnerup, M.B.; Brune, C.; Farrher, E.; Betts, M.; Fedorova, T.D.; Schaldemose, J.L.; Knudsen, K.; Ismail, R.; Seger, A.D.; et al. Regional locus coeruleus degeneration is uncoupled from noradrenergic terminal loss in Parkinson’s disease. Brain 2021, 144, 2732–2744. [Google Scholar] [CrossRef]
- Persons, A.L.; Desai Bradaric, B.; Kelly, L.P.; Kousik, S.M.; Graves, S.M.; Yamamoto, B.K.; Napier, T.C. Gut and brain profiles that resemble pre-motor and early-stage Parkinson’s disease in methamphetamine self-administering rats. Drug Alcohol. Depend. 2021, 225, 108746. [Google Scholar] [CrossRef]
- Fornai, F.; Lenzi, P.; Ferrucci, M.; Lazzeri, G.; di Poggio, A.B.; Natale, G.; Busceti, C.L.; Biagioni, F.; Giusiani, M.; Ruggieri, S.; et al. Occurrence of neuronal inclusions combined with increased nigral expression of alpha-synuclein within dopaminergic neurons following treatment with amphetamine derivatives in mice. Brain Res. Bull. 2005, 65, 405–413. [Google Scholar] [CrossRef]
- Butler, B.; Gamble-George, J.; Prins, P.; North, A.; Clarke, J.T.; Khoshbouei, H. Chronic Methamphetamine Increases Alpha-Synuclein Protein Levels in the Striatum and Hippocampus but not in the Cortex of Juvenile Mice. J. Addict. Prev. 2014, 2, 6. [Google Scholar]
- Klein, C.; König, I.R. Exploring Uncharted Territory: Genetically Determined Sex Differences in Parkinson’s Disease. Ann. Neurol. 2021, 90, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.C.; Cunningham, J.K.; Sajeev, G.; Kish, S.J. Incidence of Parkinson’s disease among hospital patients with methamphetamine-use disorders. Mov. Disord. 2010, 25, 2333–2339. [Google Scholar] [CrossRef] [PubMed]
- Callaghan, R.C.; Cunningham, J.K.; Sykes, J.; Kish, S.J. Increased risk of Parkinson’s disease in individuals hospitalized with conditions related to the use of methamphetamine or other amphetamine-type drugs. Drug Alcohol. Depend. 2012, 120, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Granado, N.; Ares-Santos, S.; Moratalla, R. Methamphetamine and Parkinson’s disease. Park. Dis. 2013, 2013, 308052. [Google Scholar] [CrossRef]
- Moratalla, R.; Khairnar, A.; Simola, N.; Granado, N.; García-Montes, J.R.; Porceddu, P.F.; Tizabi, Y.; Costa, G.; Morelli, M. Amphetamine-related drugs neurotoxicity in humans and in experimental animals: Main mechanisms. Prog. Neurobiol. 2017, 155, 149–170. [Google Scholar] [CrossRef]
- Lappin, J.M.; Darke, S. Methamphetamine and heightened risk for early-onset stroke and Parkinson’s disease: A review. Exp. Neurol. 2021, 343, 113793. [Google Scholar] [CrossRef]
- Curtin, K.; Fleckenstein, A.E.; Robison, R.J.; Crookston, M.J.; Smith, K.R.; Hanson, G.R. Methamphetamine/amphetamine abuse and risk of Parkinson’s disease in Utah: A population-based assessment. Drug Alcohol. Depend. 2015, 146, 30–38. [Google Scholar] [CrossRef]
- Moszczynska, A.; Fitzmaurice, P.; Ang, L.; Kalasinsky, K.S.; Schmunk, G.A.; Peretti, F.J.; Aiken, S.S.; Wickham, D.J.; Kish, S.J. Why is parkinsonism not a feature of human methamphetamine users? Brain 2004, 127, 363–370. [Google Scholar] [CrossRef]
- Kish, S.J.; Boileau, I.; Callaghan, R.C.; Tong, J. Brain dopamine neurone ‘damage’: Methamphetamine users vs. Parkinson’s disease—A critical assessment of the evidence. Eur. J. Neurosci. 2017, 45, 58–66. [Google Scholar] [CrossRef]
- Prince, M.; Albanese, E.; Guerchet, M.; Prina, M. World Alzheimer Report 2014. Dementia and Risk Reduction: An Analysis of Protective and Modifiable Risk Factors; Alzheimer’s Disease International: London, UK, 2014. [Google Scholar]
- Lane, C.A.; Hardy, J.; Schott, J.M. Alzheimer’s disease. Eur. J. Neurol. 2018, 25, 59–70. [Google Scholar] [CrossRef]
- Kelly, S.C.; He, B.; Perez, S.E.; Ginsberg, S.D.; Mufson, E.J.; Counts, S.E. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2017, 5, 8. [Google Scholar] [CrossRef]
- Iversen, L.L.; Rossor, M.N.; Reynolds, G.P.; Hills, R.; Roth, M.; Mountjoy, C.Q.; Foote, S.L.; Morrison, J.H.; Bloom, F.E. Loss of pigmented dopamine-beta-hydroxylase positive cells from locus coeruleus in senile dementia of Alzheimer’s type. Neurosci. Lett. 1983, 39, 95–100. [Google Scholar] [CrossRef]
- German, D.C.; Manaye, K.F.; White, C.L., III; Woodward, D.J.; McIntire, D.D.; Smith, W.K.; Kalaria, R.N.; Mann, D.M.A. Disease-specific patterns of locus coeruleus cell loss. Ann. Neurol. 1992, 32, 667–676. [Google Scholar] [CrossRef]
- Dutt, S.; Li, Y.; Mather, M.; Nation, D.A. Brainstem Volumetric Integrity in Preclinical and Prodromal Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 77, 1579–1594. [Google Scholar] [CrossRef] [PubMed]
- Hou, R.; Beardmore, R.; Holmes, C.; Osmond, C.; Darekar, A. A case-control study of the locus coeruleus degeneration in Alzheimer’s disease. Eur. Neuropsychopharmacol. 2021, 43, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Betts, M.J.; Cardenas-Blanco, A.; Kanowski, M.; Spottke, A.; Teipel, S.J.; Kilimann, I.; Jessen, F.; Düzel, E. Locus coeruleus MRI contrast is reduced in Alzheimer’s disease dementia and correlates with CSF Aβ levels. Alzheimer’s Dement 2019, 11, 281–285. [Google Scholar] [CrossRef]
- Liu, L.; Luo, S.; Zeng, L.; Wang, W.; Yuan, L.; Jian, X. Degenerative alterations in noradrenergic neurons of the locus coeruleus in Alzheimer’s disease. Neural Regen. Res. 2013, 8, 2249–2255. [Google Scholar] [CrossRef]
- O’Neil, J.N.; Mouton, P.R.; Tizabi, Y.; Ottinger, M.A.; Lei, D.L.; Ingram, D.K.; Manaye, K.F. Catecholaminergic neuronal loss in locus coeruleus of aged female dtg APP/PS1 mice. J. Chem. Neuroanat. 2007, 34, 102–107. [Google Scholar] [CrossRef]
- Jardanhazi-Kurutz, D.; Kummer, M.P.; Terwel, D.; Vogel, K.; Dyrks, T.; Thiele, A.; Heneka, M.T. Induced LC degeneration in APP/PS1 transgenic mice accelerates early cerebral amyloidosis and cognitive deficits. Neurochem. Int. 2010, 57, 375–382. [Google Scholar] [CrossRef]
- Liu, Y.; Yoo, M.J.; Savonenko, A.; Stirling, W.; Price, D.L.; Borchelt, D.R.; Mamounas, L.; Lyons, W.E.; Blue, M.E.; Lee, M.K. Amyloid pathology is associated with progressive monoaminergic neurodegeneration in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2008, 28, 13805–13814. [Google Scholar] [CrossRef]
- Kang, S.S.; Liu, X.; Ahn, E.H.; Xiang, J.; Manfredsson, F.P.; Yang, X.; Luo, H.R.; Liles, L.C.; Weinshenker, D.; Ye, K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Investig. 2020, 130, 422–437. [Google Scholar] [CrossRef] [PubMed]
- Ehrenberg, A.J.; Nguy, A.K.; Theofilas, P.; Dunlop, S.; Suemoto, C.K.; Di Lorenzo Alho, A.T.; Leite, R.P.; Diehl Rodriguez, R.; Mejia, M.B.; Rüb, U.; et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: The pathological building blocks of early Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2017, 43, 393–408. [Google Scholar] [CrossRef]
- Braun, D.J.; Van Eldik, L.J. In vivo Brainstem Imaging in Alzheimer’s Disease: Potential for Biomarker Development. Front. Aging Neurosci. 2018, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Chalermpalanupap, T.; Schroeder, J.P.; Rorabaugh, J.M.; Liles, L.C.; Lah, J.J.; Levey, A.I.; Weinshenker, D. Locus Coeruleus Ablation Exacerbates Cognitive Deficits, Neuropathology, and Lethality in P301S Tau Transgenic Mice. J. Neurosci. 2018, 38, 74–92. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Ramanathan, M.; Jacobs, A.H.; Dumitrescu-Ozimek, L.; Bilkei-Gorzo, A.; Debeir, T.; Sastre, M.; Galldiks, N.; Zimmer, A.; Hoehn, M.; et al. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J. Neurosci. 2006, 26, 1343–1354. [Google Scholar] [CrossRef]
- Kalinin, S.; Gavrilyuk, V.; Polak, P.E.; Vasser, R.; Zhao, J.; Heneka, M.T.; Feinstein, D.L. Noradrenaline deficiency in brain increases beta-amyloid plaque burden in an animal model of Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1206–1214. [Google Scholar] [CrossRef]
- Kelly, S.C.; McKay, E.C.; Beck, J.S.; Collier, T.J.; Dorrance, A.M.; Counts, S.E. Locus Coeruleus Degeneration Induces Forebrain Vascular Pathology in a Transgenic Rat Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 371–388. [Google Scholar] [CrossRef]
- Heneka, M.T.; Galea, E.; Gavriluyk, V.; Dumitrescu-Ozimek, L.; Daeschner, J.; O’Banion, M.K.; Weinberg, G.; Klockgether, T.; Feinstein, D.L. Noradrenergic depletion potentiates beta -amyloid-induced cortical inflammation: Implications for Alzheimer’s disease. J. Neurosci. 2002, 22, 2434–2442. [Google Scholar] [CrossRef]
- Gao, Z.-x.; Zhang, C.; Lu, J.-c.; Zhao, X.; Qiu, H.; Wang, H.-j. Pathological methamphetamine exposure triggers the accumulation of neuropathic protein amyloid-β by inhibiting UCHL1. NeuroToxicology 2021, 86, 19–25. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, X.; Hu, M.; Yang, T.; Xu, H.; Kang, X.; Chen, X.; Jiang, L.; Gao, R.; Wang, J. Targeting Aβ and p-Tau Clearance in Methamphetamine-Induced Alzheimer’s Disease-Like Pathology: Roles of Syntaxin 17 in Autophagic Degradation in Primary Hippocampal Neurons. Oxid. Med. Cell Longev. 2022, 2022, 3344569. [Google Scholar] [CrossRef]
- Nopparat, C.; Boontor, A.; Panmanee, J.; Govitrapong, P. Melatonin Attenuates Methamphetamine-Induced Alteration of Amyloid β Precursor Protein Cleaving Enzyme Expressions via Melatonin Receptor in Human Neuroblastoma Cells. Neurotox. Res. 2022, 40, 1086–1095. [Google Scholar] [CrossRef]
- Mercan, D.; Heneka, M.T. The Contribution of the Locus Coeruleus-Noradrenaline System Degeneration during the Progression of Alzheimer’s Disease. Biology 2022, 11, 1822. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Cai, W.; Cappellucci, L.; Ghazarian, A.; Collins, W.R.; Vienberg, S.G.; Pothos, E.N.; Kahn, C.R. Insulin resistance in brain alters dopamine turnover and causes behavioral disorders. Proc. Natl. Acad. Sci. USA 2015, 112, 3463–3468. [Google Scholar] [CrossRef] [PubMed]
- West, M.J. Space Balls Revisited: Stereological Estimates of Length With Virtual Isotropic Surface Probes. Front. Neuroanat. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Deniz, Ö.G.; Altun, G.; Kaplan, A.A.; Yurt, K.K.; von Bartheld, C.S.; Kaplan, S. A concise review of optical, physical and isotropic fractionator techniques in neuroscience studies, including recent developments. J. Neurosci. Methods 2018, 310, 45–53. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pilski, A.; Graves, S.M. Repeated Methamphetamine Administration Results in Axon Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta and Locus Coeruleus Neurons in Male but Not Female Mice. Int. J. Mol. Sci. 2023, 24, 13039. https://doi.org/10.3390/ijms241713039
Pilski A, Graves SM. Repeated Methamphetamine Administration Results in Axon Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta and Locus Coeruleus Neurons in Male but Not Female Mice. International Journal of Molecular Sciences. 2023; 24(17):13039. https://doi.org/10.3390/ijms241713039
Chicago/Turabian StylePilski, Alexander, and Steven M. Graves. 2023. "Repeated Methamphetamine Administration Results in Axon Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta and Locus Coeruleus Neurons in Male but Not Female Mice" International Journal of Molecular Sciences 24, no. 17: 13039. https://doi.org/10.3390/ijms241713039
APA StylePilski, A., & Graves, S. M. (2023). Repeated Methamphetamine Administration Results in Axon Loss Prior to Somatic Loss of Substantia Nigra Pars Compacta and Locus Coeruleus Neurons in Male but Not Female Mice. International Journal of Molecular Sciences, 24(17), 13039. https://doi.org/10.3390/ijms241713039