
Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1

 , , and
, , and
Abstract
:1. Introduction
2. Results
2.1. Selection and Mapping of GCDH Variants on the Crystallographic Structure

2.2. Impact of GA1-Associated Variants on GCDH Enzymatic Activity
2.3. GCDH Missense Variants Lead to Loss of Protein Stability and Impaired Tetramerization
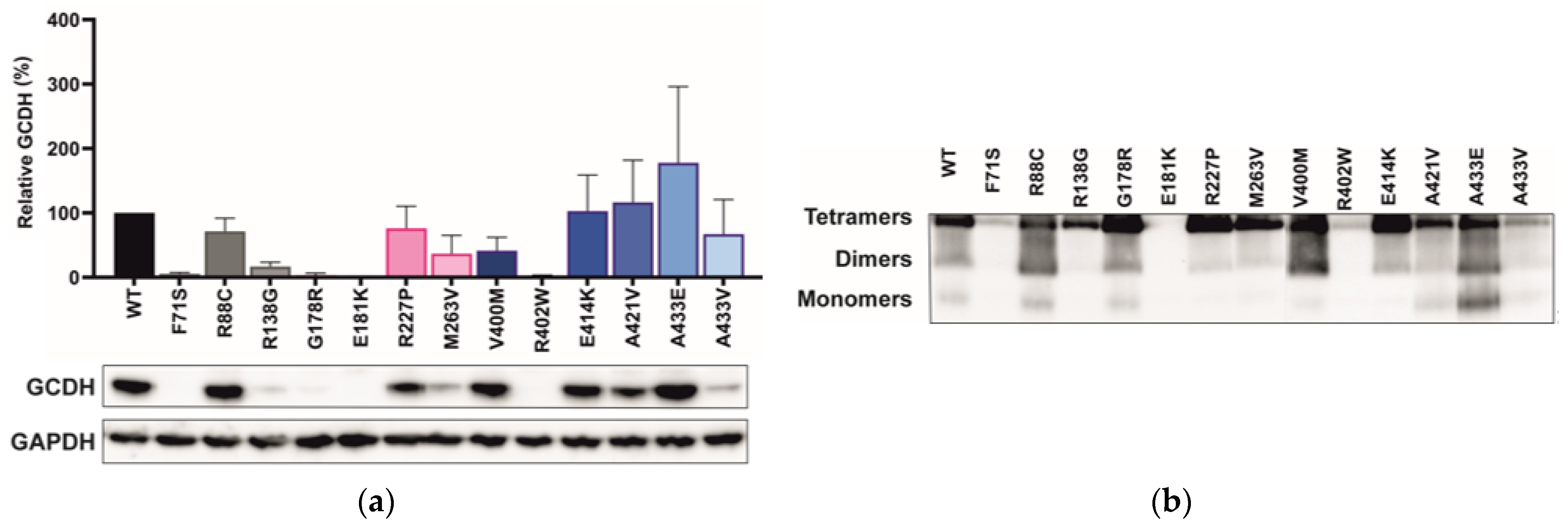
2.4. Variant Location Correlates with Cellular GCDH Levels
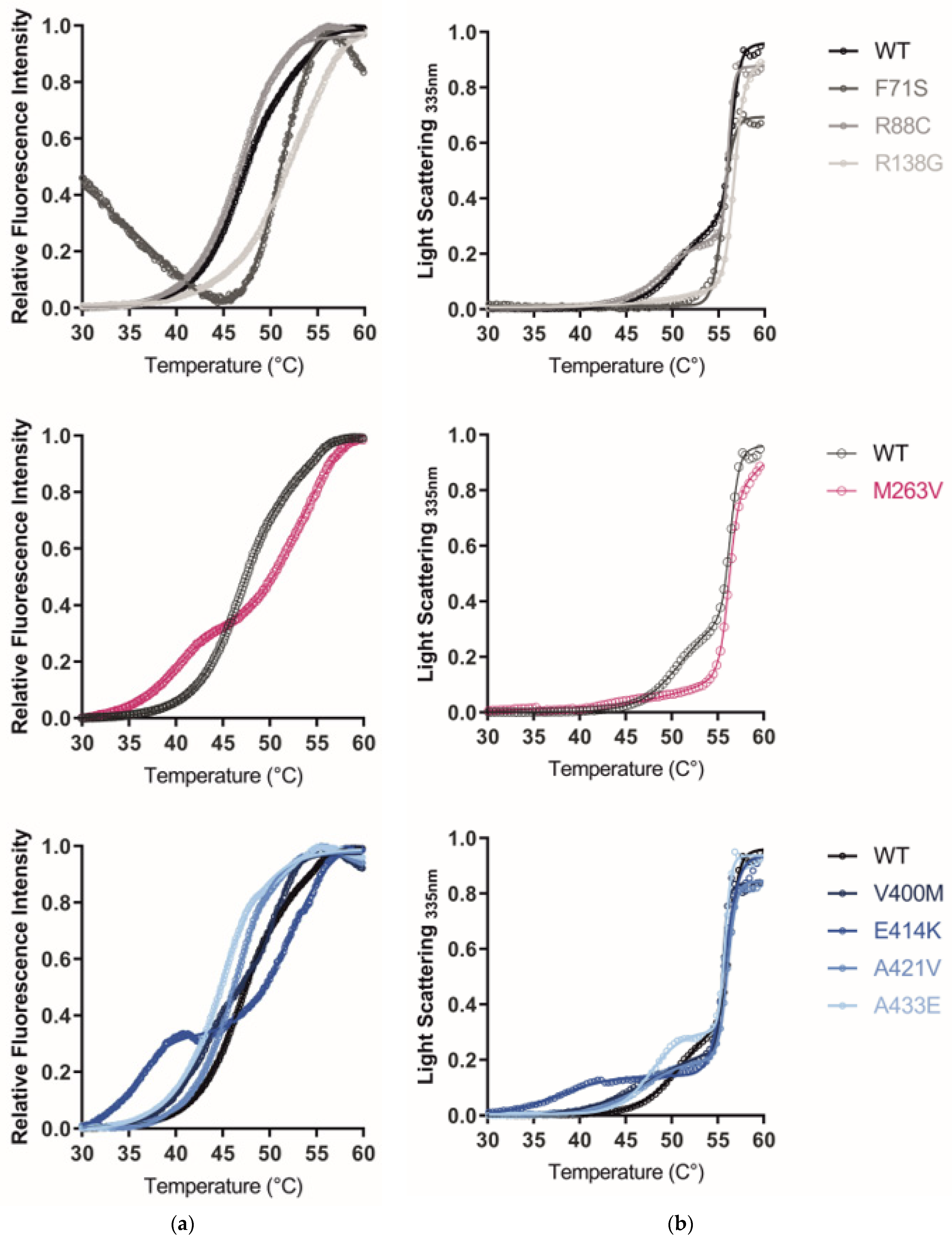
2.5. The GCDH Misfolding Phenotype Is Confirmed by Increased Hydrophobicity, Aggregation, and Loss of Thermal Stability
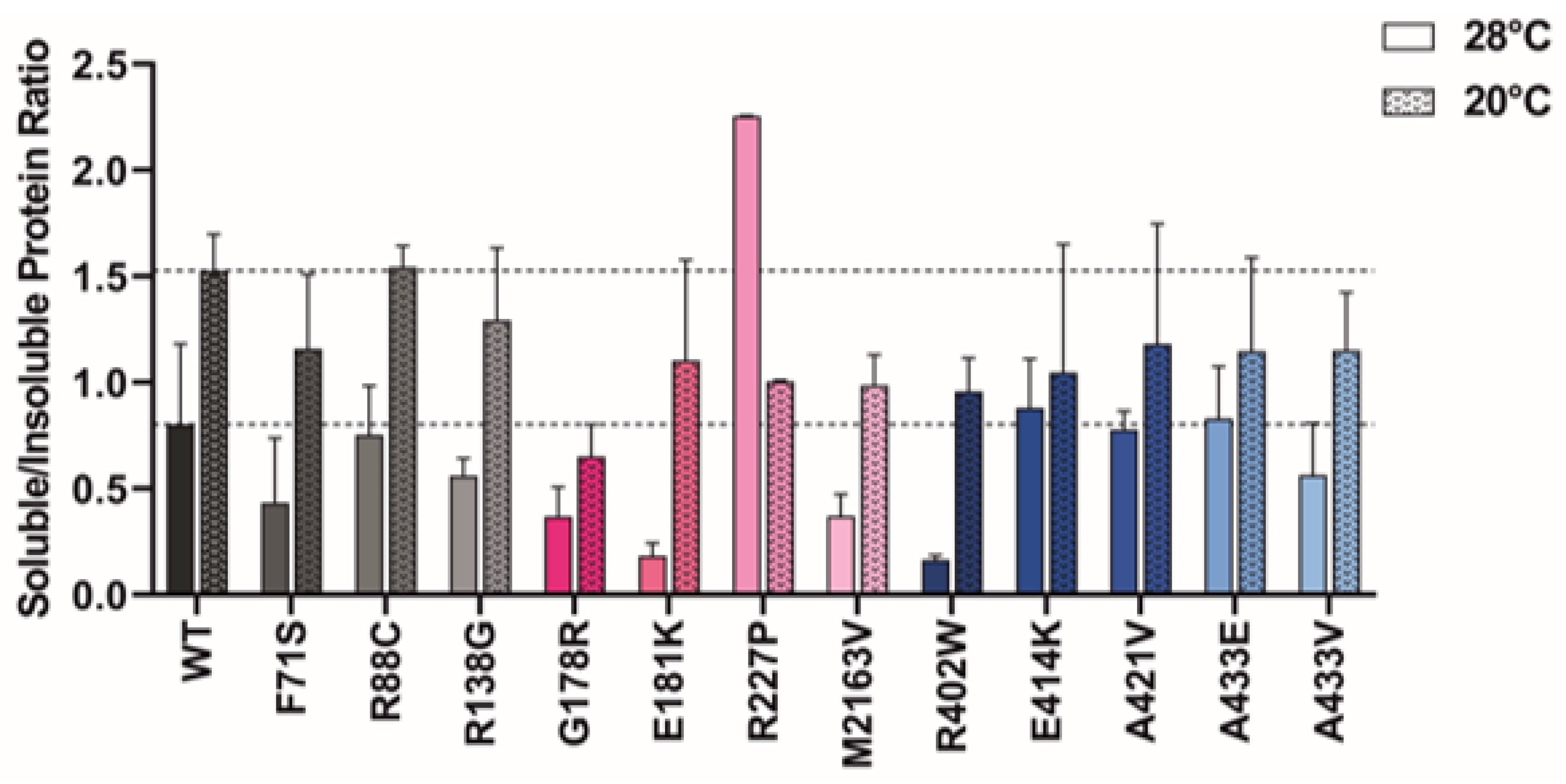
2.6. Protein Expression at Lower Temperatures Rescues Mutant GCDH Solubility
3. Discussion
3.1. Enzyme Activity Findings
3.2. Tetramerization Findings
3.3. Variant Location Correlates with Cellular GCDH Levels
3.4. The GCDH Misfolding Phenotype Is Confirmed by Increased Hydrophobicity, Aggregation, and Loss of Thermal Stability
3.5. Protein Expression at Lower Temperatures Rescues Variant GCDH
4. Materials and Methods
4.1. DNA Constructs
4.2. Prokaryotic Expression and Purification
4.3. Cellular GCDH Expression in COS-7 Cells
4.4. Polyacrylamide Gel Electrophoresis and Immunoblotting
4.5. Enzyme Activity Assays
4.6. Right-Angle Light Scattering (RALS)
4.7. Thermal Shift Assays
4.8. Structural Analysis
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bouchereau, J.; Schiff, M. Inherited disorders of lysine metabolism: A review. J. Nutr. 2020, 150, 2556S–2560S. [Google Scholar] [CrossRef] [PubMed]
- Lindner, M.; Kölker, S.; Schulze, A.; Christensen, E.; Greenberg, C.R.; Hoffmann, G.F. Neonatal screening for glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, J.V.; Lucas, T.G.; Bross, P.; Gomes, C.M.; Henriques, B.J. Potential complementation effects of two disease-associated mutations in tetrameric glutaryl-CoA dehydrogenase is due to inter subunit stability-activity counterbalance. Biochim. Biophys. Acta Proteins Proteom. 2020, 1868, 140269. [Google Scholar] [CrossRef] [PubMed]
- Sitta, A.; Guerreiro, G.; de Moura Coelho, D.; da Rocha, V.V.; Dos Reis, B.G.; Sousa, C.; Vilarinho, L.; Wajner, M.; Vargas, C.R. Clinical, biochemical and molecular findings of 24 Brazilian patients with glutaric acidemia type 1:4 novel mutations in the GCDH gene. Metab. Brain Dis. 2021, 36, 205–212. [Google Scholar] [CrossRef]
- Märtner, E.M.C.; Maier, E.M.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Santer, R.; Weinhold, N.; Marquardt, I.; Das, A.M.; et al. Impact of interventional and non-interventional variables on anthropometric long-term development in glutaric aciduria type 1: A national prospective multi-centre study. J. Inherit. Metab. Dis. 2021, 44, 629–638. [Google Scholar] [CrossRef]
- Larson, A.; Goodman, S. Glutaric Acidemia Type 1. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Schuurmans, I.M.E.; Dimitrov, B.; Schröter, J.; Ribes, A.; de la Fuente, R.P.; Zamora, B.; van Karnebeek, C.D.M.; Kölker, S.; Garanto, A. Exploring genotype-phenotype correlations in glutaric aciduria type 1. J. Inherit. Metab. Dis. 2023, 46, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Boy, N.; Mengler, K.; Thimm, E.; Schiergens, K.A.; Marquardt, T.; Weinhold, N.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. Newborn screening: A disease-changing intervention for glutaric aciduria type 1. Ann. Neurol. 2018, 83, 970–979. [Google Scholar] [CrossRef]
- Christensen, E.; Ribes, A.; Merinero, B.; Zschocke, J. Correlation of genotype and phenotype in glutaryl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2004, 27, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Dimitrov, B.; Boy, N.; Gleich, F.; Zielonka, M.; Kölker, S. Phenotypic prediction in glutaric aciduria type 1 combining in silico and in vitro modeling with real-world data. J. Inherit. Metab. Dis. 2023, 46, 391–405. [Google Scholar] [CrossRef]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Heringer, J.; Assmann, B.; Burgard, P.; Dixon, M.; Fleissner, S.; Greenberg, C.R.; Harting, I.; et al. Proposed recommendations for diagnosing and managing individuals with glutaric aciduria type I: Second revision. J. Inherit. Metab. Dis. 2017, 40, 75–101. [Google Scholar] [CrossRef]
- Märtner, E.M.C.; Thimm, E.; Guder, P.; Schiergens, K.A.; Rutsch, F.; Roloff, S.; Marquardt, I.; Das, A.M.; Freisinger, P.; Grünert, S.C.; et al. The biochemical subtype is a predictor for cognitive function in glutaric aciduria type 1: A national prospective follow-up study. Sci. Rep. 2021, 11, 19300. [Google Scholar] [CrossRef]
- Boy, N.; Mengler, K.; Heringer-Seifert, J.; Hoffmann, G.F.; Garbade, S.F.; Kölker, S. Impact of newborn screening and quality of therapy on the neurological outcome in glutaric aciduria type 1: A meta-analysis. Genet. Med. 2021, 23, 13–21. [Google Scholar] [CrossRef]
- Beauchamp, M.H.; Boneh, A.; Anderson, V. Cognitive, behavioural and adaptive profiles of children with glutaric aciduria type I detected through newborn screening. J. Inherit. Metab. Dis. 2009, 32 (Suppl. S1), S207–S213. [Google Scholar] [CrossRef]
- Keyser, B.; Mühlhausen, C.; Dickmanns, A.; Christensen, E.; Muschol, N.; Ullrich, K.; Braulke, T. Disease-causing missense mutations affect enzymatic activity, stability and oligomerization of glutaryl-CoA dehydrogenase (GCDH). Hum. Mol. Genet. 2008, 17, 3854–3863. [Google Scholar] [CrossRef]
- Westover, J.B.; Goodman, S.I.; Frerman, F.E. Pathogenic mutations in the carboxyl-terminal domain of glutaryl-CoA dehydrogenase: Effects on catalytic activity and the stability of the tetramer. Mol. Genet. Metab. 2003, 79, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Biery, B.J.; Stein, D.E.; Morton, D.H.; Goodman, S.I. Gene structure and mutations of glutaryl-coenzyme A dehydrogenase: Impaired association of enzyme subunits that is due to an A421V substitution causes glutaric acidemia type I in the Amish. Am. J. Hum. Genet. 1996, 59, 1006–1011. [Google Scholar]
- Liesert, M.; Zschocke, J.; Hoffmann, G.F.; Mühlhäuser, N.; Buckel, W. Biochemistry of glutaric aciduria type I: Activities of in vitro expressed wild-type and mutant cDNA encoding human glutaryl-CoA dehydrogenase. J. Inherit. Metab. Dis. 1999, 22, 256–258. [Google Scholar] [CrossRef]
- Pokora, P.; Jezela-Stanek, A.; Różdżyńska-Świątkowska, A.; Jurkiewicz, E.; Bogdańska, A.; Szymańska, E.; Rokicki, D.; Ciara, E.; Rydzanicz, M.; Stawiński, P.; et al. Mild phenotype of glutaric aciduria type 1 in polish patients-novel data from a group of 13 cases. Metab. Brain Dis. 2019, 34, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Busquets, C.; Coll, M.J.; Ribes, A. Evidence of a single origin for the most frequent mutation (R402W) causing glutaryl-CoA dehydrogenase deficiency: Identification of 3 novel polymorphisms and haplotype definition. Hum. Mutat. 2000, 15, 207. [Google Scholar] [CrossRef]
- Zschocke, J.; Quak, E.; Guldberg, P.; Hoffmann, G.F. Mutation analysis in glutaric aciduria type I. J. Med. Genet. 2000, 37, 177–181. [Google Scholar] [CrossRef]
- Tp, K.V.; Muntaj, S.; Devaraju, K.S.; Kamate, M.; Vedamurthy, A.B. Genetic screening of selected disease-causing mutations in glutaryl-CoA dehydrogenase gene among Indian patients with glutaric aciduria type I. J. Pediatr. Genet. 2017, 6, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Fraidakis, M.J.; Liadinioti, C.; Stefanis, L.; Dinopoulos, A.; Pons, R.; Papathanassiou, M.; Garcia-Villoria, J.; Ribes, A. Rare late-onset presentation of glutaric aciduria type I in a 16-year-old Woman with a wnovel GCDH mutation. JIMD Rep. 2015, 18, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schmiesing, J.; Lohmöller, B.; Schweizer, M.; Tidow, H.; Gersting, S.W.; Muntau, A.C.; Braulke, T.; Mühlhausen, C. Disease-causing mutations affecting surface residues of mitochondrial glutaryl-CoA dehydrogenase impair stability, heteromeric complex formation and mitochondria architecture. Hum. Mol. Genet. 2017, 26, 538–551. [Google Scholar] [CrossRef]
- Busquets, C.; Merinero, B.; Christensen, E.; Gelpí, J.L.; Campistol, J.; Pineda, M.; Fernández-Alvarez, E.; Prats, J.M.; Sans, A.; Arteaga, R.; et al. Glutaryl-CoA dehydrogenase deficiency in Spain: Evidence of two groups of patients, genetically, and biochemically distinct. Pediatr. Res. 2000, 48, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.I.; Stein, D.E.; Schlesinger, S.; Christensen, E.; Schwartz, M.; Greenberg, C.R.; Elpeleg, O.N. Glutaryl-CoA dehydrogenase mutations in glutaric acidemia (type I): Review and report of thirty novel mutations. Hum. Mutat. 1998, 12, 141–144. [Google Scholar] [CrossRef]
- Christensen, E.; Ribes, A.; Busquets, C.; Pineda, M.; Duran, M.; Poll-The, B.T.; Greenberg, C.R.; Leffers, H.; Schwartz, M. Compound heterozygosity in the glutaryl-CoA dehydrogenase gene with R227P mutation in one allele is associated with no or very low free glutarate excretion. J. Inherit. Metab. Dis. 1997, 20, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Healy, L.; O’Shea, M.; McNulty, J.; King, G.; Twomey, E.; Treacy, E.; Crushell, E.; Hughes, J.; Knerr, I.; Monavari, A.A. Glutaric aciduria type 1: Diagnosis, clinical features and long-term outcome in a large cohort of 34 Irish patients. JIMD Rep. 2022, 63, 379–387. [Google Scholar] [CrossRef]
- Kars, M.E.; Başak, A.N.; Onat, O.E.; Bilguvar, K.; Choi, J.; Itan, Y.; Çağlar, C.; Palvadeau, R.; Casanova, J.-L.; Cooper, D.N.; et al. The genetic structure of the Turkish population reveals high levels of variation and admixture. Proc. Natl. Acad. Sci. USA 2021, 118, e2026076118. [Google Scholar] [CrossRef]
- Mühlhausen, C.; Christensen, E.; Schwartz, M.; Muschol, N.; Ullrich, K.; Lukacs, Z. Severe phenotype despite high residual glutaryl-CoA dehydrogenase activity: A novel mutation in a Turkish patient with glutaric aciduria type I. J. Inherit. Metab. Dis. 2003, 26, 713–714. [Google Scholar] [CrossRef]
- Tan, J.; Chen, D.; Yan, T.; Huang, J.; Cai, R. Analysis of CGDH gene variants and clinical features in three patients with glutaric aciduria type Ⅰ. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2019, 36, 882–885. [Google Scholar] [CrossRef]
- Treacy, E.P.; Lee-Chong, A.; Roche, G.; Lynch, B.; Ryan, S.; Goodman, S. Profound neurological presentation resulting from homozygosity for a mild glutaryl-CoA dehydrogenase mutation with a minimal biochemical phenotype. J. Inherit. Metab. Dis. 2003, 26, 72–74. [Google Scholar] [CrossRef]
- Xiao, B.; Qiu, W.; Ye, J.; Zhang, H.; Zhu, H.; Wang, L.; Liang, L.; Xu, F.; Chen, T.; Xu, Y.; et al. Prenatal diagnosis of glutaric acidemia I based on amniotic fluid samples in 42 families using genetic and biochemical approaches. Front. Genet. 2020, 11, 496. [Google Scholar] [CrossRef]
- Zhang, X.; Luo, Q. Clinical and laboratory analysis of late-onset glutaric aciduria type I (GA-I) in Uighur: A report of two cases. Exp. Ther. Med. 2017, 13, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Marti-Masso, J.F.; Ruiz-Martínez, J.; Makarov, V.; López de Munain, A.; Gorostidi, A.; Bergareche, A.; Yoon, S.; Buxbaum, J.D.; Paisán-Ruiz, C. Exome sequencing identifies GCDH (glutaryl-CoA dehydrogenase) mutations as a cause of a progressive form of early-onset generalized dystonia. Hum. Genet. 2012, 131, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Kölker, S.; Garbade, S.F.; Greenberg, C.R.; Leonard, J.V.; Saudubray, J.-M.; Ribes, A.; Kalkanoglu, H.S.; Lund, A.M.; Merinero, B.; Wajner, M.; et al. Natural history, outcome, and treatment efficacy in children and adults with glutaryl-CoA dehydrogenase deficiency. Pediatr. Res. 2006, 59, 840–847. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.; Christensen, E.; Superti-Furga, A.; Brandt, N.J. The human glutaryl-CoA dehydrogenase gene: Report of intronic sequences and of 13 novel mutations causing glutaric aciduria type I. Hum. Genet. 1998, 102, 452–458. [Google Scholar] [CrossRef]
- Alaei, M.R.; Kheirkhahan, M.; Talebi, S.; Davoudi-Dehaghani, E.; Keramatipour, M. Once in a blue moon, a very rare coexistence of glutaric acidemia type I and mucopolysaccharidosis type IIIB in a patient. Iran. Biomed. J. 2020, 24, 201–205. [Google Scholar] [CrossRef]
- Boy, N.; Heringer, J.; Brackmann, R.; Bodamer, O.; Seitz, A.; Kölker, S.; Harting, I. Extrastriatal changes in patients with late-onset glutaric aciduria type I highlight the risk of long-term neurotoxicity. Orphanet J. Rare Dis. 2017, 12, 77. [Google Scholar] [CrossRef]
- Capalbo, A.; Valero, R.A.; Jimenez-Almazan, J.; Pardo, P.M.; Fabiani, M.; Jiménez, D.; Simon, C.; Rodriguez, J.M. Optimizing clinical exome design and parallel gene-testing for recessive genetic conditions in preconception carrier screening: Translational research genomic data from 14,125 exomes. PLOS Genet. 2019, 15, e1008409. [Google Scholar] [CrossRef]
- Lee, C.S.; Chien, Y.H.; Peng, S.F.; Cheng, P.W.; Chang, L.M.; Huang, A.C.; Hwu, W.L.; Lee, N.C. Promising outcomes in glutaric aciduria type I patients detected by newborn screening. Metab. Brain Dis. 2013, 28, 61–67. [Google Scholar] [CrossRef]
- Tamhankar, P.M.; Vasudevan, L.; Kondurkar, P.; Niazi, S.; Christopher, R.; Solanki, D.; Dholakia, P.; Muranjan, M.; Kamate, M.; Kalane, U.; et al. Clinical characteristics, molecular profile, and outcomes in Indian patients with glutaric aciduria type 1. J. Pediatr. Genet. 2021, 10, 213–221. [Google Scholar] [CrossRef]
- Fu, Z.; Wang, M.; Paschke, R.; Rao, K.S.; Frerman, F.E.; Kim, J.J. Crystal structures of human glutaryl-CoA dehydrogenase with and without an alternate substrate: Structural bases of dehydrogenation and decarboxylation reactions. Biochemistry 2004, 43, 9674–9684. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Niesen, F.H.; Allali-Hassani, A.; Fedorov, O.Y.; Finerty, P.J., Jr.; Wasney, G.A.; Yeung, R.; Arrowsmith, C.; Ball, L.J.; Berglund, H.; et al. Chemical screening methods to identify ligands that promote protein stability, protein crystallization, and structure determination. Proc. Natl. Acad. Sci. USA 2006, 103, 15835–15840. [Google Scholar] [CrossRef] [PubMed]
- Vedadi, M.; Arrowsmith, C.H.; Allali-Hassani, A.; Senisterra, G.; Wasney, G.A. Biophysical characterization of recombinant proteins: A key to higher structural genomics success. J. Struct. Biol. 2010, 172, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Schmiesing, J.; Schlüter, H.; Ullrich, K.; Braulke, T.; Mühlhausen, C. Interaction of glutaric aciduria type 1-related glutaryl-CoA dehydrogenase with mitochondrial matrix proteins. PLoS ONE 2014, 9, e87715. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb. Cell Factories 2005, 4, 1. [Google Scholar] [CrossRef]
- Rajan, R.; Ahmed, S.; Sharma, N.; Kumar, N.; Debas, A.; Matsumura, K. Review of the current state of protein aggregation inhibition from a materials chemistry perspective: Special focus on polymeric materials. Mater. Adv. 2021, 2, 1139–1176. [Google Scholar] [CrossRef]
- Singh, G.P.; Ganapathi, M.; Sandhu, K.S.; Dash, D. Intrinsic unstructuredness and abundance of PEST motifs in eukaryotic proteomes. Proteins 2006, 62, 309–315. [Google Scholar] [CrossRef]
- Eldeeb, M.A.; Ragheb, M.A. N-degron-mediated degradation and regulation of mitochondrial PINK1 kinase. Curr. Genet. 2020, 66, 693–701. [Google Scholar] [CrossRef]
- Timms, R.T.; Koren, I. Tying up loose ends: The N-degron and C-degron pathways of protein degradation. Biochem. Soc. Trans. 2020, 48, 1557–1567. [Google Scholar] [CrossRef]
- Camilloni, C.; Bonetti, D.; Morrone, A.; Giri, R.; Dobson, C.M.; Brunori, M.; Gianni, S.; Vendruscolo, M. Towards a structural biology of the hydrophobic effect in protein folding. Sci. Rep. 2016, 6, 28285. [Google Scholar] [CrossRef] [PubMed]
- Schein, C.H. Production of soluble recombinant proteins in bacteria. Bio/technology 1989, 7, 1141–1149. [Google Scholar] [CrossRef]
- Boy, N.; Mühlhausen, C.; Maier, E.M.; Ballhausen, D.; Baumgartner, M.R.; Beblo, S.; Burgard, P.; Chapman, K.A.; Dobbelaere, D.; Heringer-Seifert, J.; et al. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J. Inherit. Metab. Dis. 2022, 46, 482–519. [Google Scholar] [CrossRef]
- Gersting, S.W.; Lagler, F.B.; Eichinger, A.; Kemter, K.F.; Danecka, M.K.; Messing, D.D.; Staudigl, M.; Domdey, K.A.; Zsifkovits, C.; Fingerhut, R.; et al. Pahenu1 is a mouse model for tetrahydrobiopterin-responsive phenylalanine hydroxylase deficiency and promotes analysis of the pharmacological chaperone mechanism in vivo. Hum. Mol. Genet. 2010, 19, 2039–2049. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, T.M.; Rao, K.S.; Westover, J.B.; Kim, J.J.; Frerman, F.E. The function of Arg-94 in the oxidation and decarboxylation of glutaryl-CoA by human glutaryl-CoA dehydrogenase. J. Biol. Chem. 2001, 276, 133–138. [Google Scholar] [CrossRef]
- Gonzalez Melo, M.; Remacle, N.; Cudré-Cung, H.-P.; Roux, C.; Poms, M.; Cudalbu, C.; Barroso, M.; Gersting, S.W.; Feichtinger, R.G.; Mayr, J.A.; et al. The first knock-in rat model for glutaric aciduria type I allows further insights into pathophysiology in brain and periphery. Mol. Genet. Metab. 2021, 133, 157–181. [Google Scholar] [CrossRef]
- Liguori, L.; Monticelli, M.; Allocca, M.; Hay Mele, B.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Pharmacological chaperones: A therapeutic approach for diseases caused by sestabilizing missense mutations. Int. J. Mol. Sci. 2020, 21, 489. [Google Scholar] [CrossRef]
- Thies, B.; Meyer-Schwesinger, C.; Lamp, J.; Schweizer, M.; Koeller, D.M.; Ullrich, K.; Braulke, T.; Mühlhausen, C. Acute renal proximal tubule alterations during induced metabolic crises in a mouse model of glutaric aciduria type 1. Biochim. Biophys. Acta 2013, 1832, 1463–1472. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein Variant | Relative Crotonyl-CoA Formation (%) | ||
|---|---|---|---|
| rGCDH Assay | COS-7 Cellular Assay | ||
| WT | 100.0 ± 3.0 | 98.9 ± 1.1 | |
| F71S | - | n.d. | |
| R88C | 0.9 ± 0.0 * | n.d. | |
| R138G | 4.0 ± 0.2 * | n.d. | |
| G178R | - | 0.8 ± 0.5 * | |
| E181K | - | n.d. | |
| R227P | - | n.d. | |
| M263V | 30.6 ± 2.2 * | 13.1 ± 1.4 * | |
| V400M | 49.4 ± 1.3 * | 33.3 ± 4.5 * | |
| R402W | - | n.d. | |
| E414K | 1.2 ± 0.1 * | 0.9 ± 1.8 * | |
| A421V | 2.5 ± 0.4 * | 9.7 ± 2.8 * | |
| A433E | 5.6 ± 1.9 * | n.d. | |
| A433V | 18.3 ± 0.6 * | 0.2 ± 2.6 * | |
| Protein Variant | TSA | RALS | |||||
|---|---|---|---|---|---|---|---|
| F.I.25°C (AU) Mean ± SEM | Tm1/2 (°C) Mean (CI 95%) | Tm2/3 (°C) Mean (CI 95%) | Turbidity25°C (AU) Mean ± SEM | Tm1/2 (°C) Mean (CI 95%) | Tm2/3 (°C) Mean (CI 95%) | ||
| WT | 41.5 ± 0.8 | 47.1 (47.0–47.1) | 54.8 (54.6–55.0) | 21.8 ± 0.4 | 51.2 (49.3–54.1) | 56.4 (56.2–56.6) | |
| F71S | 154.1 ± 6.6 * | 51.4 (model is unfit) | – | 164.0 ± 2.1 * | – | – | |
| R88C | 85.2 ± 2.8 * | 39.2 (38.3–40.5) | 47.0 (46.9–47.1) | 39.9 ± 0.4 * | 49.5 (49.1–50.1) | 56.2 (56.0–56.1) | |
| R138G | 178.4 ± 4.6 * | 41.6 (40.4–44.6) | 52.6 (52.5–52.7) | 33.2 ± 0.2 * | – | – | |
| M263V | 72.0 ± 1.3 * | 39.7 (39.5–39.8) | 53.2 (53.1–53.3) | 21.9 ± 2.0 | – | 56.2 (56.1–56.3) | |
| V400M | 141.7 ± 5.4 * | 43.3 (43.2–43.5) | 50.7 (50.6–50.8) | 58.2 ± 0.4 * | 48.0 (47.0–49.3) | 55.6 (55.5–55.6) | |
| E414K | 96.3 ± 4.4 * | 35.9 (35.8–36.0) | 52.2 (52.1–52.3) | 59.4 ± 0.9 * | – | 55.7 (55.7–55.8) | |
| A421V | 165.0 ± 2.6 * | 39.7 (39.0–40.6) | 46.3 (46.3–46.4) | 47.4 ± 0.9 * | 47.0 (45.8–51.1) | 56.0 (55.9–56.2) | |
| A433E | 72.2 ± 4.8 * | 44.6 (44.6–44.7) | 45.0 (44.8–45.1) | 22.1 ±0.2 | 47.7 (47.2–48.4) | 55.8 (55.7–55.9) | |
| A433V # | 9.3 ± 0.8 | – | – | 27.0 ± 0.2 * | – | – | |
| WT # | 8.8 ± 0.4 | – | – | 7.9 ± 0.4 | – | – | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barroso, M.; Gertzen, M.; Puchwein-Schwepcke, A.F.; Preisler, H.; Sturm, A.; Reiss, D.D.; Danecka, M.K.; Muntau, A.C.; Gersting, S.W. Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1. Int. J. Mol. Sci. 2023, 24, 13158. https://doi.org/10.3390/ijms241713158
Barroso M, Gertzen M, Puchwein-Schwepcke AF, Preisler H, Sturm A, Reiss DD, Danecka MK, Muntau AC, Gersting SW. Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1. International Journal of Molecular Sciences. 2023; 24(17):13158. https://doi.org/10.3390/ijms241713158
Chicago/Turabian StyleBarroso, Madalena, Marcus Gertzen, Alexandra F. Puchwein-Schwepcke, Heike Preisler, Andreas Sturm, Dunja D. Reiss, Marta K. Danecka, Ania C. Muntau, and Søren W. Gersting. 2023. "Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1" International Journal of Molecular Sciences 24, no. 17: 13158. https://doi.org/10.3390/ijms241713158
APA StyleBarroso, M., Gertzen, M., Puchwein-Schwepcke, A. F., Preisler, H., Sturm, A., Reiss, D. D., Danecka, M. K., Muntau, A. C., & Gersting, S. W. (2023). Glutaryl-CoA Dehydrogenase Misfolding in Glutaric Acidemia Type 1. International Journal of Molecular Sciences, 24(17), 13158. https://doi.org/10.3390/ijms241713158