
Psoralen Alleviates Renal Fibrosis by Attenuating Inflammasome-Dependent NLRP3 Activation and Epithelial–Mesenchymal Transition in a Mouse Unilateral Ureteral Obstruction Model


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
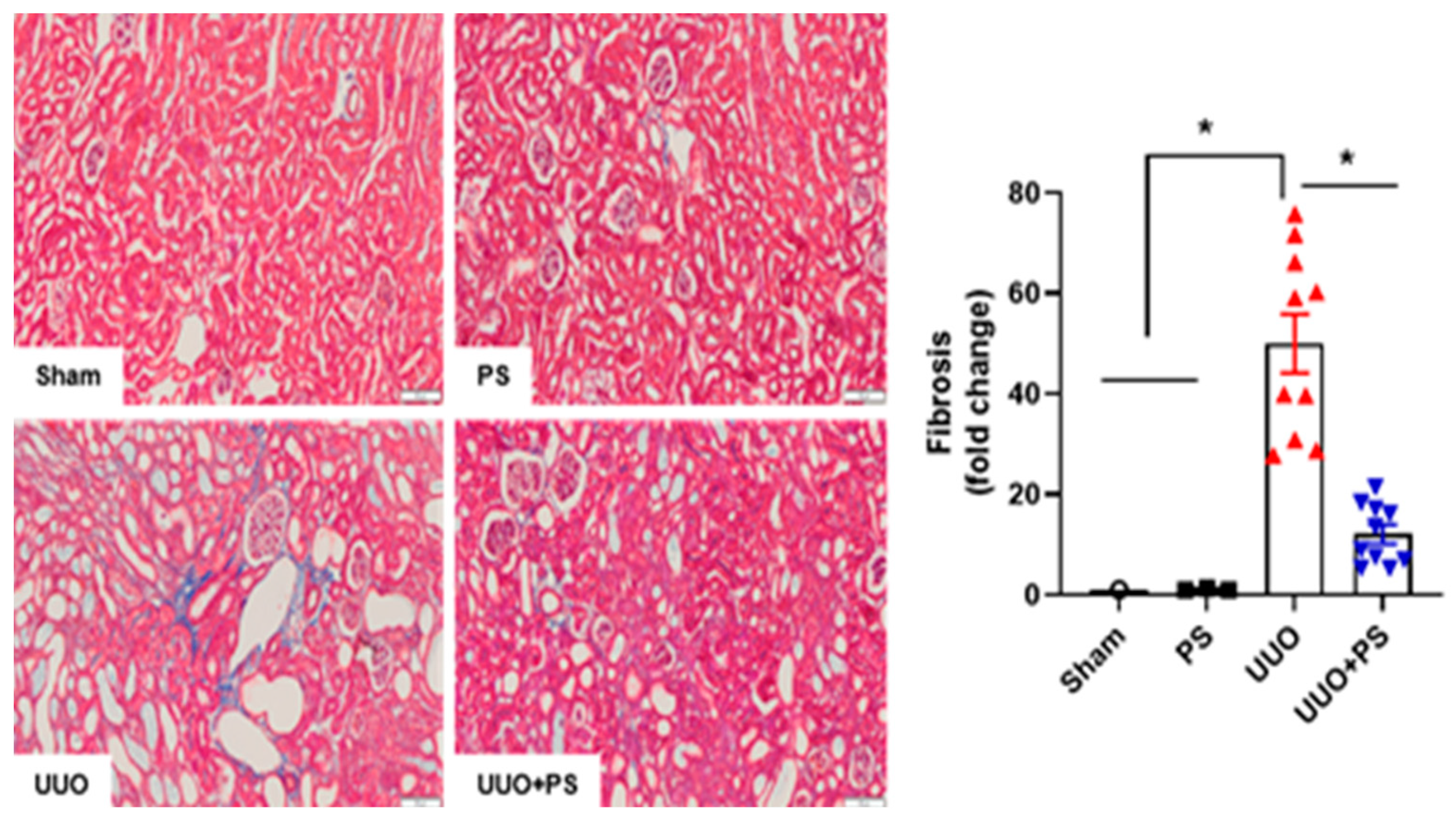
2.1. PS Improves Tissue Injury and Fibrosis in the UUO Mouse Model
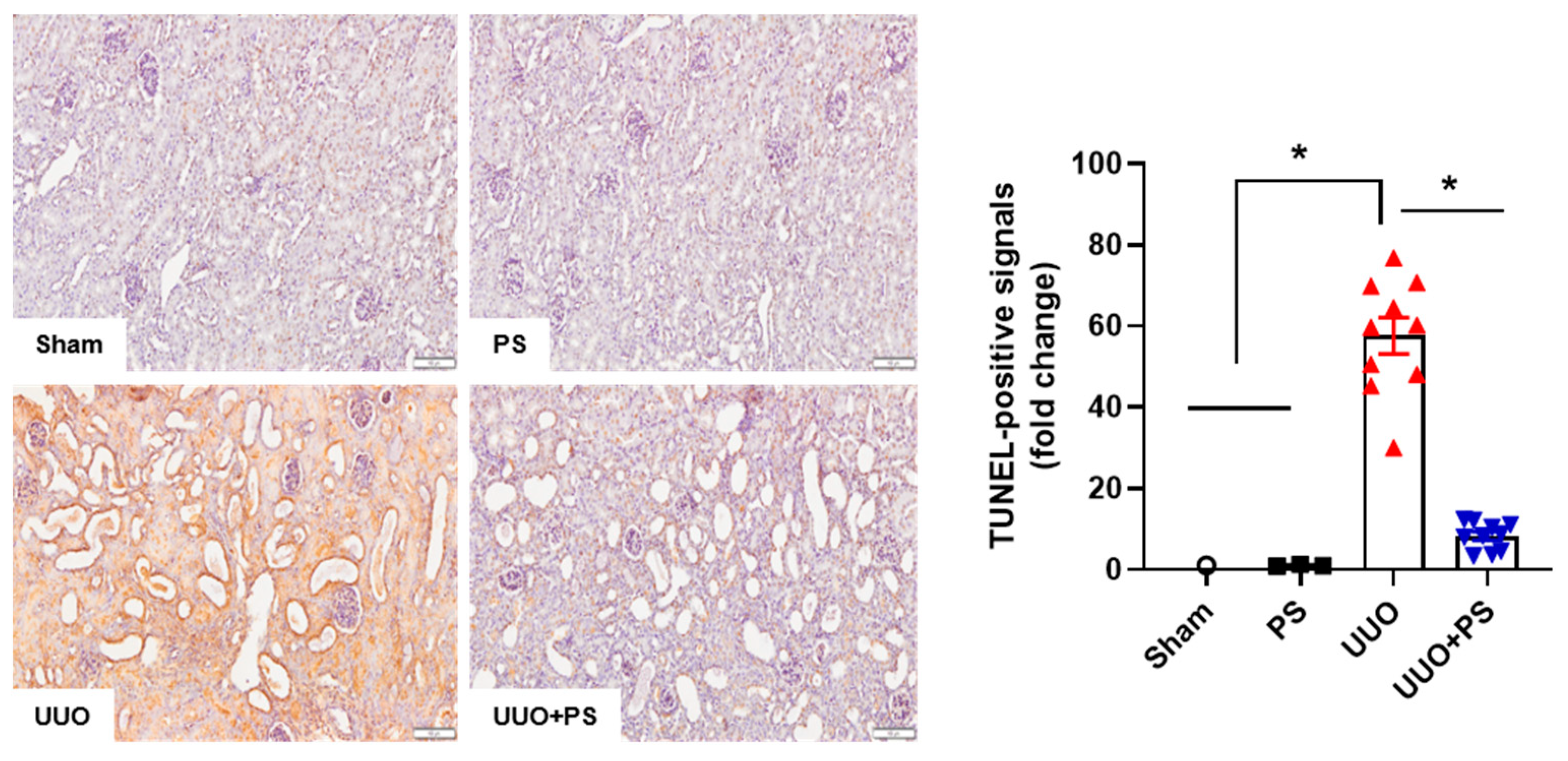
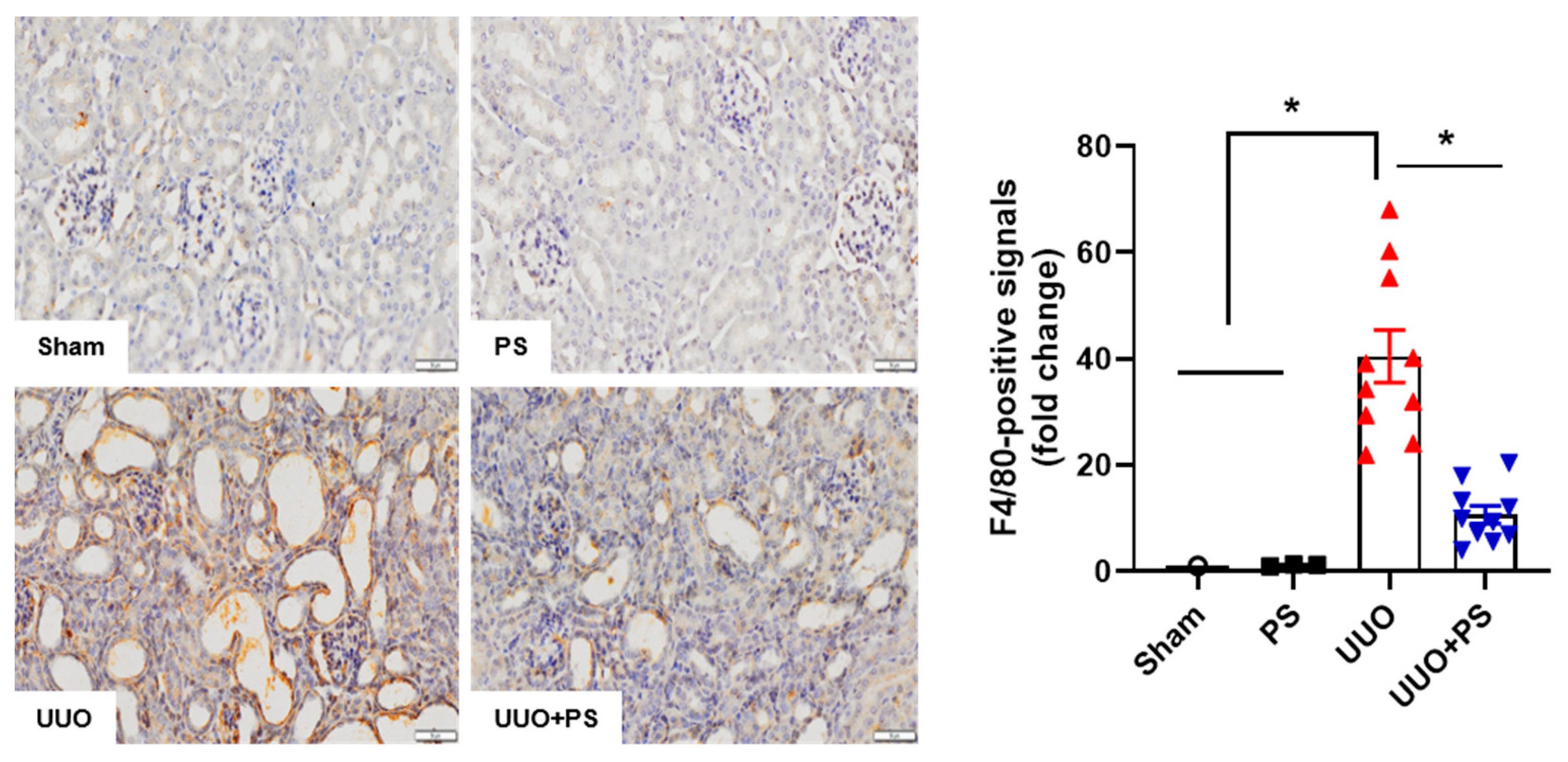
2.2. PS Reduces Tubular Epithelial Cell Apoptosis and Macrophage Infiltration Caused by UUO
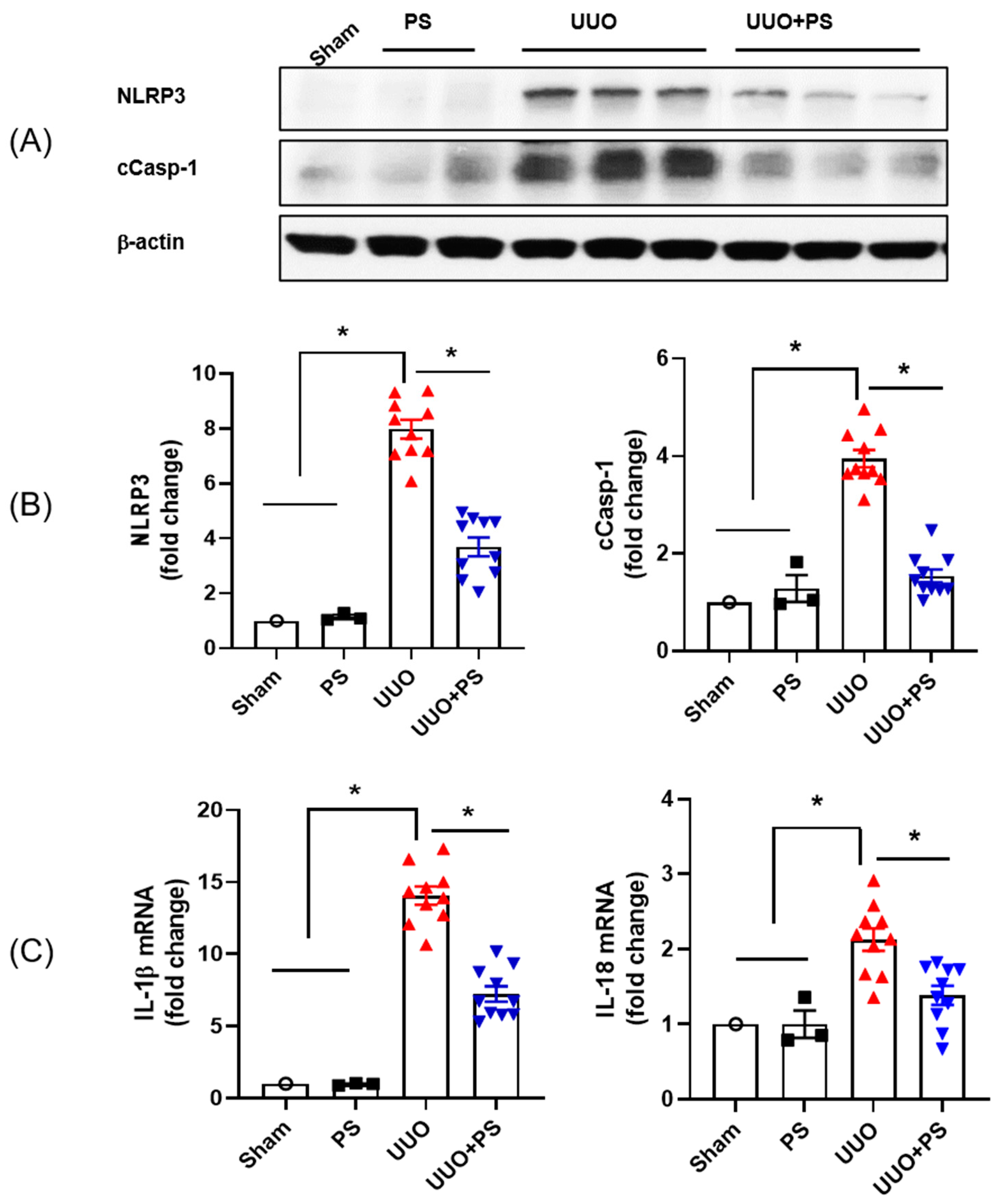
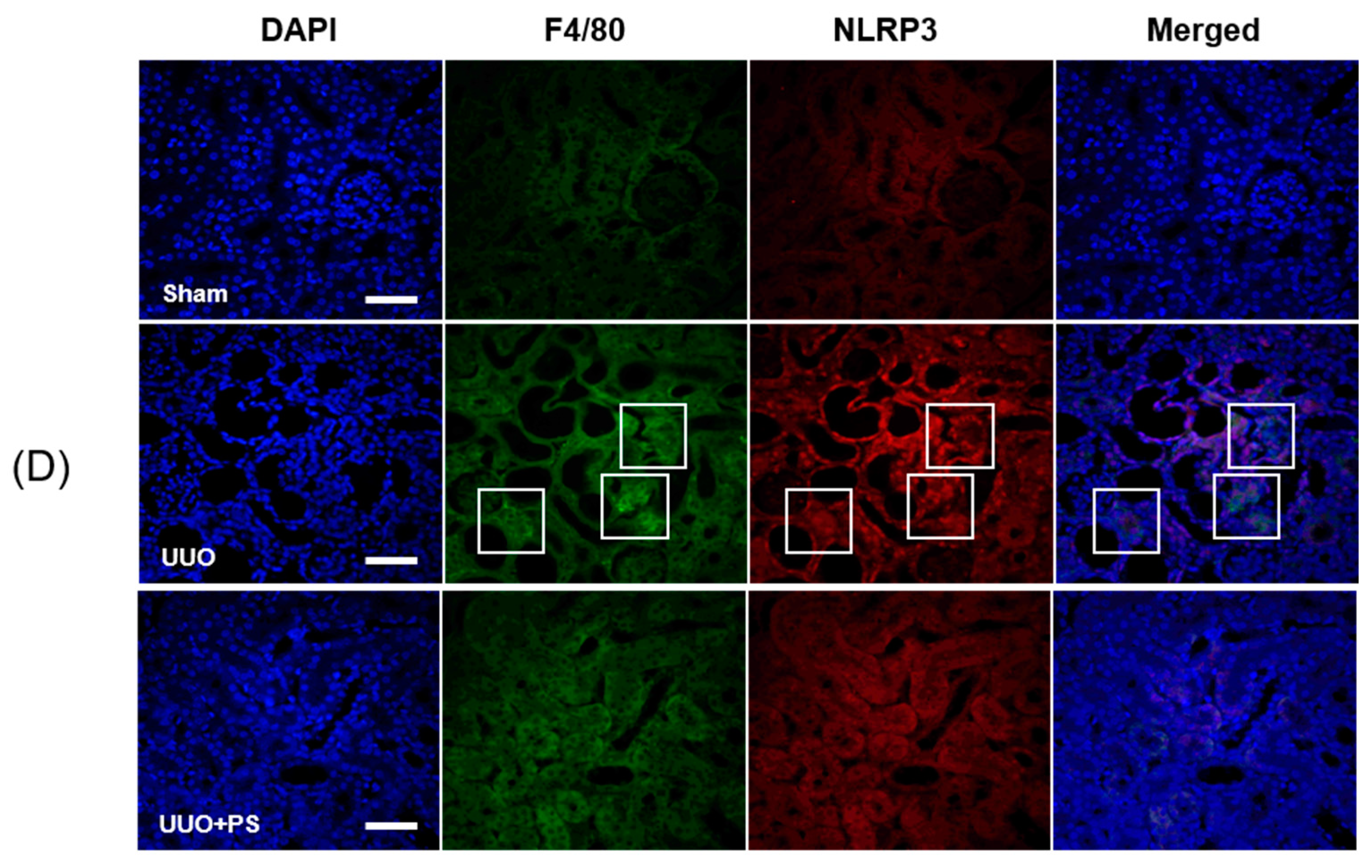
2.3. PS Attenuates the NLRP3 Inflammasome Induced by UUO
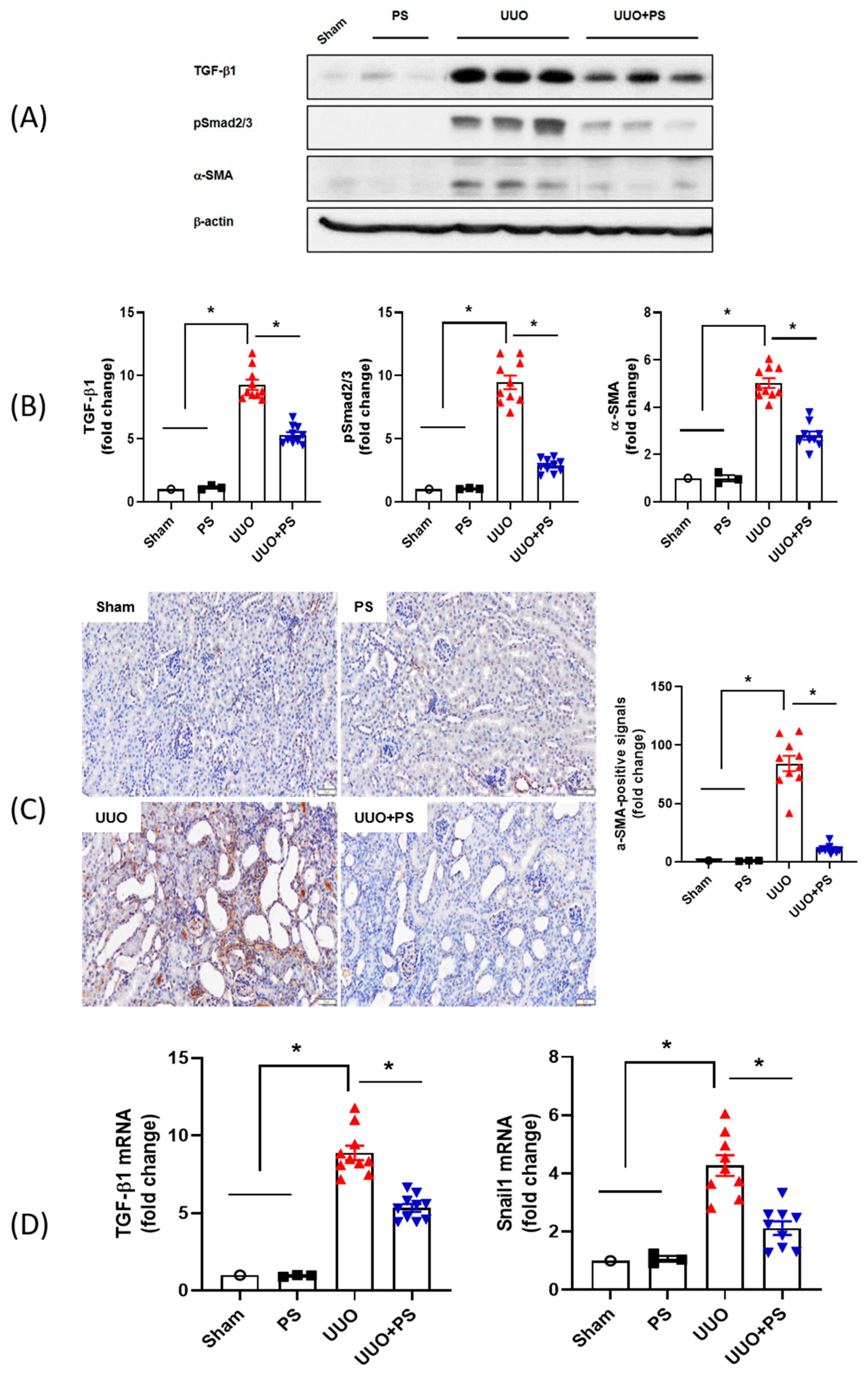
2.4. PS Inhibits Activation of TGF-β1/Smad 2/3 Signaling and EMT Pathway
3. Discussion
4. Materials and Methods
4.1. Animals, Surgery, and Tissue Preparation
4.2. Renal Pathology
4.3. Terminal Deoxynucleotidyl Transferase (TdT) dUTP Nick-End Labeling (TUNEL) Assay
4.4. Protein Preparation and Immunoblotting
4.5. Immunohistochemistry
4.6. Smad Quantitative Real-Time Polymerase Chain Reaction (PCR)
4.7. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collins, A.J.; Li, S.; Ma, J.Z.; Herzog, C. Cardiovascular disease in end-stage renal disease patients. Am. J. Kidney Dis. 2001, 38, S26–S29. [Google Scholar] [CrossRef]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Hoerger, T.J.; Simpson, S.A.; Yarnoff, B.O.; Pavkov, M.E.; Rios Burrows, N.; Saydah, S.H.; Williams, D.E.; Zhuo, X. The future burden of CKD in the United States: A simulation model for the CDC CKD Initiative. Am. J Kidney Dis. 2015, 65, 403–411. [Google Scholar] [CrossRef]
- Donadelli, R.; Abbate, M.; Zanchi, C.; Corna, D.; Tomasoni, S.; Benigni, A.; Remuzzi, G.; Zoja, C. Protein traffic activates NF-kB gene signaling and promotes MCP-1-dependent interstitial inflammation. Am. J. Kidney Dis. 2000, 36, 1226–1241. [Google Scholar] [CrossRef] [PubMed]
- Sergio, A.M.; Alejandra, M.D.; Eugenia, M.B.; Leopoldo, G.A.; Claudio, A.A.; Italo, C.; Jesus, E. Overexpression of chemokines, fibrogenic cytokines, and myofibroblasts in human membranous nephropathy. Kidney Int. 2000, 57, 147–158. [Google Scholar]
- Volker, V.; Hans-Joachim, A.; Matthias, M.; Josef, C.; Frank, S.; Manfred, S. Obstructive nephropathy in the mouse progressive fibrosis correlates with tubulointerstitial chemokine expression and accumulation of CC chemokine receptor 2- and 5-positive leukocytes. J. Am. Soc. Nephrol. 2001, 12, 1173–1187. [Google Scholar]
- Richter, K.; Konzack, A.; Pihlajaniemi, T.; Heljasvaara, R.; Kietzmann, T. Redox-fibrosis: Impact of TGFbeta1 on ROS generators, mediators and functional consequences. Redox. Biol. 2015, 6, 344–352. [Google Scholar] [CrossRef]
- Amador-Martinez, I.; Perez-Villalva, R.; Uribe, N.; Cortes-Gonzalez, C.; Bobadilla, N.A.; Barrera-Chimal, J. Reduced endothelial nitric oxide synthase activation contributes to cardiovascular injury during chronic kidney disease progression. Am. J. Physiol. Ren. Physiol. 2019, 317, F275–F285. [Google Scholar] [CrossRef]
- Guo, H.; Bi, X.; Zhou, P.; Zhu, S.; Ding, W. NLRP3 Deficiency Attenuates Renal Fibrosis and Ameliorates Mitochondrial Dysfunction in a Mouse Unilateral Ureteral Obstruction Model of Chronic Kidney Disease. Mediat. Inflamm. 2017, 2017, 8316560. [Google Scholar] [CrossRef]
- Zhang, K.; Fan, C.; Cai, D.; Zhang, Y.; Zuo, R.; Zhu, L.; Cao, Y.; Zhang, J.; Liu, C.; Chen, Y.; et al. Contribution of TGF-Beta-Mediated NLRP3-HMGB1 Activation to Tubulointerstitial Fibrosis in Rat With Angiotensin II-Induced Chronic Kidney Disease. Front. Cell Dev. Biol. 2020, 8, 1. [Google Scholar] [CrossRef]
- Decleves, A.E.; Zolkipli, Z.; Satriano, J.; Wang, L.; Nakayama, T.; Rogac, M.; Le, T.P.; Nortier, J.L.; Farquhar, M.G.; Naviaux, R.K.; et al. Regulation of lipid accumulation by AMP-activated kinase [corrected] in high fat diet-induced kidney injury. Kidney Int. 2014, 85, 611–623. [Google Scholar] [CrossRef] [PubMed]
- Meissner, M.; Viehmann, S.F.; Kurts, C. DAMPening sterile inflammation of the kidney. Kidney Int. 2019, 95, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.J. Cytokines, growth factors and renal injury: Where do we go now? Kidney Int. 1997, 63, S2–S6. [Google Scholar]
- Grandaliano, G.; Gesualdo, L.; Bartoli, F.; Ranieri, E.; Monno, R.; Leggio, A.; Paradies, G.; Caldarulo, E.; Infante, B.; Schena, F.P. MCP-1 and EGF renal expression and urine excretion in human congenital obstructive nephropathy. Kidney Int. 2000, 58, 182–192. [Google Scholar] [CrossRef]
- Ricardo, S.D.; van Goor, H.; Eddy, A.A. Macrophage diversity in renal injury and repair. J. Clin. Investig. 2008, 118, 3522–3530. [Google Scholar] [CrossRef]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Annu. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Scarpioni, R.; Obici, L. Renal involvement in autoinflammatory diseases and inflammasome-mediated chronic kidney damage. Clin. Exp. Rheumatol. 2017, 36, S54–S60. [Google Scholar]
- Romero, C.A.; Remor, A.; Latini, A.; De Paul, A.L.; Torres, A.I.; Mukdsi, J.H. Uric acid activates NRLP3 inflammasome in an in-vivo model of epithelial to mesenchymal transition in the kidney. J. Mol. Histol. 2017, 48, 209–218. [Google Scholar] [CrossRef]
- Qi, R.; Yang, C. Renal tubular epithelial cells: The neglected mediator of tubulointerstitial fibrosis after injury. Cell Death Dis. 2018, 9, 1126. [Google Scholar] [CrossRef]
- Chen, S.; Wang, Y.; Yang, Y.; Xiang, T.; Liu, J.; Zhou, H.; Wu, X. Psoralen Inhibited Apoptosis of Osteoporotic Osteoblasts by Modulating IRE1-ASK1-JNK Pathway. Biomed. Res. Int. 2017, 2017, 3524307. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Al-Ani, M.K.; Sha, Y.; Chi, Q.; Dong, N.; Yang, L.; Xu, K. Psoralen Protects Chondrocytes, Exhibits Anti-Inflammatory Effects on Synoviocytes, and Attenuates Monosodium Iodoacetate-Induced Osteoarthritis. Int. J. Biol. Sci. 2019, 15, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.; Lee, E.K.; Lee, C.S.; Chun, K.H.; Lee, M.Y.; Jun, H.S. Psoralea corylifolia L. seed extract ameliorates streptozotocin-induced diabetes in mice by inhibition of oxidative stress. Oxid. Med. Cell. Longe. V 2014, 2014, 897296. [Google Scholar]
- Seo, E.; Oh, Y.S.; Kim, D.; Lee, M.Y.; Chae, S.; Jun, H.S. Protective Role of Psoralea corylifolia L. Seed Extract against Hepatic Mitochondrial Dysfunction Induced by Oxidative Stress or Aging. Evid. Based Complement Alternat. Med. 2013, 2013, 678028. [Google Scholar] [CrossRef]
- Du, M.Y.; Duan, J.X.; Zhang, C.Y.; Yang, H.H.; Guan, X.X.; Zhong, W.J.; Liu, Y.Z.; Li, Z.M.; Cheng, Y.R.; Zhou, Y.; et al. Psoralen attenuates bleomycin-induced pulmonary fibrosis in mice through inhibiting myofibroblast activation and collagen deposition. Cell Biol. Int. 2020, 44, 98–107. [Google Scholar] [CrossRef]
- Lan, H.Y. Diverse Roles of TGF-β/Smads in Renal Fibrosis and Inflammation. Int. J. Biol. Sci. 2017, 7, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Ma, T.T.; Meng, X.M. TGF-beta/Smad and Renal Fibrosis. Adv. Exp. Med. Biol. 2019, 1165, 347–364. [Google Scholar]
- Vilaysane, A.; Chun, J.; Seamone, M.E.; Wang, W.; Chin, R.; Hirota, S.; Li, Y.; Clark, S.A.; Tschopp, J.; Trpkov, K.; et al. The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. J. Am. Soc. Nephrol. 2010, 21, 1732–1744. [Google Scholar] [CrossRef]
- Komada, T.; Usui, F.; Kawashima, A.; Kimura, H.; Karasawa, T.; Inoue, Y.; Kobayashi, M.; Mizushina, Y.; Kasahara, T.; Taniguchi, S.; et al. Role of NLRP3 Inflammasomes for Rhabdomyolysis-induced Acute Kidney Injury. Sci. Rep. 2015, 5, 10901. [Google Scholar] [CrossRef]
- Shahzad, K.; Bock, F.; Dong, W.; Wang, H.; Kopf, S.; Kohli, S.; Al-Dabet, M.M.; Ranjan, S.; Wolter, J.; Wacker, C.; et al. Nlrp3-inflammasome activation in non-myeloid-derived cells aggravates diabetic nephropathy. Kidney Int. 2015, 87, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, S.M.; Dowling, J.K.; Ling, Y.H.; Chan, C.T.; Ferens, D.; Kett, M.; Pinar, A.; Samuel, C.S.; Vinh, A.; Arumugam, T.V.; et al. Inflammasome activity is essential for one kidney deoxycorticosterone acetate salt-induced hypertension in mice. Br. J. Pharmacol. 2016, 173, 752–765. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Kim, Y.G.; Kim, D.J.; Park, S.H.; Jeong, K.H.; Lee, Y.H.; Lim, S.J.; Lee, S.H.; Moon, J.Y. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front. Immunol. 2018, 9, 2563. [Google Scholar] [CrossRef]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.J.; Suarez-Alvarez, B.; Grigorescu, M.; Foresto-Neto, O.; Steiger, S.; Desai, J.; Marschner, J.A.; Honarpisheh, M.; Shi, C.; Jordan, J.; et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int. 2018, 93, 656–669. [Google Scholar] [CrossRef]
- Kim, Y.G.; Kim, S.M.; Kim, K.P.; Lee, S.H.; Moon, J.Y. The Role of Inflammasome-Dependent and Inflammasome-Independent NLRP3 in the Kidney. Cells 2019, 8, 1389. [Google Scholar] [CrossRef]
- Shigeoka, A.A.; Mueller, J.L.; Kambo, A.; Mathison, J.C.; King, A.J.; Hall, W.F.; Correia Jda, S.; Ulevitch, R.J.; Hoffman, H.M.; McKay, D.B. An inflammasome-independent role for epithelial-expressed Nlrp3 in renal ischemia-reperfusion injury. J. Immunol. 2010, 185, 6277–6285. [Google Scholar] [CrossRef]
- Wang, W.; Wang, X.; Chun, J.; Vilaysane, A.; Clark, S.; French, G.; Bracey, N.A.; Trpkov, K.; Bonni, S.; Duff, H.J.; et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J. Immunol. 2013, 190, 1239–1249. [Google Scholar] [CrossRef]
- Bracey, N.A.; Gershkovich, B.; Chun, J.; Vilaysane, A.; Meijndert, H.C.; Wright, J.R.; Fedak, P.W., Jr.; Beck, P.L.; Muruve, D.A.; Duff, H.J. Mitochondrial NLRP3 protein induces reactive oxygen species to promote Smad protein signaling and fibrosis independent from the inflammasome. J. Biol. Chem. 2014, 289, 19571–19584. [Google Scholar] [CrossRef]
- Chung, H.; Vilaysane, A.; Lau, A.; Stahl, M.; Morampudi, V.; Bondzi-Simpson, A.; Platnich, J.M.; Bracey, N.A.; French, M.C.; Beck, P.L.; et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ. 2016, 23, 1331–1346. [Google Scholar] [CrossRef]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.M.; Huang, X.R.; Ng, Y.Y.; Nikolic-Paterson, D.J.; Mu, W.; Atkins, R.C.; Lan, H.Y. Interleukin-1 induces tubular epithelial-myofibroblast transdifferentiation through a transforming growth factor-beta1-dependent mechanism in vitro. Am. J. Kidney Dis. 2001, 37, 820–831. [Google Scholar] [CrossRef] [PubMed]
- Burns, W.C.; Kantharidis, P.; Thomas, M.C. The role of tubular epithelial-mesenchymal transition in progressive kidney disease. Cells Tissues Organs 2007, 185, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Chuang, P.Y.; Menon, M.C.; He, J.C. Molecular targets for treatment of kidney fibrosis. J. Mol. Med. 2013, 91, 549–559. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, G.; Darisipudi, M.N.; Anders, H.J. Canonical and non-canonical effects of the NLRP3 inflammasome in kidney inflammation and fibrosis. Nephrol. Dial. Transplant. 2014, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- El-Deeb, O.S.; Atef, M.M.; Hafez, Y.M. The interplay between microbiota-dependent metabolite trimethylamine N-oxide, Transforming growth factor beta/SMAD signaling and inflammasome activation in chronic kidney disease patients: A new mechanistic perspective. J. Cell Biochem. 2019, 120, 14476–14485. [Google Scholar] [CrossRef]
- Wang, Y.J.; Chen, Y.Y.; Hsiao, C.M.; Pan, M.H.; Wang, B.J.; Chen, Y.C.; Ho, C.T.; Huang, K.C.; Chen, R.J. Induction of Autophagy by Pterostilbene Contributes to the Prevention of Renal Fibrosis via Attenuating NLRP3 Inflammasome Activation and Epithelial-Mesenchymal Transition. Front. Cell. Dev. Biol. 2020, 8, 436. [Google Scholar] [CrossRef]
- Mei, Y.; Fang, C.; Ding, S.; Liu, X.; Hu, J.; Xu, J.; Mei, Q. PAP-1 ameliorates DSS-induced colitis with involvement of NLRP3 inflammasome pathway. Int. Immunopharmacol. 2019, 75, 105776. [Google Scholar] [CrossRef]
- Mizuno, S.; Matsumoto, K.; Nakamura, T. Hepatocyte growth factor suppresses interstitial fibrosis in a mouse model of obstructive nephropathy. Kidney Int. 2001, 59, 1304–1314. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, T.W.; Bae, E.; Kim, J.H.; Jung, M.H.; Park, D.J. Psoralen Alleviates Renal Fibrosis by Attenuating Inflammasome-Dependent NLRP3 Activation and Epithelial–Mesenchymal Transition in a Mouse Unilateral Ureteral Obstruction Model. Int. J. Mol. Sci. 2023, 24, 13171. https://doi.org/10.3390/ijms241713171
Lee TW, Bae E, Kim JH, Jung MH, Park DJ. Psoralen Alleviates Renal Fibrosis by Attenuating Inflammasome-Dependent NLRP3 Activation and Epithelial–Mesenchymal Transition in a Mouse Unilateral Ureteral Obstruction Model. International Journal of Molecular Sciences. 2023; 24(17):13171. https://doi.org/10.3390/ijms241713171
Chicago/Turabian StyleLee, Tae Won, Eunjin Bae, Jin Hyun Kim, Myeong Hee Jung, and Dong Jun Park. 2023. "Psoralen Alleviates Renal Fibrosis by Attenuating Inflammasome-Dependent NLRP3 Activation and Epithelial–Mesenchymal Transition in a Mouse Unilateral Ureteral Obstruction Model" International Journal of Molecular Sciences 24, no. 17: 13171. https://doi.org/10.3390/ijms241713171
APA StyleLee, T. W., Bae, E., Kim, J. H., Jung, M. H., & Park, D. J. (2023). Psoralen Alleviates Renal Fibrosis by Attenuating Inflammasome-Dependent NLRP3 Activation and Epithelial–Mesenchymal Transition in a Mouse Unilateral Ureteral Obstruction Model. International Journal of Molecular Sciences, 24(17), 13171. https://doi.org/10.3390/ijms241713171