
“Neuroimmunoendocrinology” in Children with Rheumatic Diseases: How Glucocorticoids Are the Orchestra Director

Abstract
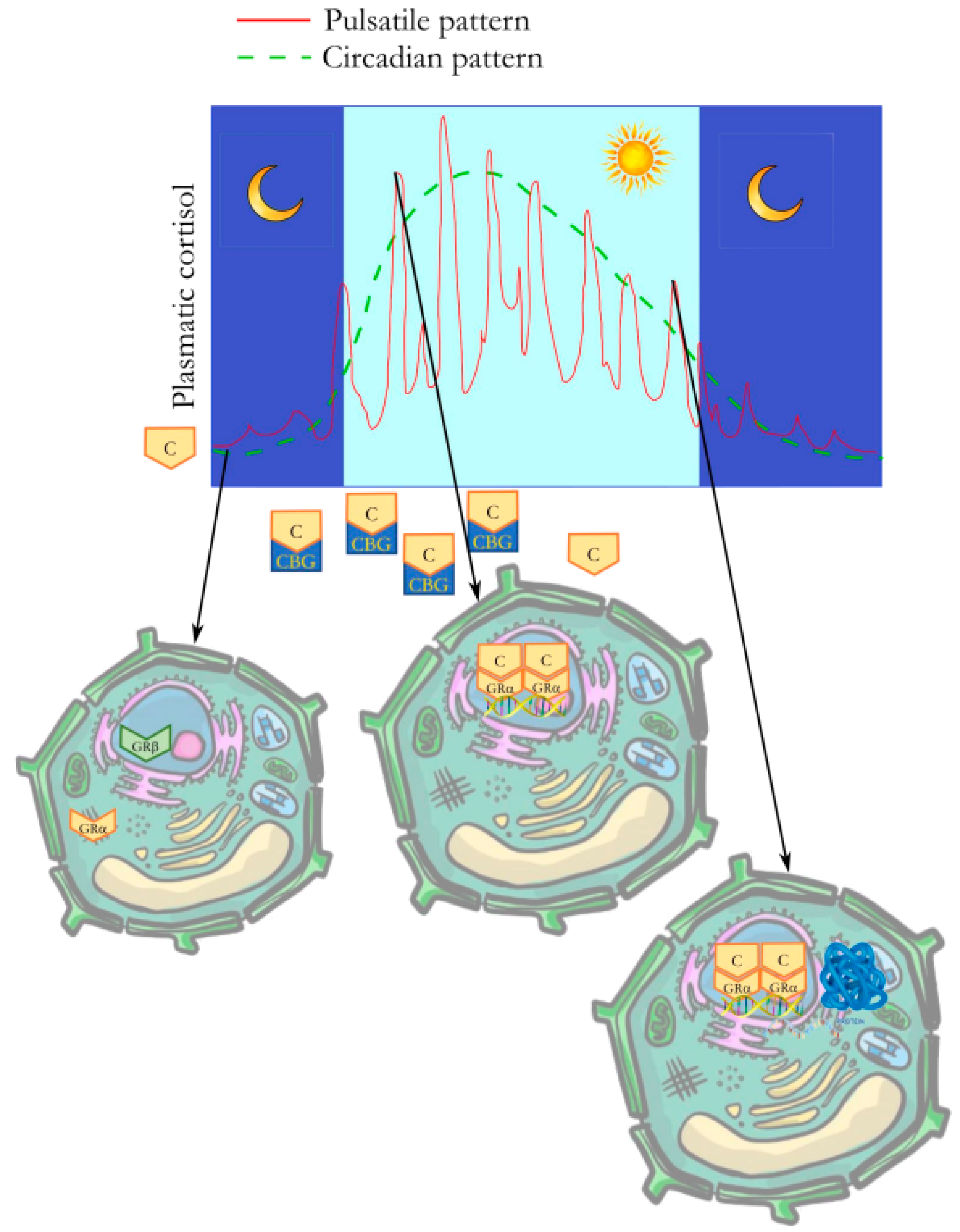
:1. Introduction
2. A Strict Dialog among Neurotransmitters, Hormones, and Cytokines
3. Corticosteroids, Exogenous Glucocorticoids, and Rheumatic Diseases in Children and Adolescents
4. Growth Delay and Short Stature
4.1. Glucocorticoids and Growth
4.2. Glucocorticoids and the Bone
4.3. Glucocorticoids and the Teeth
4.4. Iatrogenic Cushing’s Syndrome
4.5. Glucocorticoids and Inflammation
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Muller, P.H.; Brinkman, D.M.C.; Schonenberg-Meinema, D.; van den Bosch, W.B.; Koopman-Keemink, Y.; Brederije, I.C.J.; Bekkering, P.W.; Kuijpers, T.W.; Van Rossum, M.; van Suijlekom-Smit, L.W.; et al. Treat to target (drug-free) inactive disease in DMARD-naive juvenile idiopathic arthritis: 24-month clinical outcomes of a three-armed randomised trial. Ann. Rheum. Dis. 2018, 78, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar] [PubMed]
- Stojanovich, L.; Marisavljevich, D. Stress as a trigger of autoimmune disease. Autoimmun. Rev. 2008, 7, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Pilipović, I.; Stojić-Vukanić, Z.; Prijić, I.; Leposavić, G. Role of the End-Point Mediators of Sympathoadrenal and Sympathoneural Stress Axes in the Pathogenesis of Experimental Autoimmune Encephalomyelitis and Multiple Sclerosis. Front. Endocrinol. 2020, 10, 921. [Google Scholar] [CrossRef] [PubMed]
- Chhatar, S.; Lal, G. Role of adrenergic receptor signalling in neuroimmune communication. Curr. Res. Immunol. 2021, 2, 202–217. [Google Scholar] [CrossRef]
- Yanase, T.; Nawata, H.; Kato, K.-I.; Ibayashi, H. In vivo evidence of regulation by pituitary-adrenal axis of urinary epinephrine excretion in men. Endocrinol. Jpn. 1986, 33, 449–455. [Google Scholar] [CrossRef]
- Härle, P.; Straub, R.H.; Wiest, R.; Mayer, A.; Schölmerich, J.; Atzeni, F.; Carrabba, M.; Cutolo, M.; Sarzi-Puttini, P. Increase of sympathetic outflow measured by neuropeptide Y and decrease of the hypothalamic-pituitary-adrenal axis tone in patients with systemic lupus erythematosus and rheumatoid arthritis: Another example of uncoupling of response systems. Ann. Rheum. Dis. 2006, 65, 51–56. [Google Scholar] [CrossRef]
- Ferrara, G.; Petrillo, M.G.; Giani, T.; Marrani, E.; Filippeschi, C.; Oranges, T.; Simonini, G.; Cimaz, R. Clinical Use and Molecular Action of Corticosteroids in the Pediatric Age. Int. J. Mol. Sci. 2019, 20, 444. [Google Scholar] [CrossRef]
- Consolaro, A.; Negro, G.; Lanni, S.; Solari, N.; Martini, A.; Ravelli, A. Toward a treat-to-target approach in the management of juvenile idiopathic arthritis. Clin. Exp. Rheumatol. 2012, 30 (Suppl. 73), S157–S162. [Google Scholar]
- National Guideline Centre (UK). Treat-to-Target: Rheumatoid Arthritis in Adults: Diagnosis and Management: Evidence Review C; National Institute for Health and Care Excellence (NICE): London, UK, 2018. [Google Scholar]
- Lightman, S.L.; Birnie, M.T.; Conway-Campbell, B.L. Dynamics of ACTH and Cortisol Secretion and Implications for Disease. Endocr. Rev. 2020, 41, bnaa002. [Google Scholar] [CrossRef]
- Rosol, T.J.; Yarrington, J.T.; Latendresse, J.; Capen, C.C. Adrenal Gland: Structure, Function, and Mechanisms of Toxicity. Toxicol. Pathol. 2001, 29, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Talabér, G.; Jondal, M.; Okret, S. Extra-adrenal glucocorticoid synthesis: Immune regulation and aspects on local organ homeostasis. Mol. Cell. Endocrinol. 2013, 380, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Evans, R.M. The steroid and thyroid hormone receptor superfamily. Science 1988, 240, 889–895. [Google Scholar] [CrossRef]
- Pujols, L.; Mullol, J.; Roca-Ferrer, J.; Torrego, A.; Xaubet, A.; Cidlowski, J.A.; Picado, C. Expression of glucocorticoid receptor α- and β-isoforms in human cells and tissues. Am. J. Physiol. Cell Physiol. 2002, 283, C1324–C1331. [Google Scholar] [CrossRef]
- Smoak, K.A.; Cidlowski, J.A. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech. Ageing Dev. 2004, 125, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Bae, Y.J.; Kratzsch, J. Corticosteroid-binding globulin: Modulating mechanisms of bioavailability of cortisol and its clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2015, 29, 761–772. [Google Scholar] [CrossRef]
- Chapman, K.; Holmes, M.; Seckl, J.; Clemmer, J.S.; Pruett, W.A.; Coleman, T.G.; Hall, J.E.; Hester, R.L.; Rossier, B.C.; Baker, M.E.; et al. 11β-Hydroxysteroid Dehydrogenases: Intracellular Gate-Keepers of Tissue Glucocorticoid Action. Physiol. Rev. 2013, 93, 1139–1206. [Google Scholar] [CrossRef]
- Ramamoorthy, S.; Cidlowski, J.A. Corticosteroids: Mechanisms of Action in Health and Disease. Rheum. Dis. Clin. N. Am. 2016, 42, 15–31. [Google Scholar] [CrossRef]
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Steven, K. Adrenal Cortical Steroids. Drug Facts and Comparisons, 5th ed.; Facts and Comparisons, Inc.: St. Louis, MI, USA, 1997; pp. 122–128. [Google Scholar]
- Stavreva, D.A.; Wiench, M.; John, S.; Conway-Campbell, B.L.; McKenna, M.A.; Pooley, J.R.; Johnson, T.A.; Voss, T.C.; Lightman, S.L.; Hager, G.L. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat. Cell Biol. 2009, 11, 1093–1102. [Google Scholar] [CrossRef]
- Shanks, N.; Windle, R.J.; Perks, P.A.; Harbuz, M.S.; Jessop, D.S.; Ingram, C.D.; Lightman, S.L. Early-life exposure to endotoxin alters hypothalamic–pituitary–adrenal function and predisposition to inflammation. Proc. Natl. Acad. Sci. USA 2000, 97, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- D’angelo, D.M.; Di Donato, G.; Breda, L.; Chiarelli, F. Growth and puberty in children with juvenile idiopathic arthritis. Pediatr. Rheumatol. 2021, 19, 28. [Google Scholar] [CrossRef] [PubMed]
- Ravelli, A.; Davì, S.; Bracciolini, G.; Pistorio, A.; Consolaro, A.; van Dijkhuizen, E.H.P.; Lattanzi, B.; Filocamo, G.; Verazza, S.; Gerloni, V.; et al. Intra-articular corticosteroids versus intra-articular corticosteroids plus methotrexate in oligoarticular juvenile idiopathic arthritis: A multicentre, prospective, randomised, open-label trial. Lancet 2017, 389, 909–916. [Google Scholar] [CrossRef]
- Simon, D.; Fernando, C.; Czernichow, P.; Prieur, A.-M. Linear growth and final height in patients with systemic juvenile idi-opathic arthritis treated with longterm glucocorticoids. J. Rheumatol. 2002, 29, 1296–1300. [Google Scholar]
- Wong, S.C.; Dobie, R.; Altowati, M.A.; Werther, G.A.; Farquharson, C.; Ahmed, S.F. Growth and the Growth Hormone-Insulin Like Growth Factor 1 Axis in Children with Chronic Inflammation: Current Evidence, Gaps in Knowledge, and Future Directions. Endocr. Rev. 2015, 37, 62–110. [Google Scholar] [CrossRef] [PubMed]
- Machado, S.H.; Xavier, R.M.; Lora, P.S.; Gonçalves, L.M.K.; Trindade, L.R.; Marostica, P.J.C. Height and sexual maturation in girls with juvenile idiopathic arthritis. J. Pediatr. 2018, 96, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Giancane, G.; Muratore, V.; Marzetti, V.; Quilis, N.; Benavente, B.S.; Bagnasco, F.; Alongi, A.; Civino, A.; Quartulli, L.; Consolaro, A.; et al. Disease activity and damage in juvenile idiopathic arthritis: Methotrexate era versus biologic era. Thromb. Haemost. 2019, 21, 168. [Google Scholar] [CrossRef] [PubMed]
- Guerne, P.A.; Carson, D.A.; Lotz, M. IL-6 production by human articular chondrocytes. Modulation of its synthesis by cytokines, growth factors, and hormones in vitro. J. Immunol. 1990, 144, 499–505. [Google Scholar] [CrossRef]
- Polito, C.; Strano, C.G.; Olivieri, A.N.; Alessio, M.; Lammarrone, C.S.; Todisco, N.; Papale, M.R. Growth Retardation in Non-steroid Treated Juvenile Rheumatoid Arthritis. Scand. J. Rheumatol. 1997, 26, 99–103. [Google Scholar] [CrossRef]
- McErlane, F.; Carrasco, R.; Kearsley-Fleet, L.; Baildam, E.M.; Wedderburn, L.R.; Foster, H.E.; Ioannou, Y.; Chieng, S.A.; Davidson, J.E.; Thomson, W.; et al. Growth patterns in early juvenile idiopathic arthritis: Results from the Childhood Arthritis Prospective Study (CAPS). Semin. Arthritis Rheum. 2017, 48, 53–60. [Google Scholar] [CrossRef]
- Patti, A.; Maggio, M.C.; Corsello, G.; Messina, G.; Iovane, A.; Palma, A. Evaluation of Fitness and the Balance Levels of Children with a Diagnosis of Juvenile Idiopathic Arthritis: A Pilot Study. Int. J. Environ. Res. Public Health 2017, 14, 806. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, J.; Pistorio, A.; Aldera, E.; Puzone, L.; El Miedany, Y.; Pal, P.; Giri, P.P.; De, H.; Khubchandani, R.; Chavan, P.P.; et al. Development and initial validation of a composite disease activity score for systemic juvenile idiopathic arthritis. Rheumatology 2020, 59, 3505–3514. [Google Scholar] [CrossRef] [PubMed]
- Señarís, R.M.; Lago, F.; Coya, R.; Pineda, J.; Diéguez, C. Regulation of hypothalamic somatostatin, growth hormone-releasing hormone, and growth hormone receptor messenger ribonucleic acid by glucocorticoids. Endocrinology 1996, 137, 5236–5241. [Google Scholar] [CrossRef] [PubMed]
- Bechtold, S.; Simon, D. Growth abnormalities in children and adolescents with juvenile idiopathic arthritis. Rheumatol. Int. 2014, 34, 1483–1488. [Google Scholar] [CrossRef]
- Vakili, H.; Cattini, P.A. The hidden but positive role for glucocorticoids in the regulation of growth hormone-producing cells. Mol. Cell. Endocrinol. 2012, 363, 1–9. [Google Scholar] [CrossRef]
- Agha, A.; Monson, J.P. Modulation of glucocorticoid metabolism by the growth hormone? IGF-1 axis. Clin. Endocrinol. 2007, 66, 459–465. [Google Scholar] [CrossRef]
- Burguera, B.; Muruais, C.; Peñalva, A.; Dieguez, C.; Casanueva, F.F. Dual and Selective Actions of Glucocorticoids upon Basal and Stimulated Growth Hormone Release in Man. Neuroendocrinology 1990, 51, 51–58. [Google Scholar] [CrossRef]
- Giustina, A.; Veldhuis, J.D. Pathophysiology of the Neuroregulation of Growth Hormone Secretion in Experimental Animals and the Human. Endocr. Rev. 1998, 19, 717–797. [Google Scholar] [CrossRef]
- Cannavò, S.; Cappa, M.; Ferone, D.; Isidori, A.M.; Loche, S.; Salerno, M.; Maghnie, M.; Aimaretti, G.; Ambrosio, M.R.; Bellone, S.; et al. Appropriate management of growth hormone deficiency during the age of transition: An Italian Delphi consensus statement. J. Endocrinol. Investig. 2022, 46, 189–200. [Google Scholar] [CrossRef]
- Iughetti, L.; Vannelli, S.; Street, M.E.; Pirazzoli, P.; Bertelloni, S.; Radetti, G.; Capone, L.; Stasiowska, B.; Mazzanti, L.; Gastaldi, R.; et al. Impaired GH Secretion in Patients with SHOX Deficiency and Efficacy of Recombinant Human GH Therapy. Horm. Res. Paediatr. 2012, 78, 279–287. [Google Scholar] [CrossRef]
- Isakova, T.; Nickolas, T.L.; Denburg, M.; Yarlagadda, S.; Weiner, D.E.; Gutiérrez, O.M.; Bansal, V.; Rosas, S.E.; Nigwekar, S.; Yee, J.; et al. KDOQI US Commentary on the 2017 KDIGO Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease–Mineral and Bone Disorder (CKD-MBD). Am. J. Kidney Dis. 2017, 70, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, Z.; Klein, G.L. Overview of pediatric bone problems and related osteoporosis. J. Musculoskelet. Neuronal Interact. 2012, 12, 174–182. [Google Scholar] [PubMed]
- Canalis, E.; Mazziotti, G.; Giustina, A.; Bilezikian, J.P. Glucocorticoid-induced osteoporosis: Pathophysiology and therapy. Osteoporos. Int. 2007, 18, 1319–1328. [Google Scholar] [CrossRef] [PubMed]
- Sota, J.; Rigante, D.; Ruscitti, P.; Insalaco, A.; Sfriso, P.; de Vita, S.; Cimaz, R.; Lopalco, G.; Emmi, G.; La Torre, F.; et al. Anakinra Drug Retention Rate and Predictive Factors of Long-Term Response in Systemic Juvenile Idiopathic Arthritis and Adult Onset Still Disease. Front. Pharmacol. 2019, 10, 918. [Google Scholar] [CrossRef]
- Sota, J.; Insalaco, A.; Cimaz, R.; Alessio, M.; Cattalini, M.; Gallizzi, R.; Maggio, M.C.; Lopalco, G.; La Torre, F.; Fabiani, C.; et al. Drug Retention Rate and Predictive Factors of Drug Survival for Interleukin-1 Inhibitors in Systemic Juvenile Idiopathic Arthritis. Front. Pharmacol. 2019, 9, 1526. [Google Scholar] [CrossRef]
- 49 Bianchi, M.L.; Cimaz, R.; Galbiati, E.; Corona, F.; Cherubini, R.; Bardare, M. Bone Mass Change During Methotrexate Treatment in Patients with Juvenile Rheumatoid Arthritis. Osteoporos. Int. 1999, 10, 20–25. [Google Scholar] [CrossRef]
- Maggio, M.C.; Cimaz, R. Metabolic Bone Disease and Osteoporosis in Children. In Pediatric Rheumatology; Sawhney, S., Aggarwal, A., Eds.; Springer: Singapore, 2017. [Google Scholar] [CrossRef]
- Rousseau-Nepton, I.; Lang, B.; Rodd, C. Long-Term Bone Health in Glucocorticoid-Treated Children with Rheumatic Diseases. Curr. Rheumatol. Rep. 2013, 15, 315. [Google Scholar] [CrossRef]
- Lilleby, V. Bone status in juvenile systemic lupus erythematosus. Lupus 2007, 16, 580–586. [Google Scholar] [CrossRef]
- Zhang, Y.; Milojevic, D. Protecting Bone Health in Pediatric Rheumatic Diseases: Pharmacological Considerations. Pediatr. Drugs 2017, 19, 193–211. [Google Scholar] [CrossRef]
- Cimaz, R. Osteoporosis in childhood rheumatic diseases: Prevention and therapy. Best Pract. Res. Clin. Rheumatol. 2002, 16, 397–409. [Google Scholar] [CrossRef]
- Ward, L.M.; Ma, J.; Robinson, M.-E.; Scharke, M.; Ho, J.; Houghton, K.; Huber, A.; Scuccimarri, R.; Barsalou, J.; Roth, J.; et al. Osteoporotic Fractures and Vertebral Body Reshaping in Children with Glucocorticoid-Treated Rheumatic Disorders. J. Clin. Endocrinol. Metab. 2021, 106, e5195–e5207. [Google Scholar] [CrossRef] [PubMed]
- van Staa, T.P.; Leufkens, H.G.M.; Abenhaim, L.; Zhang, B.; Cooper, C. Oral corticosteroids and fracture risk: Relationship to daily and cumulative doses. Rheumatology 2000, 39, 1383–1389. [Google Scholar] [CrossRef] [PubMed]
- Di Marcello, F.; Di Donato, G.; D’angelo, D.M.; Breda, L.; Chiarelli, F. Bone Health in Children with Rheumatic Disorders: Focus on Molecular Mechanisms, Diagnosis, and Management. Int. J. Mol. Sci. 2022, 23, 5725. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-W.; Jung, J.-Y.; Kim, H.-A.; Suh, C.-H. Anti-Inflammatory Effects of Low-Dose Glucocorticoids Compensate for Their Detrimental Effects on Bone Mineral Density in Patients with Rheumatoid Arthritis. J. Clin. Med. 2021, 10, 2944. [Google Scholar] [CrossRef]
- Wiebe, E.; Huscher, D.; Schaumburg, D.; Palmowski, A.; Hermann, S.; Buttgereit, T.; Biesen, R.; Burmester, G.-R.; Palmowski, Y.; Boers, M.; et al. Optimising both disease control and glucocorticoid dosing is essential for bone protection in patients with rheumatic disease. Ann. Rheum. Dis. 2022, 81, 1313–1322. [Google Scholar] [CrossRef]
- Stagi, S.; Masi, L.; Capannini, S.; Cimaz, R.; Tonini, G.; Matucci-Cerinic, M.; de Martino, M.; Falcini, F. Cross-sectional and Longitudinal Evaluation of Bone Mass in Children and Young Adults with Juvenile Idiopathic Arthritis: The Role of Bone Mass Determinants in a Large Cohort of Patients. J. Rheumatol. 2010, 37, 1935–1943. [Google Scholar] [CrossRef]
- Stagi, S.; Cavalli, L.; Bertini, F.; Signorini, C.; Cerinic, M.M.; de Martino, M.; Brandi, M.L.; Falcini, F. Comparison of bone mass and quality determinants in adolescents and young adults with juvenile systemic lupus erythematosus (JSLE) and juvenile idiopathic arthritis (JIA). Lupus 2014, 23, 1392–1406. [Google Scholar] [CrossRef]
- Brusa, J.; Maggio, M.C.; Giustino, V.; Thomas, E.; Zangla, D.; Iovane, A.; Palma, A.; Corsello, G.; Messina, G.; Bellafiore, M. Upper and Lower Limb Strength and Body Posture in Children with Congenital Hypothyroidism: An Observational Case-Control Study. Int. J. Environ. Res. Public Health 2020, 17, 4830. [Google Scholar] [CrossRef]
- Bianco, A.; Patti, A.; Thomas, E.; Palma, R.; Maggio, M.C.; Paoli, A.; Palma, A. Evaluation of fitness levels of children with a diagnosis of acute leukemia and lymphoma after completion of chemotherapy and autologous hematopoietic stem cell transplantation. Cancer Med. 2014, 3, 385–389. [Google Scholar] [CrossRef]
- Boyce, B.F.; Schwarz, E.M.; Xing, L. Osteoclast precursors: Cytokine-stimulated immunomodulators of inflammatory bone disease. Curr. Opin. Rheumatol. 2006, 18, 427–432. [Google Scholar] [CrossRef]
- Devlin, R.D.; Reddy, S.V.; Savino, R.; Ciliberto, G.; Roodman, G.D.; Roodman, G.D. IL-6 Mediates the Effects of IL-1 or TNF, but Not PTHrP or 1,25(OH)2D3, on Osteoclast-like Cell Formation in Normal Human Bone Marrow Cultures. J. Bone Miner. Res. 1998, 13, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Vega, D.; Maalouf, N.M.; Sakhaee, K. CLINICAL Review: The role of receptor activator of nuclear factor-kappaB (RANK)/RANK ligand/osteoprotegerin: Clinical implications. J. Clin. Endocrinol. Metab. 2007, 92, 4514–4521. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Billiau, A.D.; Loop, M.; Le, P.-Q.; Berthet, F.; Philippet, P.; Kasran, A.; Wouters, C.H. Etanercept improves linear growth and bone mass acquisition in MTX-resistant polyarticular-course juvenile idiopathic arthritis. Rheumatology 2010, 49, 1550–1558. [Google Scholar] [CrossRef] [PubMed]
- Heidt, C.; Grueberger, N.; Grisch, D.; Righini-Grunder, F.; Rueger, M.; Ramseier, L. The Assessment of Steroid Injections as a Potential Risk Factor for Osteochondral Lesions in Children with Juvenile Idiopathic Arthritis. Cartilage 2020, 13, 894S–899S. [Google Scholar] [CrossRef] [PubMed]
- Hemke, R.; Nusman, C.M.; Van Der Heijde, D.M.F.M.; Doria, A.S.; Kuijpers, T.W.; Maas, M.; Van Rossum, M.A.J. Frequency of joint involvement in juvenile idiopathic arthritis during a 5-year follow-up of newly diagnosed patients: Implications for MR imaging as outcome measure. Rheumatol. Int. 2014, 35, 351–357. [Google Scholar] [CrossRef]
- Civino, A.; Alighieri, G.; Prete, E.; Caroleo, A.M.; Magni-Manzoni, S.; Vinti, L.; Romano, M.; Santoro, N.; Filocamo, G.; Belotti, T.; et al. Musculoskeletal manifestations of childhood cancer and differential diagnosis with juvenile idiopathic arthritis (ONCOREUM): A multicentre, cross-sectional study. Lancet Rheumatol. 2021, 3, e507–e516. [Google Scholar] [CrossRef]
- Kaneko, K.; Chen, H.; Kaufman, M.; Sverdlov, I.; Stein, E.M.; Park-Min, K.H. Glucocorticoid-induced osteonecrosis in systemic lupus erythematosus patients. Clin. Transl. Med. 2021, 11, e526. [Google Scholar] [CrossRef]
- Nakamura, J.; Saisu, T.; Yamashita, K.; Suzuki, C.; Kamegaya, M.; Takahashi, K. Age at time of corticosteroid administration is a risk factor for osteonecrosis in pediatric patients with systemic lupus erythematosu: A prospective MagnetIc resonance imaging study. Arthritis Rheum. 2010, 62, 609–615. [Google Scholar] [CrossRef]
- Houari, S.; Loiodice, S.; Jedeon, K.; Berdal, A.; Babajko, S. Expression of Steroid Receptors in Ameloblasts during Amelogenesis in Rat Incisors. Front. Physiol. 2016, 7, 503. [Google Scholar] [CrossRef]
- Babajko, S.; Jedeon, K.; Houari, S.; Loiodice, S.; Berdal, A. Disruption of Steroid Axis, a New Paradigm for Molar Incisor Hypomineralization (MIH). Front. Physiol. 2017, 8, 343. [Google Scholar] [CrossRef]
- Firetto, M.C.; Abbinante, A.; Barbato, E.; Bellomi, M.; Biondetti, P.; Borghesi, A.; Bossu’, M.; Cascone, P.; Corbella, D.; Di Candido, V.; et al. National guidelines for dental diagnostic imaging in the developmental age. Radiol. Med. 2019, 124, 887–916. [Google Scholar] [CrossRef]
- Güven, A. Different Potent Glucocorticoids, Different Routes of Exposure but the Same Result: Iatrogenic Cushing’s Syndrome and Adrenal Insufficiency. J. Clin. Res. Pediatr. Endocrinol. 2020, 12, 383–392. [Google Scholar] [CrossRef]
- McCann, S.M.; Antunes-Rodrigues, J.; Franci, C.R.; Anselmo-Franci, J.A.; Karanth, S.; Rettori, V. Role of the hypothalamic pituitary adrenal axis in the control of the response to stress and infection. Braz. J. Med. Biol. Res. 2000, 33, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, S.R.; Ziegler, C.G.; Krug, A.W.; Kanczkowski, W.; Rettori, V.; McCANN, S.M.; Wirth, M.; Zacharowski, K. The Role of Toll-like Receptors in the Immune-Adrenal Crosstalk. Ann. N. Y. Acad. Sci. 2006, 1088, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Cain, D.W.; Cidlowski, J.A. Immune regulation by glucocorticoids. Nat. Rev. Immunol. 2017, 17, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Kunnathully, V.; Gomez-Lira, M.; Bassi, G.; Poli, F.; Zoratti, E.; La Verde, V.; Idolazzi, L.; Gatti, D.; Viapiana, O.; Adami, S.; et al. CD14++ CD16− monocytes are the main source of 11β-HSD type 1 after IL-4 stimulation. Int. Immunopharmacol. 2017, 43, 156–163. [Google Scholar] [CrossRef]
- Barnes, P.J. Anti-inflammatory Actions of Glucocorticoids: Molecular Mechanisms. Clin. Sci. 1998, 94, 557–572. [Google Scholar] [CrossRef]
- Berrebi, D.; Bruscoli, S.; Cohen, N.; Foussat, A.; Migliorati, G.; Bouchet-Delbos, L.; Maillot, M.-C.; Portier, A.; Couderc, J.; Galanaud, P.; et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: An anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood 2003, 101, 729–738. [Google Scholar] [CrossRef]
- Hardy, R.S.; Fenton, C.; Croft, A.P.; Naylor, A.J.; Begum, R.; Desanti, G.; Buckley, C.D.; Lavery, G.; Cooper, M.S.; Raza, K. 11 Beta-hydroxysteroid dehydrogenase type 1 regulates synovitis, joint destruction, and systemic bone loss in chronic polyarthritis. J. Autoimmun. 2018, 92, 104–113. [Google Scholar] [CrossRef]
- Dimitrov, S.; Benedict, C.; Heutling, D.; Westermann, J.; Born, J.; Lange, T. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 2009, 113, 5134–5143. [Google Scholar] [CrossRef] [PubMed]
- Jabara, H.H.; Ahern, D.J.; Vercelli, D.; Geha, R.S. Hydrocortisone and IL-4 induce IgE isotype switching in human B cells. J. Immunol. 1991, 147, 1557–1560. [Google Scholar] [CrossRef] [PubMed]
- Dragoş, D.; Tănăsescu, M.D. The effect of stress on the defense systems. J. Med. Life 2010, 3, 10–18. [Google Scholar] [PubMed]
- Margaryan, S.; Hyusyan, A.; Martirosyan, A.; Sargsian, S.; Manukyan, G. Differential modulation of innate immune response by epinephrine and estradiol. Horm. Mol. Biol. Clin. Investig. 2017, 30, 20160046. [Google Scholar] [CrossRef] [PubMed]
- Dhabhar, F.S.; Malarkey, W.B.; Neri, E.; McEwen, B.S. Stress-induced redistribution of immune cells—From barracks to boulevards to battlefields: A tale of three hormones—Curt Richter Award Winner. Psychoneuroendocrinology 2012, 37, 1345–1368. [Google Scholar] [CrossRef] [PubMed]
- Krueger, J.M.; Obál, F.J.; Fang, J.D.; Kubota, T.; Taishi, P. The role of cytokines in physiological sleep regulation. Ann. N. Y. Acad. Sci. 2001, 933, 211–221. [Google Scholar] [CrossRef]
- Giancane, G.; Swart, J.F.; Castagnola, E.; Groll, A.H.; Horneff, G.; Huppertz, H.-I.; Lovell, D.J.; Wolfs, T.; Herlin, T.; Dolezalova, P.; et al. Opportunistic infections in immunosuppressed patients with juvenile idiopathic arthritis: Analysis by the Pharmachild Safety Adjudication Committee. Arthritis Res. Ther. 2020, 22, 71. [Google Scholar] [CrossRef]
- Maggio, M.C.; Corsello, G. FMF is not always “fever”: From clinical presentation to “treat to target”. Ital. J. Pediatr. 2020, 46, 7. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Potency Relative to Hydrocortisone | Half-Life | ||||
|---|---|---|---|---|---|
| Equivalent Glucocorticoid Dose (mg) | Anti-Inflammatory | Mineralocorticoid | Plasma (minutes) | Duration of Action (hours) | |
| GLUCOCORTICOIDS | |||||
| Short acting | |||||
| Hydrocortisone | 20 | 1 | 1 | 90 | 8–12 |
| Cortisone acetate | 25 | 0.8 | 0.8 | 30 | 8–12 |
| Intermediate acting | |||||
| Prednisone | 5 | 4 | 0.8 | 60 | 12–36 |
| Prednisolone | 5 | 4 | 0.8 | 200 | 12–36 |
| Triamcinolone | 4 | 5 | 0 | 300 | 12–36 |
| Methylprednisolone | 4 | 5 | 0.5 | 180 | 12–36 |
| Long acting | |||||
| Dexamethasone | 0.75 | 30 | 0 | 200 | 36–54 |
| Betamethasone | 0.6 | 30 | 0 | 300 | 36–54 |
| MINERALCORTICOIDS | |||||
| Fludrocortisone | 0 | 15 | 150 | 240 | 24–36 |
| Aldosterone | 0 | 0 | >400 | 20 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maggio, M.C.; Miniaci, A.; Gallizzi, R.; Civino, A. “Neuroimmunoendocrinology” in Children with Rheumatic Diseases: How Glucocorticoids Are the Orchestra Director. Int. J. Mol. Sci. 2023, 24, 13192. https://doi.org/10.3390/ijms241713192
Maggio MC, Miniaci A, Gallizzi R, Civino A. “Neuroimmunoendocrinology” in Children with Rheumatic Diseases: How Glucocorticoids Are the Orchestra Director. International Journal of Molecular Sciences. 2023; 24(17):13192. https://doi.org/10.3390/ijms241713192
Chicago/Turabian StyleMaggio, Maria Cristina, Angela Miniaci, Romina Gallizzi, and Adele Civino. 2023. "“Neuroimmunoendocrinology” in Children with Rheumatic Diseases: How Glucocorticoids Are the Orchestra Director" International Journal of Molecular Sciences 24, no. 17: 13192. https://doi.org/10.3390/ijms241713192