
Molecular Modelling of Polychlorinated Dibenzo-p-Dioxins Non-Covalent Interactions with β and γ-Cyclodextrins

Abstract
:1. Introduction
2. Results and Discussion
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- White, S.S.; Birnbaum, L.S. An overview of the effects of dioxins and dioxin-like compounds on vertebrates, as documented in human and ecological epidemiology. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2009, 27, 197–211. [Google Scholar] [CrossRef]
- Van Den Heuvel, J.P.; Lucier, G. Environmental toxicology of polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans. Environ. Health Perspect. 1993, 100, 189–200. [Google Scholar] [CrossRef]
- Hites, R.A. Dioxins: An overview and history. Environ. Sci. Technol. 2011, 45, 16–20. [Google Scholar] [CrossRef] [PubMed]
- Sobol, Ł.; Dyjakon, A.; Soukup, K. Dioxins and furans in biochars, hydrochars and torreficates produced by thermochemical conversion of biomass: A review. Environ. Chem. Lett. 2023, 21, 2225–2249. [Google Scholar] [CrossRef]
- Kirkok, S.K.; Kibet, J.K.; Kinyanjui, T.K.; Okanga, F.I. A review of persistent organic pollutants: Dioxins, furans, and their associated nitrogenated analogues. SN Appl. Sci. 2020, 2, 1729. [Google Scholar] [CrossRef]
- Backhaus, T.; Faust, M. Predictive environmental risk assessment of chemical mixtures: A conceptual framework. Environ. Sci. Technol. 2012, 46, 2564–2573. [Google Scholar] [CrossRef] [PubMed]
- Rossberg, M.; Lendle, W.; Pfleiderer, G.; Tögel, A.; Dreher, E.-L.; Langer, E.; Rassaerts, H.; Kleinschmidt, P.; Strack, H.; Cook, R.; et al. Chlorinated Hydrocarbons. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley: Chicester, UK, 2006. [Google Scholar] [CrossRef]
- Mandal, P.K. Dioxin: A review of its environmental effects and its aryl hydrocarbon receptor biology. J. Comp. Physiol. B Biochem. Syst. Environ. Physiol. 2005, 175, 221–230. [Google Scholar] [CrossRef]
- Nikiema, J.; Wielgosiński, G. The Possibilities of Reduction of Polychlorinated Dibenzo-P-Dioxins and Polychlorinated Dibenzofurans Emission. Int. J. Chem. Eng. 2010, 2010, 392175. [Google Scholar] [CrossRef]
- Seidel, S.D.; Winters, G.M.; Rogers, W.J.; Ziccardi, M.H.; Li, V.; Keser, B.; Denison, M.S. Activation of the Ah Receptor Signaling Pathway by Prostaglandins. J. Biochem. Mol. Toxicol. 2001, 15, 187–196. [Google Scholar] [CrossRef]
- Sorg, O. AhR signalling and dioxin toxicity. Toxicol. Lett. 2013, 230, 225–233. [Google Scholar] [CrossRef]
- Van den Berg, M.; Birnbaum, L.S.; Denison, M.; De Vito, M.; Farland, W.; Feeley, M.; Fiedler, H.; Hakansson, H.; Hanberg, A.; Haws, L.; et al. The 2005 World Health Organization reevaluation of human and Mammalian toxic equivalency factors for dioxins and dioxin-like compounds. Toxicol. Sci. 2006, 93, 223–241. [Google Scholar] [CrossRef] [PubMed]
- Saokham, P.; Muankaew, C.; Jansook, P.; Loftsson, T. Solubility of Cyclodextrins and Drug/Cyclodextrin Complexes. Molecules 2018, 23, 1161. [Google Scholar] [CrossRef] [PubMed]
- Crumling, M.A.; King, K.A.; Duncan, R.K. Cyclodextrins and Iatrogenic Hearing Loss: New Drugs with Significant Risk. Front. Cell. Neurosci. 2017, 11, 355. [Google Scholar] [CrossRef]
- Winkler, R.; Fioravanti, S.; Ciccott, G.; Margheritis, C.; Villa, M. Hydration of β-cyclodextrin: A molecular dynamics simulation study. J. Comput.-Aided Mol. Des. 2000, 14, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Gidwani, B.; Vyas, A. A Comprehensive Review on Cyclodextrin-Based Carriers for Delivery of Chemotherapeutic Cytotoxic Anticancer Drugs. Biomed Res. Int. 2015, 2015, 198268. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, G.; Tiwari, R.; Rai, A.K. Cyclodextrins in delivery systems: Applications. J. Pharm. Bioallied Sci. 2010, 2, 72–79. [Google Scholar] [CrossRef]
- Bethanis, K.; Christoforides, E.; Tsorteki, F.; Fourtaka, K.; Mentzafos, D. Structural studies of the inclusion compounds of α-naphthaleneacetic acid in heptakis(2,6-di-O-methyl)-β-Cyclodextrin and heptakis(2,3,6-tri-O-methyl)-β-Cyclodextrin by X-ray crystallography and molecular dynamics. J. Incl. Phenom. Macrocycl. Chem. 2018, 92, 157–171. [Google Scholar] [CrossRef]
- Liu, P.; Zhang, D.; Zhan, J. Investigation on the inclusions of PCB52 with cyclodextrins by performing DFT calculations and molecular dynamics simulations. J. Phys. Chem. A 2010, 114, 13122–13128. [Google Scholar] [CrossRef]
- Cid-Samamed, A.; Rakmai, J.; Mejuto, J.C.; Simal-Gandara, J.; Astray, G. Cyclodextrins inclusion complex: Preparation methods, analytical techniques and food industry applications. Food Chem. 2022, 384, 132467. [Google Scholar] [CrossRef]
- Zhang, H.; He, W.; Luo, X.; Lin, X.; Lu, X. Adsorption of 2,3,7,8-tetrochlorodibenzo-p-dioxins on intrinsic, defected, and Ti (N, Ag) doped graphene: A DFT study. J. Mol. Model. 2014, 20, 2238. [Google Scholar] [CrossRef]
- Izakmehri, Z.; Davish Ganji, M.; Ardjmand, M. Adsorption of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin (TCDD) on pristine, defected and Al-doped carbon nanotube: A dispersion corrected DFT study. Vacuum 2017, 136, 51–59. [Google Scholar] [CrossRef]
- Paustenbach, D.J.; Wenning, R.J.; Lau, V.; Harrington, N.W.; Rennix, D.K.; Parsons, A.H. Recent developments on the hazards posed by 2,3,7,8-tetrachlorodibenzo-p-dioxin in soil: Implications for setting risk-based cleanup levels at residential and industrial sites. J. Toxicol. Environ. Health 1992, 36, 103–1049. [Google Scholar] [CrossRef] [PubMed]
- Nhung, N.T.H.; Nguyen, X.-T.T.; Long, V.D.; Wei, Y.; Fujita, T. A Review of Soil Contaminated with Dioxins and Biodegradation Technologies: Current Status and Future Prospects. Toxics 2022, 10, 278. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Zhang, D.; Zhan, J. Theoretical investigation on the inclusion of TCDD with β-cyclodextrin by performing QM calculations and MD simulations. J. Hazard. Mater. 2011, 192, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Mandravel, C.; Vergoten, G.; Stanculescu, I. Interactions Moleculaires; Applications aux Medicaments, Editura Universitatii din Bucuresti: Bucharest, Romania, 2007. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graph. Mod. 1999, 17, 57–61. [Google Scholar] [CrossRef]
- Liu, L.; Guo, Q.X. The Driving Forces in the Inclusion Complexation of Cyclodextrins. J. Incl. Phenom. 2002, 42, 1–14. [Google Scholar] [CrossRef]
- Kollman, P. A general analysis of noncovalent intermolecular interactions. J. Am. Chem. Soc. 1977, 99, 4875–4894. [Google Scholar] [CrossRef]
- Brinck, T.; Murray, J.S.; Politzer, P. Surface electrostatic potentials of halogenated methanes as indicators of directional intermolecular interactions. Int. J. Quantum Chem. 1992, 44, 57–64. [Google Scholar] [CrossRef]
- Borovina, M.; Kodrin, I.; Đaković, M. Testing the limits of halogen bonding in coordination chemistry. Cryst. Eng. Comm. 2018, 20, 539–549. [Google Scholar] [CrossRef]
- Kashina, M.V.; Kinzhalov, M.A.; Smirnov, A.S.; Ivanov, D.M.; Novikov, A.S.; Kukushkin, V.Y. Dihalomethanes as bent bifunctional XB/XB-Donating building blocks for construction of metal-involving halogen bonded hexagons. Chem.—Asian J. 2019, 14, 3915–3920. [Google Scholar] [CrossRef]
- Bulatova, M.; Melekhova, A.A.; Novikov, A.S.; Ivanov, D.M.; Bokach, N.A. Redox reactive (RNC)CuII species stabilized in the solid state via halogen bond with I2. Z. Kristallogr.—Crystal. Mater. 2018, 233, 371–377. [Google Scholar] [CrossRef]
- Gamekkanda, J.C.; Sinha, A.S.; Desper, J.; Ðaković, M.; Aakeröy, C.B. The Role of Halogen Bonding in Controlling Assembly and Organization of Cu (II)-Acac Based Coordination Complexes. Crystals 2017, 7, 226. [Google Scholar] [CrossRef]
- Anisimova, T.B.; Kinzhalov, M.A.; da Silva, M.F.C.G.; Novikov, A.S.; Kukushkin, V.Y.; Pombeiro, A.J.; Luzyanin, K.V. Addition of N-nucleophiles to gold (III)-bound isocyanides leading to short-lived gold (III) acyclic diaminocarbene complexes. New J. Chem. 2017, 41, 3246–3250. [Google Scholar] [CrossRef]
- Khavasi, H.R.; Norouzi, F.; Azhdari Tehrani, A. Halogen bonding synthon modularity in coordination compounds. Crystal Growth Des. 2015, 15, 2579–2583. [Google Scholar] [CrossRef]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T.; Resnati, G. The σ-hole revisited. Phys. Chem. Chem. Phys. 2017, 19, 32166–32178. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, G.; Milani, R.; Pilati, T.; Priimagi, A.; Resnati, G.; Terraneo, G. The Halogen Bond. Chem. Rev. 2016, 116, 2478–2601. [Google Scholar] [CrossRef]
- Sirimulla, S.; Bailey, J.B.; Vegesna, R.; Narayan, M. Halogen Interactions in Protein−Ligand Complexes: Implications of Halogen Bonding for Rational Drug Design. J. Chem. Inf. Model. 2013, 53, 2781–2791. [Google Scholar] [CrossRef]
- Lu, Y.; Li, H.; Zhu, X.; Zhu, W.; Liu, H. How does halogen bonding behave in solution? A theoretical study using implicit solvation model. J. Phys. Chem. A 2011, 115, 4467–4475. [Google Scholar] [CrossRef]
- Murray, J.S.; Evans, P.; Politzer, P. A comparative analysis of the electrostatic potentials of some structural analogues of 2,3,7,8-tetrachlorodibenzo-p-dioxin and of related aromatic systems. Int. J. Quant. Chem. 1990, 37, 271–289. [Google Scholar] [CrossRef]
- Sjoberg, P.; Murray, J.S.; Brinck, T.; Evans, P.; Politzer, P. The use of the electrostatic potential at the molecular surface in recognition interactions: Dibenzo-p-dioxins and related systems. J. Mol. Graph. 1990, 8, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Murray, J.S.; Clark, T. Mathematical modeling and physical reality in noncovalent interactions. J. Mol. Model. 2015, 21, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Pinjari, R.V.; Joshi, K.A.; Gejji, S.P. Molecular Electrostatic Potentials and Hydrogen Bonding in α-, β-, and γ-Cyclodextrins. J. Phys. Chem. A 2006, 110, 13073–13080. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Politzer, P. Hydrogen Bonding: A Coulombic σ-Hole Interaction. J. Indian. Inst. Sci. 2020, 100, 21–30. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Geometry of C-H···O hydrogen bonds in carbohydrate crystal structures. Analysis of neutron diffraction data. J. Am. Chem. Soc. 1992, 114, 10146–10154. [Google Scholar] [CrossRef]
- Desiraju, G.R. The C-H···O hydrogen bond: Structural implications and supramolecular design. Acc. Chem. Res. 1996, 29, 441–449. [Google Scholar] [CrossRef]
- Yan, C.; Xiu, Z.; Li, X.; Hao, C. Molecular modeling study of β-cyclodextrin complexes with (+)-catechin and (−)-epicatechin. J. Mol. Graph. Model. 2007, 26, 420–428. [Google Scholar] [CrossRef]
- Hyperchem Program, Version 6.01 for Windows; Hypercube Inc.: Gainesville, FL, USA, 2000.
- Available online: http://www.rcsb.org/pdb/home/home.do (accessed on 16 December 2022).
- Camacho, C.J.; Vajda, S. Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. USA 2001, 98, 10636–10641. [Google Scholar] [CrossRef]
- Hobza, P.; Zahradnic, R. Intermolecular Complexes; Akademia: Praha, Czech Republic, 1988. [Google Scholar]
- Lino, A.C.S.; Takahata, Y.; Jaime, C. Alpha- and beta-cyclodextrin complexes with n-alkyl carboxylic acids and n-alkyl p-hydroxy benzoates. A molecular mechanics study of 1:1 and 1:2 associations. J. Mol. Struct. THEOCHEM 2002, 594, 207–213. [Google Scholar] [CrossRef]
- Scheiner, S. (Ed.) Molecular Interactions: From Van der Waals to Strongly Bound Complexes; Willey: Chicester, UK, 1997. [Google Scholar]
- Mindrila, G.; Mandravel, C.; Dobrica, I.; Bugheanu, P.; Stanculescu, I.R. Theoretical study of beta and gama-cyclodextrins inclusion complexes with nineteen atropisomeric polychlorobiphenyls. J. Incl. Phenom. Macrocycl. Chem. 2012, 74, 137–143. [Google Scholar] [CrossRef]
- Murray, J.S.; Lane, P.; Politzer, P. Expansion of the σ-hole concept. J. Mol. Model. 2009, 15, 723–729. [Google Scholar] [CrossRef] [PubMed]
- Masetti, M.; Rocchia, W. Molecular mechanics and dynamics: Numerical tools to sample the configuration space. Front. Biosci. 2014, 19, 578–604. [Google Scholar] [CrossRef] [PubMed]
- Poltev, V. Molecular Mechanics: Principles, History, and Current Status. In Handbook of Computational Chemistry; Leszczynski, J., Ed.; Springer: Dordrecht, The Netherlands, 2015. [Google Scholar] [CrossRef]

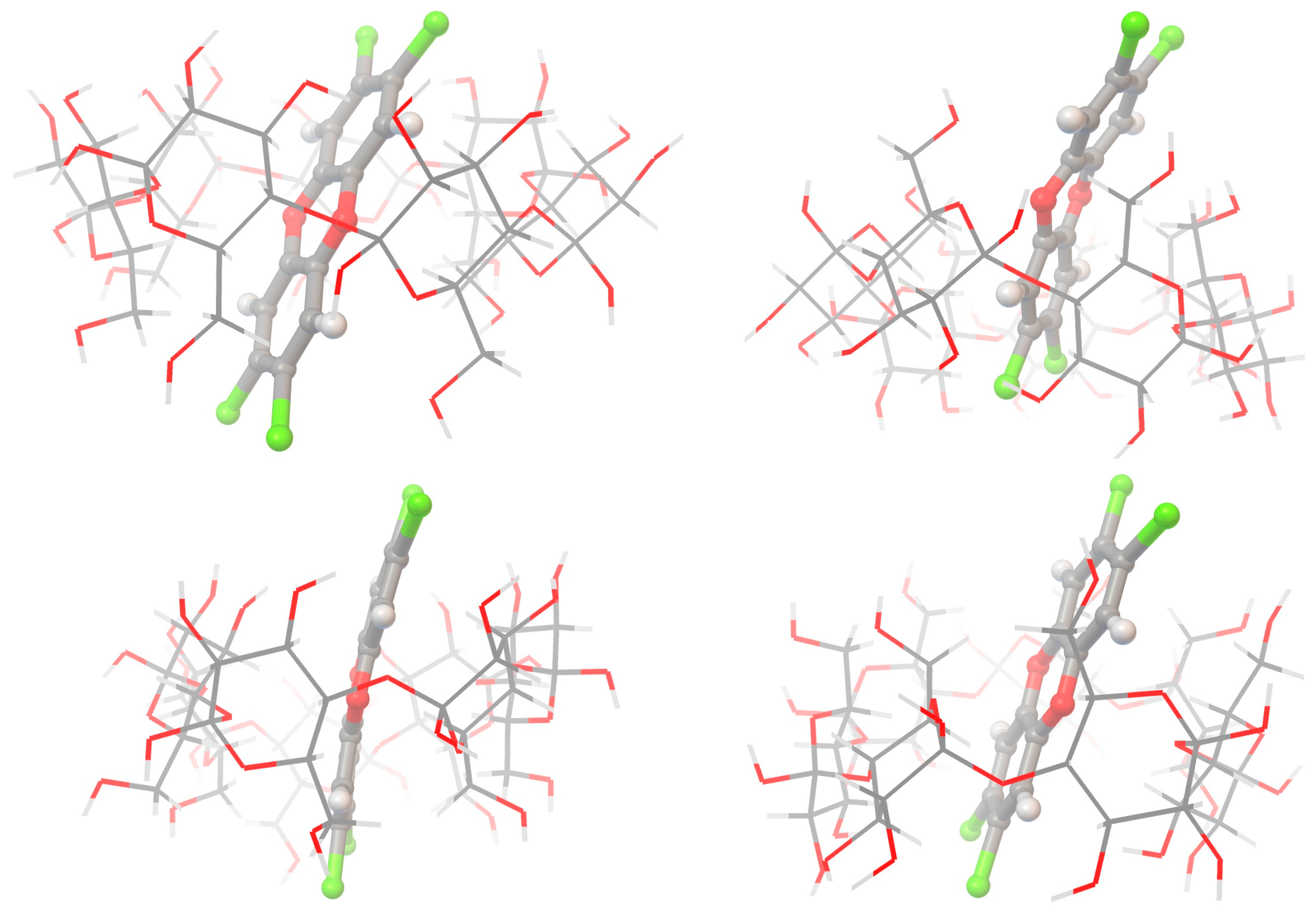


{kind=link}
{kind=link}
{kind=link}
{kind=link}
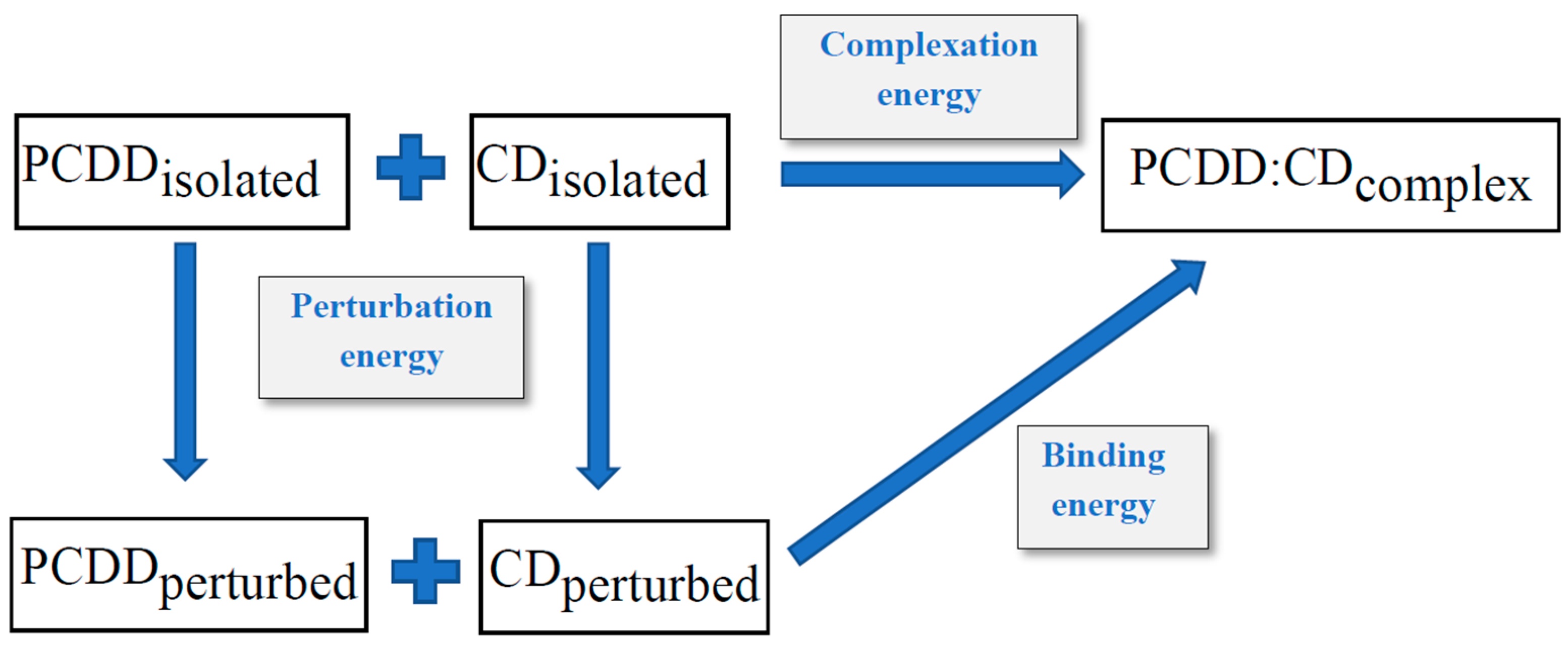
| MM+ | Ecomplex (1) | EvdWcomplex (1) | Eperturbed (1) | EvdWperturbed (1) | Ecomplex (2) | EvdWcomplex (2) | Eperturbed (2) | EvdWperturbed (2) |
|---|---|---|---|---|---|---|---|---|
| TCDD2378 | 52.30 | 9.21 | 77.63 | 34.69 | 70.96 | 16.27 | 107.05 | 46.03 |
| PCDD12378-123 | 58.43 | 10.12 | 82.79 | 34.35 | 76.29 | 24.11 | 95.08 | 42.33 |
| PCDD12378-78 | 55.45 | 7.90 | 84.04 | 35.25 | 72.09 | 20.04 | 96.95 | 42.41 |
| H6CDD123478-1234 | 65.27 | 16.84 | 88.07 | 36.74 | 78.16 | 23.64 | 99.43 | 43.89 |
| H6CDD123478-78 | 63.77 | 13.90 | 87.65 | 37.60 | 77.10 | 21.25 | 99.71 | 43.16 |
| H6CDD123678 | 63.80 | 15.10 | 91.05 | 40.99 | 81.29 | 22.55 | 108.66 | 48.22 |
| H6CDD123789 | 65.79 | 16.38 | 92.52 | 41.63 | 81.46 | 24.61 | 107.58 | 48.36 |
| H7CDD1234678-1234 | 66.58 | 16.77 | 87.98 | 37.25 | 79.74 | 23.51 | 103.61 | 44.80 |
| H7CDD1234678-678 | 61.58 | 10.21 | 88.25 | 36.38 | 79.08 | 24.54 | 100.77 | 44.92 |
| OCDD12346789 | 69.48 | 17.44 | 91.59 | 38.43 | 82.16 | 24.48 | 107.05 | 46.03 |
| MM+ | Ecomplex (1) | EvdWcomplex (1) | Eperturbed (1) | EvdWperturbed (1) | Ecomplex (2) | EvdWcomplex (2) | Eperturbed (2) | EvdWperturbed (2) |
|---|---|---|---|---|---|---|---|---|
| TCDD2378 | 54.04 | 11.07 | 77.98 | 34.39 | 76.40 | 24.75 | 93.43 | 40.70 |
| PCDD12378-123 | 58.66 | 15.88 | 80.37 | 35.51 | 72.15 | 18.61 | 90.11 | 34.21 |
| PCDD12378-78 | 55.33 | 10.29 | 81.59 | 35.02 | 74.94 | 25.27 | 95.63 | 42.38 |
| H6CDD123478-1234 | 64.47 | 18.87 | 84.53 | 35.60 | 80.69 | 27.01 | 100.38 | 43.54 |
| H6CDD123478-78 | 57.79 | 10.64 | 84.90 | 36.80 | 81.95 | 27.50 | 99.99 | 43.45 |
| H6CDD123678 | 69.16 | 18.90 | 92.32 | 41.50 | 84.23 | 27.02 | 107.46 | 48.54 |
| H6CDD123789 | 74.42 | 27.62 | 90.60 | 41.75 | 80.21 | 23.61 | 103.20 | 43.73 |
| H7CDD1234678-1234 | 66.17 | 17.00 | 88.74 | 38.03 | 79.55 | 24.34 | 102.83 | 44.74 |
| H7CDD1234678-678 | 64.32 | 13.89 | 90.18 | 37.77 | 79.97 | 23.30 | 104.23 | 44.82 |
| OCDD12346789 | 69.04 | 16.93 | 93.03 | 39.02 | 80.56 | 23.31 | 104.23 | 40.82 |
| OPLS | Ecomplex (1) | EvdWcomplex (1) | Eperturbed (1) | EvdWperturbed (1) | Ecomplex (2) | EvdWcomplex (2) | Eperturbed (2) | EvdWperturbed (2) |
|---|---|---|---|---|---|---|---|---|
| TCDD2378 | 52.72 | −41.72 | 85.95 | −8.49 | 77.97 | −32.22 | 100.39 | −9.80 |
| PCDD12378-123 | 58.95 | −37.93 | 87.18 | −9.69 | 76.95 | −33.64 | 100.46 | −10.13 |
| PCDD12378-78 | 57.34 | −38.31 | 86.87 | −8.78 | 74.63 | −37.65 | 103.77 | −8.51 |
| H6CDD123478-1234 | 59.19 | −37.29 | 87.20 | −9.39 | 74.43 | −35.77 | 103.38 | −6.82 |
| H6CDD123478-78 | 56.33 | −39.99 | 86.96 | −9.39 | 76.36 | −36.89 | 107.98 | −5.27 |
| H6CDD123678 | 53.50 | −43.28 | 87.62 | −9.15 | 73.72 | −36.66 | 101.55 | −8.83 |
| H6CDD123789 | 52.50 | −43.02 | 86.38 | −9.15 | 74.85 | −34.63 | 105.06 | −4.42 |
| H7CDD1234678-1234 | 56.78 | −41.52 | 88.17 | −10.13 | 75.83 | −37.60 | 105.20 | −8.23 |
| H7CDD1234678-678 | 52.71 | −44.75 | 87.87 | −9.59 | 73.28 | −37.60 | 101.88 | −9.01 |
| OCDD12346789 | 56.96 | −42.90 | 89.10 | −10.77 | 72.39 | −37.64 | 105.19 | −4.84 |
| OPLS | Ecomplex (1) | EvdWcomplex (1) | Eperturbed (1) | EvdWperturbed (1) | Ecomplex (2) | EvdWcomplex (2) | Eperturbed (2) | EvdWperturbed (2) |
|---|---|---|---|---|---|---|---|---|
| TCDD2378 | 56.07 | −39.07 | 86.67 | −8.47 | 81.46 | −27.05 | 100.29 | −8.22 |
| PCDD12378-123 | 57.97 | −38.71 | 87.36 | −9.33 | 80.82 | −30.40 | 101.85 | −9.37 |
| PCDD12378-78 | 52.58 | −40.34 | 84.66 | −8.26 | 74.70 | −32.03 | 102.61 | −4.11 |
| H6CDD123478-1234 | 59.18 | −11.31 | 84.77 | −13.66 | 80.36 | −29.02 | 104.26 | −5.12 |
| H6CDD123478-78 | 56.33 | −39.99 | 85.66 | −9.17 | 55.41 | −39.42 | 85.66 | −9.17 |
| H6CDD123678 | 60.61 | −33.65 | 85.03 | −9.22 | 76.10 | −33.67 | 104.20 | −5.57 |
| H6CDD123789 | 57.38 | −38.27 | 86.25 | −9.41 | 82.23 | −27.11 | 104.68 | −4.66 |
| H7CDD1234678-1234 | 61.63 | −32.62 | 85.05 | −9.19 | 78.77 | −30.61 | 100.61 | −8.77 |
| H7CDD1234678-678 | 58.81 | −37.93 | 86.96 | −9.77 | 82.36 | −27.65 | 105.04 | −4.97 |
| OCDD12346789 | 61.42 | −33.35 | 85.17 | −9.60 | 78.81 | −34.37 | 96.69 | −16.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghetu, M.-C.; Virgolici, M.; Tirsoaga, A.; Stanculescu, I. Molecular Modelling of Polychlorinated Dibenzo-p-Dioxins Non-Covalent Interactions with β and γ-Cyclodextrins. Int. J. Mol. Sci. 2023, 24, 13214. https://doi.org/10.3390/ijms241713214
Ghetu M-C, Virgolici M, Tirsoaga A, Stanculescu I. Molecular Modelling of Polychlorinated Dibenzo-p-Dioxins Non-Covalent Interactions with β and γ-Cyclodextrins. International Journal of Molecular Sciences. 2023; 24(17):13214. https://doi.org/10.3390/ijms241713214
Chicago/Turabian StyleGhetu, Maria-Cristina, Marian Virgolici, Alina Tirsoaga, and Ioana Stanculescu. 2023. "Molecular Modelling of Polychlorinated Dibenzo-p-Dioxins Non-Covalent Interactions with β and γ-Cyclodextrins" International Journal of Molecular Sciences 24, no. 17: 13214. https://doi.org/10.3390/ijms241713214