
Altered Expression of Autophagy Biomarkers in Hippocampal Neurons in a Multiple Sclerosis Animal Model
 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. The Beclin-1, LC3 II, and p62 Expression Levels Decreased in the Hippocampus of EAE Mice
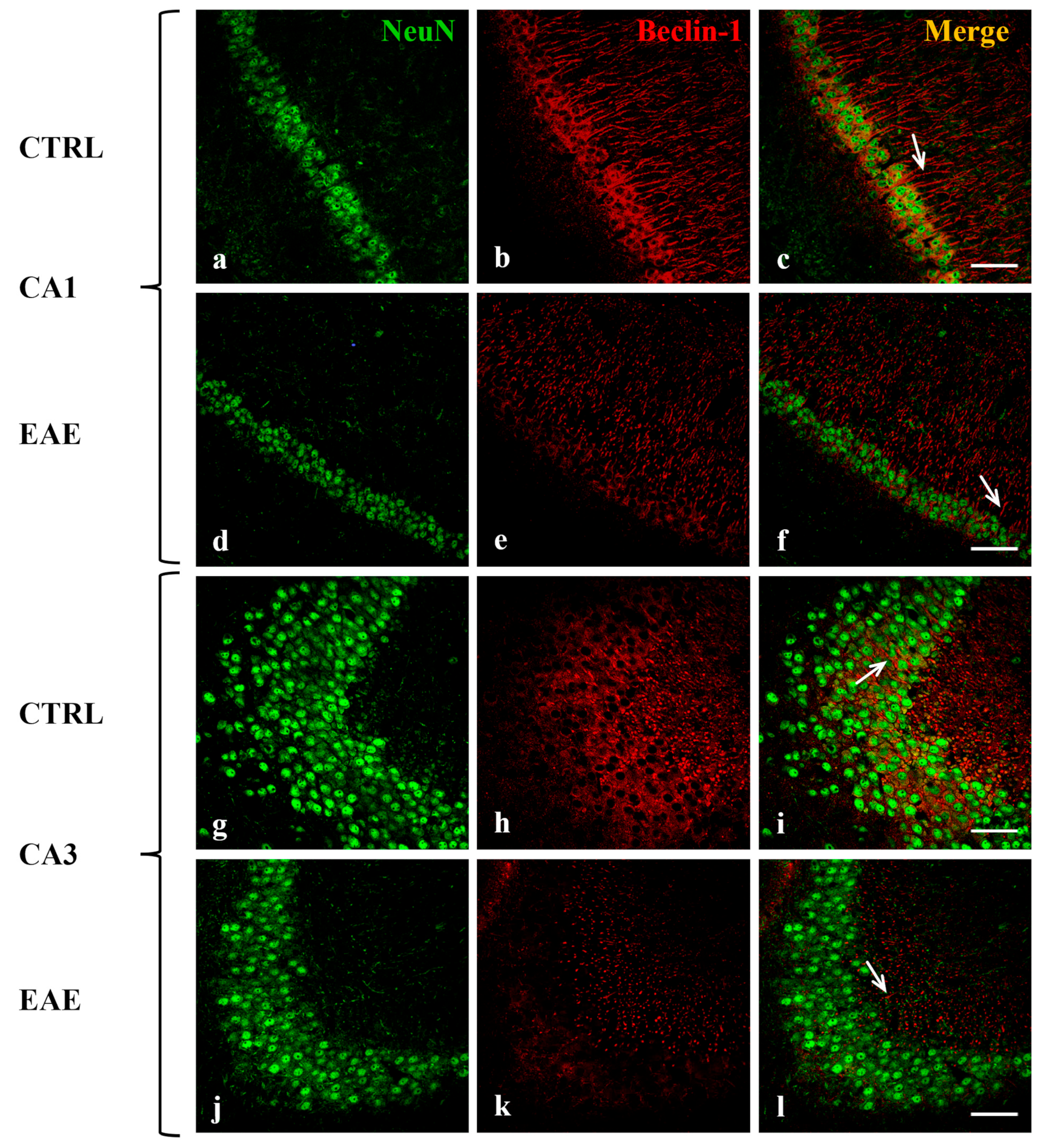
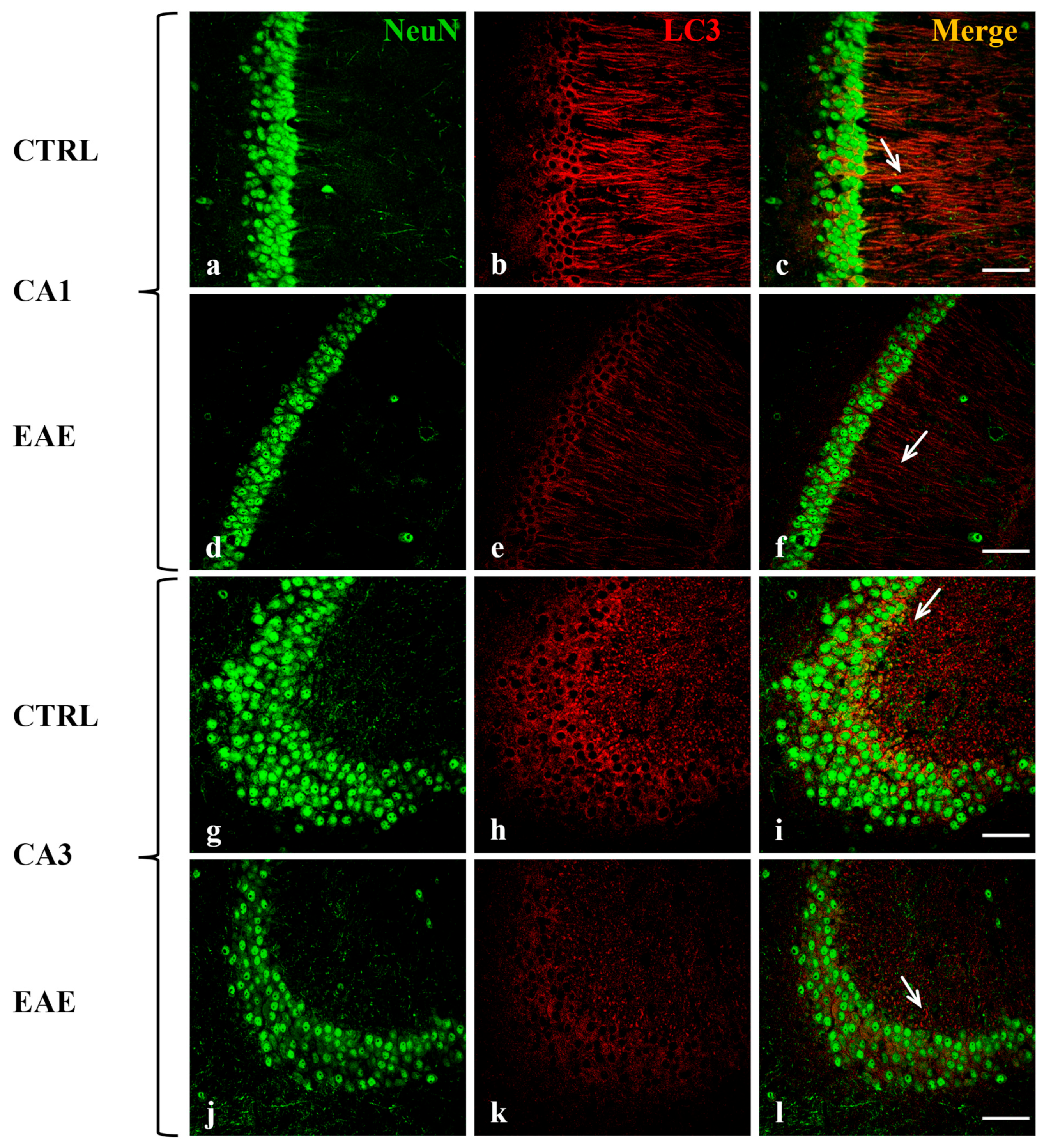
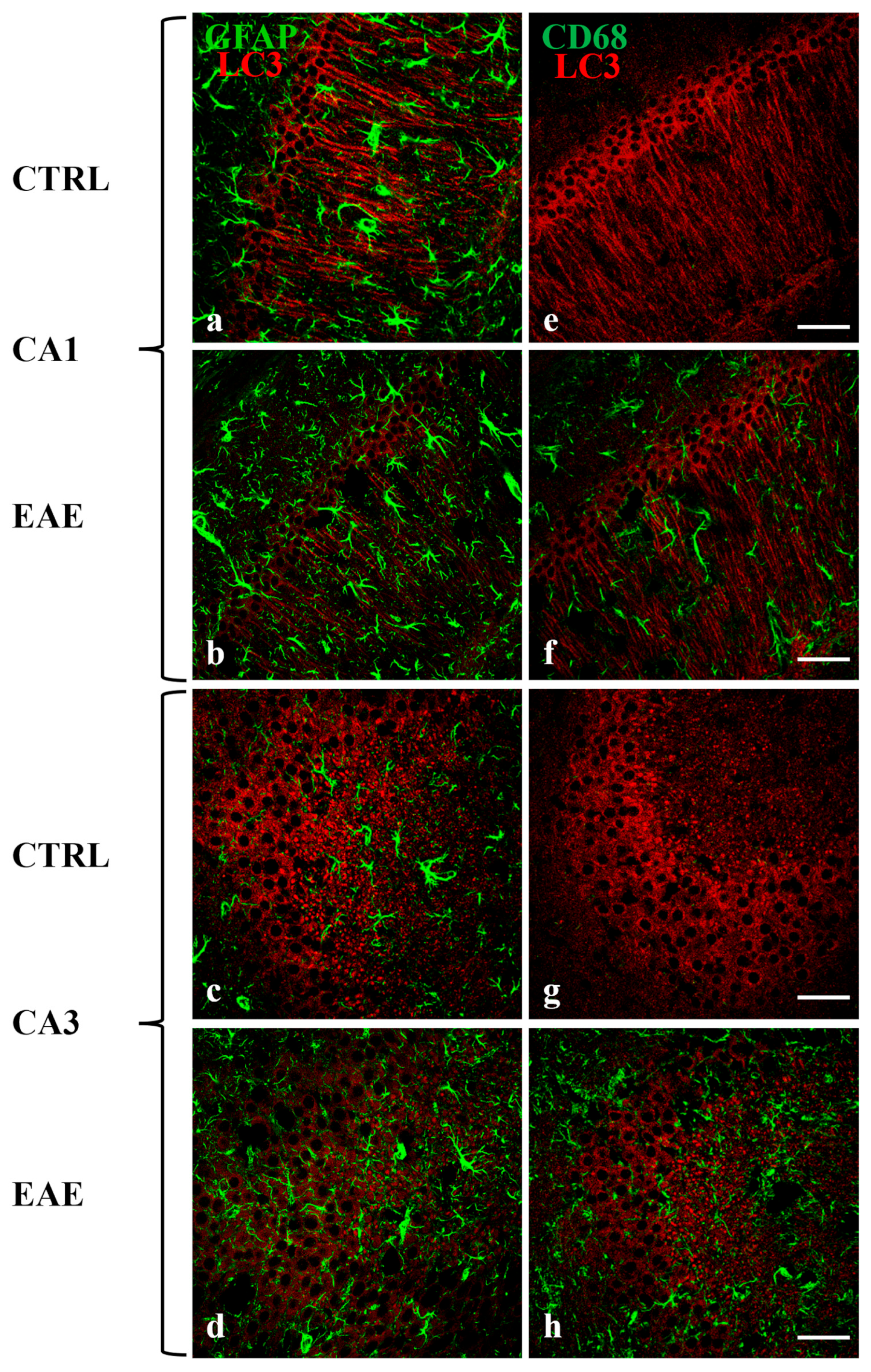
2.2. Beclin-1, LC3, and p62 Were Localized in Hippocampal Neurons of Control and EAE Mice
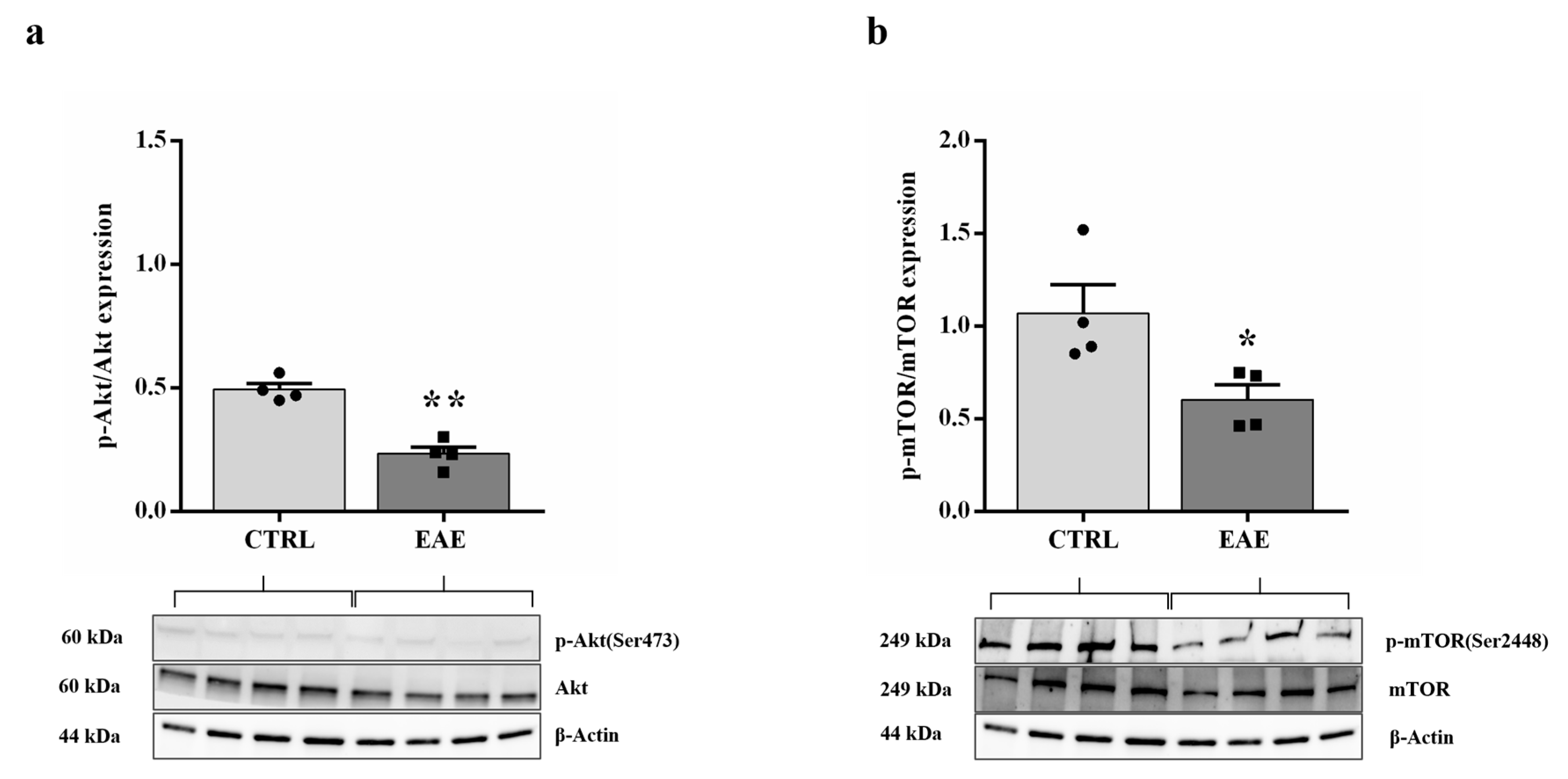
2.3. Akt/mTOR Pathway Is Not Activated in the Hippocampus of the EAE Mice
3. Discussion
4. Materials and Methods
4.1. EAE Induction and Monitoring
4.2. Ethical Considerations
4.3. Western Blot Experiments
4.4. Immunofluorescence Cellular Localization of Autophagic Markers
4.5. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lassmann, H.; Brück, W.; Lucchinetti, C.F. The immunopathology of multiple sclerosis: An overview. Brain Pathol. 2007, 17, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solari, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. Dis. Primers 2018, 4, 43. [Google Scholar] [CrossRef] [PubMed]
- Voskuhl, R.R.; MacKenzie-Graham, A. Chronic experimental autoimmune encephalomyelitis is an excellent model to study neuroaxonal degeneration in multiple sclerosis. Front. Mol. Neurosci. 2022, 15, 1024058. [Google Scholar] [CrossRef]
- Gangitano, C.; Falasca, C.; Del Fà, A.; Corvino, V.; Ceccariglia, S.; Zelano, G.; Geloso, M.; Monego, G.; Michetti, F. Hippocampal calretinin-containing neurons cultured in vitro are resistant to trimethyltin-induced neurodegeneration. Calcium Bind. Proteins 2006, 1, 120–124. [Google Scholar]
- Piacentini, R.; Gangitano, C.; Ceccariglia, S.; Del Fà, A.; Azzena, G.B.; Michetti, F.; Grassi, C. Dysregulation of intracellular calcium homeostasis is responsible for neuronal death in an experimental model of selective hippocampal degeneration induced by trimethyltin. J. Neurochem. 2008, 105, 2109–2121. [Google Scholar] [CrossRef]
- Ceccariglia, S.; D’Altocolle, A.; Del Fa’, A.; Pizzolante, F.; Caccia, E.; Michetti, F.; Gangitano, C. Cathepsin D plays a crucial role in the trimethyltin-induced hippocampal neurodegeneration process. Neuroscience 2011, 174, 160–170. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, T.; Wulff, P. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience 2015, 309, 1–16. [Google Scholar] [CrossRef]
- Ceccariglia, S.; Alvino, A.; Del Fà, A.; Parolini, O.; Michetti, F.; Gangitano, C. Autophagy is activated in vivo during trimethyltin-induced apoptotic neurodegeneration: A study in the rat hippocampus. Int. J. Mol. Sci. 2020, 21, 175. [Google Scholar] [CrossRef]
- Rocca, M.A.; Barkhof, F.; De Luca, J.; Frisén, J.; Geurts, J.J.G.; Hulst, H.E.; Sastre-Garriga, J.; Filippi, M.; Ciccarelli, O.; De Stefano, N.; et al. The hippocampus in multiple sclerosis. Lancet Neurol. 2018, 17, 918–926. [Google Scholar] [CrossRef]
- Ziehn, M.O.; Avedisian, A.A.; Tiwari-Woodruff, S.; Voskuhl, R.R. Hippocampal CA1 atrophy and synaptic loss during experimental autoimmune encephalomyelitis, EAE. Lab. Investig. 2010, 90, 774–786. [Google Scholar] [CrossRef]
- Ziehn, M.O.; Avedisian, A.A.; Dervin, S.M.; O’Dell, T.J.; Voskuhl, R.R. Estriol preserves synaptic transmission in the hippocampus during autoimmune demyelinating disease. Lab. Investig. 2012, 92, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, A.M.; Forkert, N.D.; Yang, R.; Wu, Y.; Rogers, J.A.; Yong, V.W.; Dunn, J.F. Central nervous system targeted autoimmunity causes regional atrophy: A 9.4T MRI study of the EAE mouse model of Multiple Sclerosis. Sci. Rep. 2019, 9, 8488. [Google Scholar] [CrossRef]
- Hou, B.; Zhang, Y.; Liang, P.; He, Y.; Peng, B.; Liu, W.; Han, S.; Yin, J.; He, X. Inhibition of the NLRP3-inflammasome prevents cognitive deficits in experimental autoimmune encephalomyelitis mice via the alteration of astrocyte phenotype. Cell Death Dis. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, A.; Di Sante, G.; Rivignani Vaccari, L.; Tredicine, M.; Ria, F.; Bonvissuto, D.; Corvino, V.; Sette, C.; Geloso, M.C. Regionally restricted modulation of Sam68 expression and Arhgef9 alternative splicing in the hippocampus of a murine model of multiple sclerosis. Front. Mol. Neurosci. 2023, 15, 1073627. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef]
- Guo, F.; Liu, X.; Cai, H.; Le, W. Autophagy in neurodegenerative diseases: Pathogenesis and therapy. Brain Pathol. 2018, 28, 3–13. [Google Scholar] [CrossRef]
- DeVorkin, L.; Pavey, N.; Carleton, G.; Comber, A.; Ho, C.; Lim, J.; McNamara, E.; Huang, H.; Kim, P.; Zacharias, L.G.; et al. Autophagy Regulation of Metabolism Is Required for CD8+ T Cell Anti-tumor Immunity. Cell Rep. 2019, 27, 502–513. [Google Scholar] [CrossRef]
- Towers, C.G.; Wodetzki, D.; Thorburn, A. Autophagy and cancer: Modulation of cell death pathways and cancer cell adaptations. J. Cell Biol. 2020, 219, e201909033. [Google Scholar] [CrossRef]
- Lu, Q.; Yokoyama, C.C.; Williams, J.W.; Baldridge, M.T.; Jin, X.; Desrochers, B.; Bricker, T.; Wilen, C.B.; Bagaitkar, J.; Loginicheva, E.; et al. Homeostatic Control of Innate Lung Inflammation by Vici Syndrome Gene Epg5 and Additional Autophagy Genes Promotes Influenza Pathogenesis. Cell Host Microbe 2016, 19, 102–113. [Google Scholar] [CrossRef]
- Park, S.; Buck, M.D.; Desai, C.; Zhang, X.; Loginicheva, E.; Martinez, J.; Freeman, M.L.; Saitoh, T.; Akira, S.; Guan, J.L.; et al. Autophagy Genes Enhance Murine Gammaherpesvirus 68 Reactivation from Latency by Preventing Virus-Induced Systemic Inflammation. Cell Host Microbe 2016, 19, 91–101. [Google Scholar] [CrossRef]
- Gassen, N.C.; Papies, J.; Bajaj, T.; Emanuel, J.; Dethloff, F.; Chua, R.L.; Trimpert, J.; Heinemann, N.; Niemeyer, C.; Weege, F.; et al. SARS-CoV-2-mediated dysregulation of metabolism and autophagy uncovers host-targeting antivirals. Nat. Commun. 2021, 12, 3818. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Chen, S.; Du, F.; Li, S.; Zhao, L.; Wang, X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proc. Natl. Acad. Sci. USA 2011, 108, 4788–4793. [Google Scholar] [CrossRef] [PubMed]
- Ceccariglia, S.; Cargnoni, A.; Silini, A.R.; Parolini, O. Autophagy: A potential key contributor to the therapeutic action of mesenchymal stem cells. Autophagy 2020, 16, 28–37. [Google Scholar] [CrossRef]
- Yang, G.; Van Kaer, L. Therapeutic Targeting of Immune Cell Autophagy in Multiple Sclerosis: Russian Roulette or Silver Bullet? Front. Immunol. 2021, 12, 724108. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Liu, K.; Wang, H.; Wang, H. Autophagy modulation in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2022, 209, 140–150. [Google Scholar] [CrossRef]
- Bankston, A.N.; Forston, M.D.; Howard, R.M.; Andres, K.R.; Smith, A.E.; Ohri, S.S.; Bates, M.L.; Bunge, M.B.; Whittemore, S.R. Autophagy is essential for oligodendrocyte differentiation, survival, and proper myelination. Glia 2019, 67, 1745–1759. [Google Scholar] [CrossRef]
- Smith, C.M.; Mayer, J.A.; Duncan, I.D. Autophagy promotes oligodendrocyte survival and function following dysmyelination in a long-lived myelin mutant. J. Neurosci. 2013, 33, 8088–8100. [Google Scholar] [CrossRef]
- Alirezaei, M.; Fox, H.S.; Flynn, C.T.; Moore, C.S.; Hebb, A.L.O.; Frausto, R.F.; Bhan, V.; Kiosses, W.B.; Whitton, J.L.; Robertson, G.S.; et al. Elevated ATG5 expression in autoimmune demyelination and multiple sclerosis. Autophagy 2009, 5, 152–158. [Google Scholar] [CrossRef]
- Yang, Z.; Goronzy, J.J.; Weyand, C.M. Autophagy in Autoimmune Disease. J. Mol. Med. 2015, 93, 707–717. [Google Scholar] [CrossRef]
- Feng, X.; Hou, H.; Zou, Y.; Guo, L. Defective autophagy is associated with neuronal injury in a mouse model of multiple sclerosis. Bosn. J. Basic Med. Sci. 2017, 17, 95–103. [Google Scholar] [CrossRef]
- Yuan, B.; Sun, M.; Bao, C.; Li, X.; Liu, Z.; Wang, M. Dynamic expression of autophagy-related factors in autoimmune encephalomyelitis and exploration of curcumin therapy. J. Neuroimmunol. 2019, 337, 577067. [Google Scholar]
- Yu, X.; Long, Y.C.; Shen, H.M. Differential regulatory functions of three classes of phosphatidylinositol and phosphoinositide 3-kinases in autophagy. Autophagy 2015, 11, 1711–1728. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologuen of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yamamoto, A.; Matsui, M.; Yoshimori, T.; Yoshinori, O. In Vivo Analysis of Autophagy in Response to Nutrient Starvation Using Transgenic Mice Expressing a Fluorescent Autophagosome Marker. Mol. Biol. Cell 2004, 15, 1101–1111. [Google Scholar] [CrossRef]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Chapter 12 Monitoring Autophagic Degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar]
- Xu, F.; Na, L.; Li, Y.; Chen, L. Retraction Note to: Roles of the PI3K/AKT/mTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci. 2021, 11, 13578. [Google Scholar] [CrossRef]
- Bockaert, J.; Marin, P. mTOR in brain physiology and pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Chiang, G.G.; Abraham, R.T. Phosphorylation of mammalian target of rapamycin (mTOR) at Ser-2448 is mediated by p70S6 kinase. J. Biol. Chem. 2005, 280, 25485–25490. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.L.B. Cognitive deficits in multiple sclerosis: A systematic review. Arq. De Neuro-Psiquiatr. 2010, 68, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Acharjee, S.; Nayani, N.; Tsutsui, M.; Hill, M.N.; Ousman, S.S.; Pittman, Q.J. Altered cognitive-emotional behavior in early experimental autoimmune encephalitis—Cytokine and hormonal correlates. Brain Behav. Immun. 2013, 33, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.J.; Ren, Q.G.; Xu, L.; Zhang, Z.J. LINGO-1 antibody ameliorates myelin impairment and spatial memory deficits in experimental autoimmune encephalomyelitis mice. Sci. Rep. 2015, 5, 14235. [Google Scholar] [CrossRef]
- Aharoni, R.; Schottlender, N.; Bar-Lev, D.D.; Eilam, R.; Sela, M.; Tsoory, M.; Arnon, R. Cognitive impairment in an animal model of multiple sclerosis and its amelioration by glatiramer acetate. Sci. Rep. 2019, 9, 4140. [Google Scholar] [CrossRef]
- Marchese, E.; Valentini, M.; Di Sante, G.; Cesari, E.; Adinolfi, A.; Corvino, V.; Ria, F.; Sette, C.; Geloso, M.C. Alternative splicing of neurexins 1–3 is modulated by neuroinflammation in the prefrontal cortex of a murine model of multiple sclerosis. Exp. Neurol. 2021, 335, 113497. [Google Scholar] [CrossRef]
- Glatigny, M.; Moriceau, S.; Rivagorda, M.; Ramos-Brossier, M.; Nascimbeni, A.C.; Lante, F.; Shanley, M.R.; Boudarene, N.; Rousseaud, A.; Friedman, A.K.; et al. Autophagy Is Required for Memory Formation and Reverses Age-Related Memory Decline. Curr. Biol. 2019, 29, 435–448.e8. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Katsuragi, Y.; Ichimura, Y.; Komatsu, M. P62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015, 282, 4672–4678. [Google Scholar] [CrossRef]
- Larsen, K.B.; Lamark, T.; Øvervatn, A.; Harneshaug, I.; Johansen, T.; Bjørkøy, G. A reporter cell system to monitor autophagy based on p62/SQSTM1. Autophagy 2010, 6, 784–793. [Google Scholar] [CrossRef]
- Kamarehei, M.; Kabudanian Ardestani, S.; Firouzi, M.; Zahednasab, H.; Keyvani, H.; Harirchian, M.H. Increased expression of endoplasmic reticulum stress-related caspase-12 and CHOP in the hippocampus of EAE mice. Brain Res. Bull. 2019, 147, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Rashid, H.O.; Yadav, R.K.; Kim, H.R.; Chae, H.J. ER stress: Autophagy induction, inhibition and selection. Autophagy 2015, 11, 1956–1977. [Google Scholar] [CrossRef] [PubMed]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes initiate distally and mature during transport toward the cell soma in primary neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Stavoe, A.K.H.; Holzbaur, E.L.F. Axonal autophagy: Mini-review for autophagy in the CNS. Neurosci. Lett. 2019, 697, 17–23. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Jung, C.H.; Ro, S.H.; Cao, J.; Otto, N.M.; Kim, D.H. MTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef]
- Perluigi, M.; Di Domenico, F.; Butterfield, D.A. mTOR signaling in aging and neurodegeneration: At the crossroad between metabolism dysfunction and impairment of autophagy. Neurobiol. Dis. 2015, 84, 39–49. [Google Scholar] [CrossRef]
- Vartak, R.S.; Rodin, A.; Oddo, S. Differential activation of the mTOR/autophagy pathway predicts cognitive performance in APP/PS1 mice. Neurobiol. Aging 2019, 83, 105–113. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhou, Y.; Ma, H.; Cao, X.; Li, Z.; Chen, F.; Wang, H. Autophagy Dysfunction and mTOR Hyperactivation Is Involved in Surgery: Induced Behavioral Deficits in Aged C57BL/6J Mice. Neurochem. Res. 2020, 45, 331–344. [Google Scholar] [CrossRef]
- Wang, J.; Song, X.; Tan, G.; Sun, P.; Guo, L.; Zhang, N.; Wang, J.; Li, B. NAD+ improved experimental autoimmune encephalomyelitis by regulating SIRT1 to inhibit PI3K/Akt/mTOR signaling pathway. Aging 2021, 13, 25931–25943. [Google Scholar] [CrossRef]
- Nutma, E.; Marzin, M.C.; Cillessen, S.A.; Amor, S. Autophagy in white matter disorders of the CNS: Mechanisms and therapeutic opportunities. J. Pathol. 2021, 253, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Nicolò, C.; Di Sante, G.; Orsini, M.; Rolla, S.; Columba-Cabezas, S.; Spica, V.R.; Ricciardi, G.; Chan, B.M.C.; Ria, F. Mycobacterium tuberculosis in the adjuvant modulates the balance of Th immune response to self-antigen of the CNS without influencing a “core” repertoire of specific T cells. Int. Immunol. 2006, 18, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Nicolò, C.; Sali, M.; Di Sante, G.; Geloso, M.C.; Signori, E.; Penitente, R.; Uniyal, S.; Rinaldi, M.; Ingrosso, L.; Fazio, V.M.; et al. Mycobacterium smegmatis Expressing a Chimeric Protein MPT64-Proteolipid Protein (PLP) 139–151 Reorganizes the PLP-Specific T Cell Repertoire Favoring a CD8-Mediated Response and Induces a Relapsing Experimental Autoimmune Encephalomyelitis. J. Immunol. 2010, 184, 222–235. [Google Scholar] [CrossRef]
- Camponeschi, C.; De Carluccio, M.; Amadio, S.; Clementi, M.E.; Sampaolese, B.; Volonté, C.; Tredicine, M.; Spica, V.R.; Di Liddo, R.; Ria, F.; et al. S100B protein as a therapeutic target in multiple sclerosis: The S100B inhibitor arundic acid protects from chronic experimental autoimmune encephalomyelitis. Int. J. Mol. Sci. 2021, 22, 13558. [Google Scholar] [CrossRef] [PubMed]
- Pandolfi, F.; Cianci, R.; Casciano, F.; Pagliari, D.; De Pasquale, T.; Landolfi, R.; Di Sante, G.; Kurnick, J.T.; Ria, F. Skewed T-cell receptor repertoire: More than a marker of malignancy, a tool to dissect the immunopathology of inflammatory diseases. J. Biol. Regul. Homeost. Agents 2011, 25, 153–161. [Google Scholar]
- Nicolò, C.; Di Sante, G.; Procoli, A.; Migliara, G.; Piermattei, A.; Valentini, M.; Delogu, G.; Cittadini, A.; Constantin, G.; Ria, F. M tuberculosis in the Adjuvant Modulates Time of Appearance of CNS-Specific Effector T Cells in the Spleen through a Polymorphic Site of TLR2. PLoS ONE 2013, 8, e55819. [Google Scholar] [CrossRef] [PubMed]
- Piermattei, A.; Migliara, G.; Di Sante, G.; Foti, M.; Hayrabedyan, S.B.; Papagna, A.; Geloso, M.C.; Corbi, M.; Valentini, M.; Sgambato, A.; et al. Toll-like receptor 2 mediates in vivo pro- and anti-inflammatory effects of Mycobacterium Tuberculosis and modulates autoimmune encephalomyelitis. Front. Immunol. 2016, 7, 191. [Google Scholar] [CrossRef]
- Di Sante, G.; Amadio, S.; Sampaolese, B.; Clementi, M.E.; Valentini, M.; Volonté, C.; Casalbore, P.; Ria, F.; Michetti, F. The S100B inhibitor pentamidine ameliorates clinical score and neuropathology of relapsing—Remitting multiple sclerosis mouse model. Cells 2020, 9, 748. [Google Scholar] [CrossRef]
- Cortés-Ríos, J.; Zárate, A.M.; Figueroa, J.D.; Medina, J.; Fuentes-Lemus, E.; Rodríguez-Fernández, M.; Aliaga, M.; López-Alarcón, C. Protein quantification by bicinchoninic acid (BCA) assay follows complex kinetics and can be performed at short incubation times. Anal. Biochem. 2020, 608, 113904. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccariglia, S.; Sibilia, D.; Parolini, O.; Michetti, F.; Di Sante, G. Altered Expression of Autophagy Biomarkers in Hippocampal Neurons in a Multiple Sclerosis Animal Model. Int. J. Mol. Sci. 2023, 24, 13225. https://doi.org/10.3390/ijms241713225
Ceccariglia S, Sibilia D, Parolini O, Michetti F, Di Sante G. Altered Expression of Autophagy Biomarkers in Hippocampal Neurons in a Multiple Sclerosis Animal Model. International Journal of Molecular Sciences. 2023; 24(17):13225. https://doi.org/10.3390/ijms241713225
Chicago/Turabian StyleCeccariglia, Sabrina, Diego Sibilia, Ornella Parolini, Fabrizio Michetti, and Gabriele Di Sante. 2023. "Altered Expression of Autophagy Biomarkers in Hippocampal Neurons in a Multiple Sclerosis Animal Model" International Journal of Molecular Sciences 24, no. 17: 13225. https://doi.org/10.3390/ijms241713225