
Protein Disulfide Isomerase A3 (PDIA3): A Pharmacological Target in Glioblastoma?
 , , , , and
, , , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
2.1. Inhibition and Knockdown of PDIA3 Decrease Cell Viability and Cell Proliferation in the T98G and U-87 MG Cell Lines
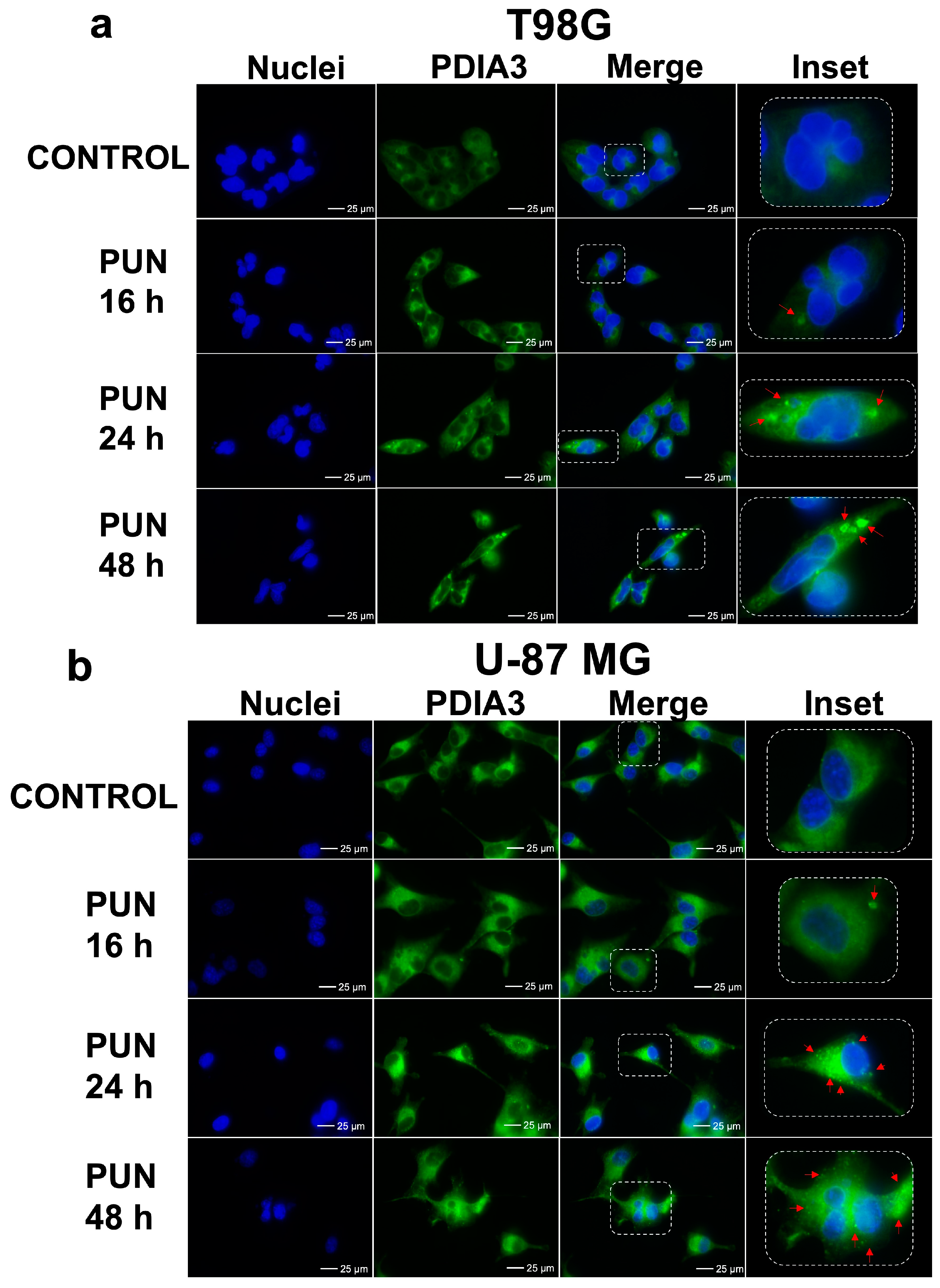
2.2. PDIA3 Inhibition Leads to PDIA3 Aggregation and Cell Cycle Arrest
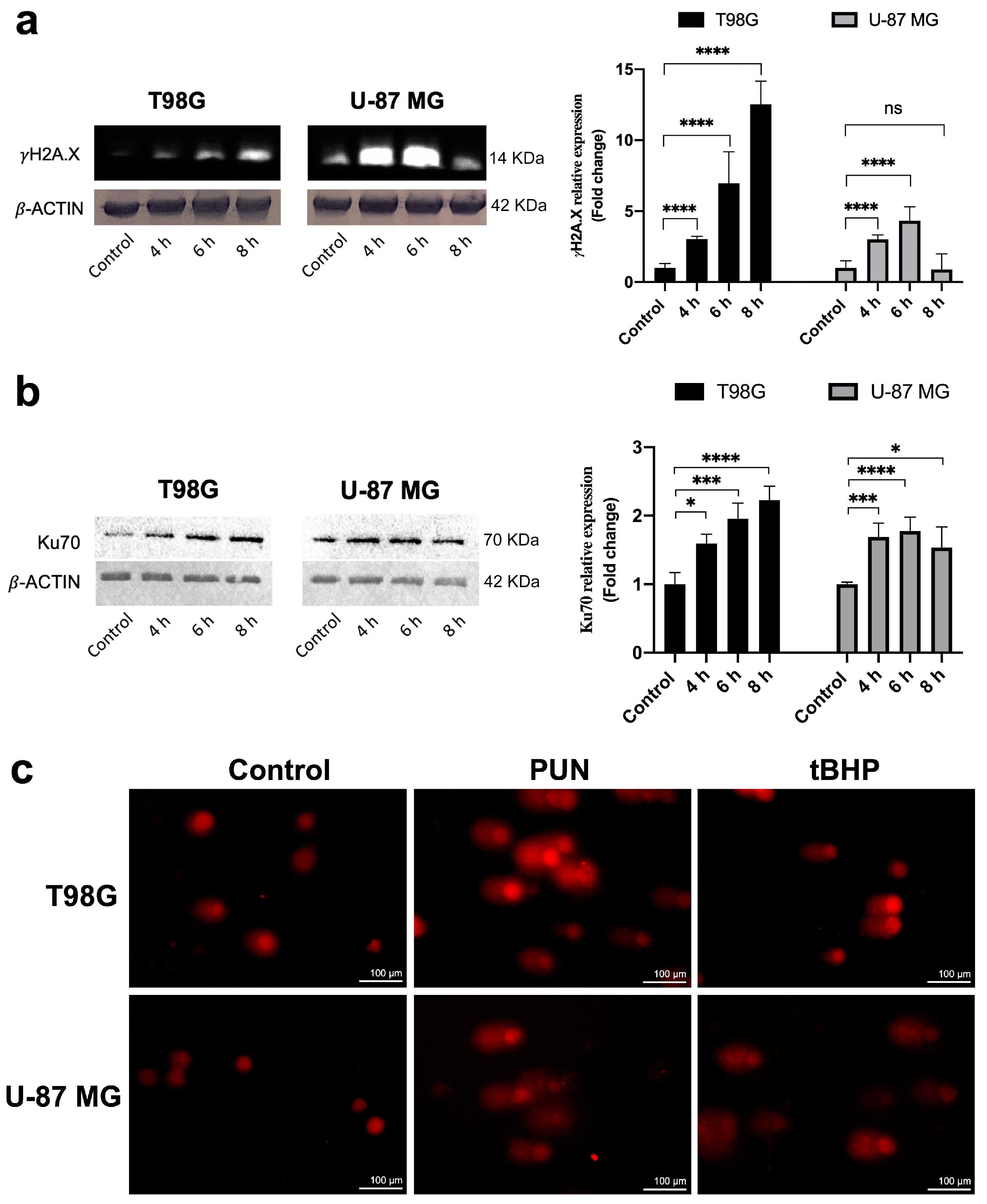
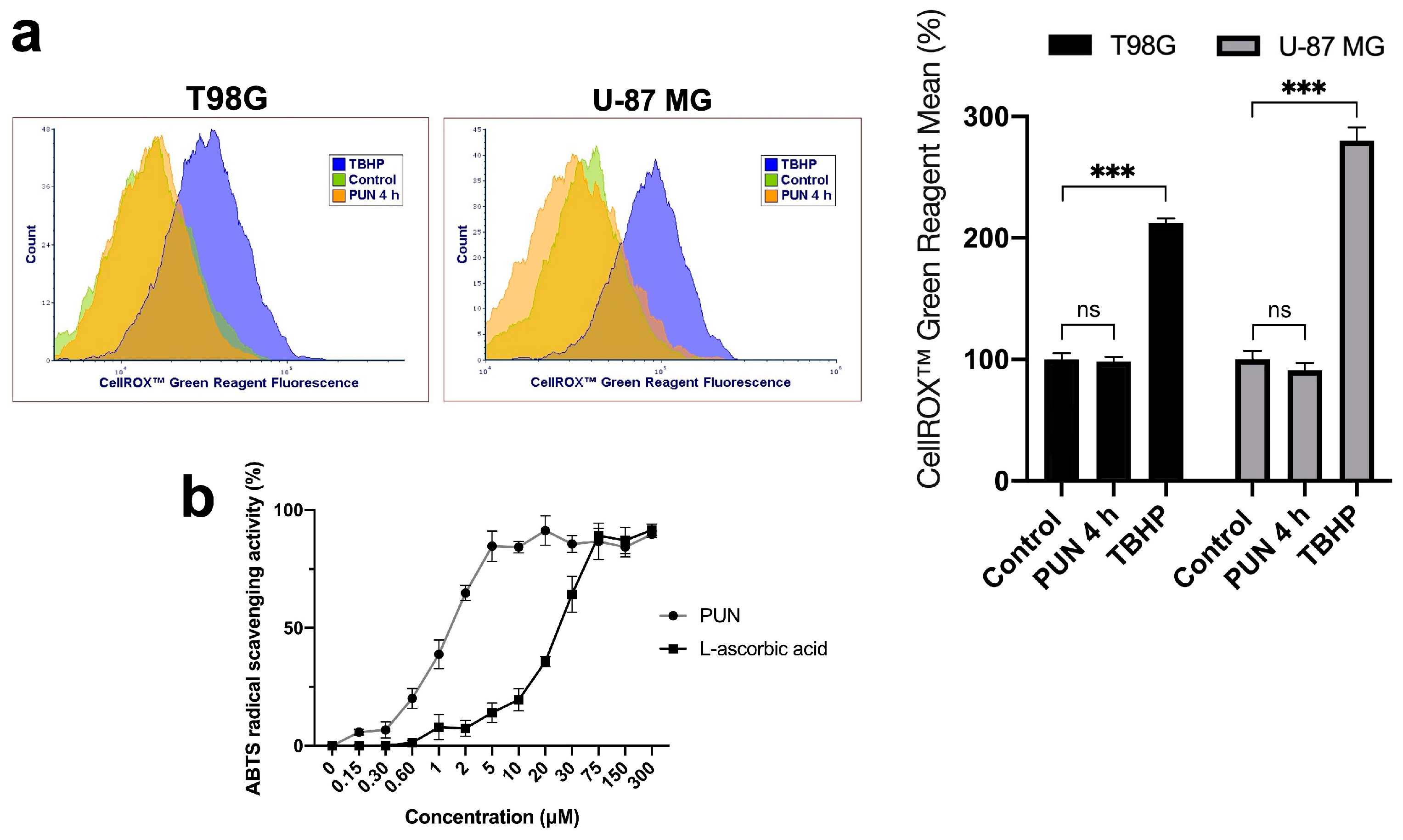
2.3. PDIA3 Inhibition Causes Activation of γ-H2AX and NHEJ Repair Pathway in a ROS-Independent Manner
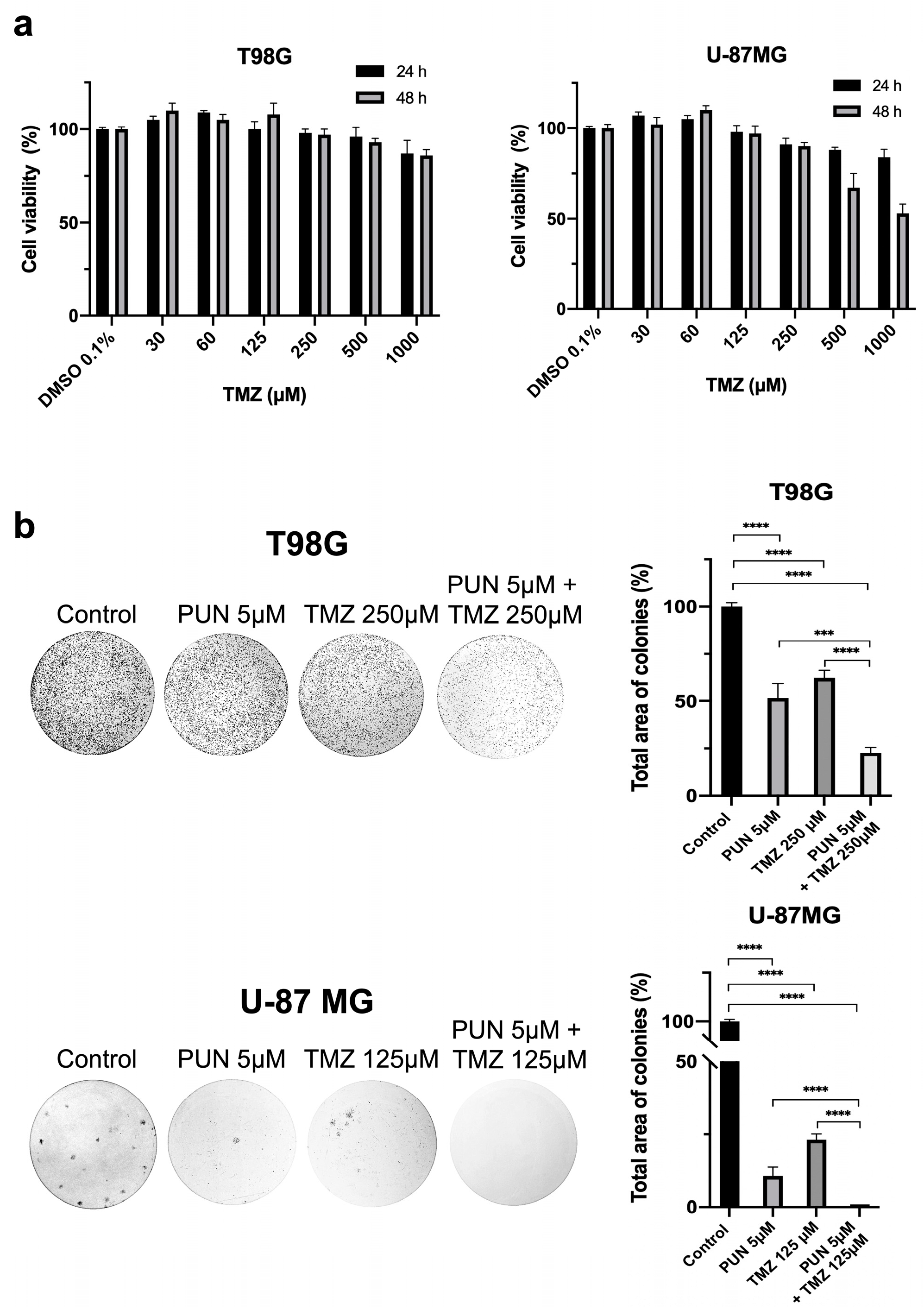
2.4. PDIA3 Inhibition as an Adjuvant for Temozolomide Chemotherapy Treatment in Glioblastoma
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Viability Assay
4.3. PDIA3 Knockdown Procedure
4.4. Cell Imaging
4.5. Cell Counting
4.6. Protein Extraction and Immunoblotting Analysis
4.7. Cell Cycle Analysis
4.8. Clonogenic Potential Assay
4.9. Immunofluorescence
4.10. Comet Assay
4.11. CellROXTM Green Flow Cytometry Assay
4.12. ABTS Radical Scavenging Assay of Punicalagin
4.13. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chichiarelli, S.; Altieri, F.; Paglia, G.; Rubini, E.; Minacori, M.; Eufemi, M. ERp57/PDIA3: New Insight. Cell Mol. Biol. Lett. 2022, 27, 12. [Google Scholar] [CrossRef]
- Mahmood, F.; Xu, R.; Awan, M.U.N.; Song, Y.; Han, Q.; Xia, X.; Zhang, J. PDIA3: Structure, Functions and Its Potential Role in Viral Infections. Biomed. Pharmacother. 2021, 143, 112110. [Google Scholar] [CrossRef] [PubMed]
- Hettinghouse, A.; Liu, R.; Liu, C. Multifunctional Molecule ERp57: From Cancer to Neurodegenerative Diseases. Pharmacol. Ther. 2018, 181, 34–48. [Google Scholar] [CrossRef]
- Essex, D.W.; Wu, Y. Multiple Protein Disulfide Isomerases Support Thrombosis. Curr. Opin. Hematol. 2018, 25, 395–402. [Google Scholar] [CrossRef]
- Song, D.; Liu, H.; Wu, J.; Gao, X.; Hao, J.; Fan, D. Insights into the Role of ERp57 in Cancer. J. Cancer 2021, 12, 2456–2464. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, K.; Hainisayimu, T.; Li, H. Pan-Cancer Analysis of PDIA3: Identifying It as a Potential Biomarker for Tumor Prognosis and Immunotherapy. Oxid. Med. Cell Longev. 2022, 2022, e9614819. [Google Scholar] [CrossRef]
- Tu, Z.; Ouyang, Q.; Long, X.; Wu, L.; Li, J.; Zhu, X.; Huang, K. Protein Disulfide-Isomerase A3 Is a Robust Prognostic Biomarker for Cancers and Predicts the Immunotherapy Response Effectively. Front. Immunol. 2022, 13, 837512. [Google Scholar] [CrossRef]
- Kaneya, Y.; Takata, H.; Wada, R.; Kure, S.; Ishino, K.; Kudo, M.; Kondo, R.; Taniai, N.; Ohashi, R.; Yoshida, H.; et al. Inhibitor for Protein Disulfide isomerase Family A Member 3 Enhances the Antiproliferative Effect of Inhibitor for Mechanistic Target of Rapamycin in Liver Cancer: An in Vitro Study on Combination Treatment with Everolimus and 16F16. Oncol. Lett. 2020, 21, 28. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Gittleman, H.; Liao, P.; Rouse, C.; Chen, Y.; Dowling, J.; Wolinsky, Y.; Kruchko, C.; Barnholtz-Sloan, J. CBTRUS Statistical Report: Primary Brain and Central Nervous System Tumors Diagnosed in the United States in 2007–2011. Neuro Oncol. 2014, 16 (Suppl. S4), iv1–iv63. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A Summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef]
- Sharma, P.; Aaroe, A.; Liang, J.; Puduvalli, V.K. Tumor Microenvironment in Glioblastoma: Current and Emerging Concepts. Neurooncol Adv. 2023, 5, vdad009. [Google Scholar] [CrossRef]
- Kyani, A.; Tamura, S.; Yang, S.; Shergalis, A.; Samanta, S.; Kuang, Y.; Ljungman, M.; Neamati, N. Discovery and Mechanistic Elucidation of a Class of Protein Disulfide Isomerase Inhibitors for the Treatment of Glioblastoma. ChemMedChem 2018, 13, 164–177. [Google Scholar] [CrossRef]
- Mouawad, R.; Neamati, N. Inhibition of Protein Disulfide Isomerase (PDIA1) Leads to Proteasome-Mediated Degradation of Ubiquitin-like PHD and RING Finger Domain-Containing Protein 1 (UHRF1) and Increased Sensitivity of Glioblastoma Cells to Topoisomerase II Inhibitors. ACS Pharmacol. Transl. Sci. 2023, 6, 100–114. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The Role of the Unfolded Protein Response in Cancer Progression: From Oncogenesis to Chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, W.; Shergalis, A.; Xu, J.; Delaney, A.M.; Calcaterra, A.; Pal, A.; Ljungman, M.; Neamati, N.; Rehemtulla, A. Activation of the Unfolded Protein Response via Inhibition of Protein Disulfide Isomerase Decreases the Capacity for DNA Repair to Sensitize Glioblastoma to Radiotherapy. Cancer Res. 2019, 79, 2923–2932. [Google Scholar] [CrossRef] [PubMed]
- Kiang, K.M.-Y.; Tang, W.; Song, Q.; Liu, J.; Li, N.; Lam, T.-L.; Shum, H.C.; Zhu, Z.; Leung, G.K.-K. Targeting Unfolded Protein Response Using Albumin-Encapsulated Nanoparticles Attenuates Temozolomide Resistance in Glioblastoma. Br. J. Cancer 2023, 128, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Sun, S.; Ho, A.S.W.; Kiang, K.M.Y.; Zhang, X.Q.; Xu, F.F.; Leung, G.K.K. Hyperoxia Resensitizes Chemoresistant Glioblastoma Cells to Temozolomide through Unfolded Protein Response. Anticancer. Res. 2014, 34, 2957–2966. [Google Scholar]
- Sun, S.; Lee, D.; Ho, A.S.W.; Pu, J.K.S.; Zhang, X.Q.; Lee, N.P.; Day, P.J.R.; Lui, W.M.; Fung, C.F.; Leung, G.K.K. Inhibition of Prolyl 4-Hydroxylase, Beta Polypeptide (P4HB) Attenuates Temozolomide Resistance in Malignant Glioma via the Endoplasmic Reticulum Stress Response (ERSR) Pathways. Neuro Oncol. 2013, 15, 562–577. [Google Scholar] [CrossRef]
- Salaroglio, I.C.; Panada, E.; Moiso, E.; Buondonno, I.; Provero, P.; Rubinstein, M.; Kopecka, J.; Riganti, C. PERK Induces Resistance to Cell Death Elicited by Endoplasmic Reticulum Stress and Chemotherapy. Mol. Cancer 2017, 16, 91. [Google Scholar] [CrossRef]
- Zou, H.; Wen, C.; Peng, Z.; Shao, Y.; Hu, L.; Li, S.; Li, C.; Zhou, H. P4HB and PDIA3 Are Associated with Tumor Progression and Therapeutic Outcome of Diffuse Gliomas. Oncol. Rep. 2017, 39, 501–510. [Google Scholar] [CrossRef]
- Chiavari, M.; Ciotti, G.M.P.; Canonico, F.; Altieri, F.; Lacal, P.M.; Graziani, G.; Navarra, P.; Lisi, L. PDIA3 Expression in Glioblastoma Modulates Macrophage/Microglia Pro-Tumor Activation. Int. J. Mol. Sci. 2020, 21, 8214. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, R.; Zhang, L.; Meng, X.; Sun, W. Primary glioblastoma transcriptome data analysis for screening survival-related genes. J. Cell Biochem. 2020, 121, 1901–1910. [Google Scholar] [CrossRef]
- Li, T.; Xu, J.; Liu, Y. A Novel Circular RNA CircRFX3 Serves as a Sponge for MicroRNA-587 in Promoting Glioblastoma Progression via Regulating PDIA3. Front. Cell Dev. Biol. 2021, 9, 757260. [Google Scholar] [CrossRef]
- Kochanowski, P.; Catapano, J.; Pudełek, M.; Wróbel, T.; Madeja, Z.; Ryszawy, D.; Czyż, J. Temozolomide Induces the Acquisition of Invasive Phenotype by O6-Methylguanine-DNA Methyltransferase (MGMT)+ Glioblastoma Cells in a Snail-1/Cx43-Dependent Manner. Int. J. Mol. Sci. 2021, 22, 4150. [Google Scholar] [CrossRef]
- Giamogante, F.; Marrocco, I.; Cervoni, L.; Eufemi, M.; Chichiarelli, S.; Altieri, F. Punicalagin, an Active Pomegranate Component, Is a New Inhibitor of PDIA3 Reductase Activity. Biochimie 2018, 147, 122–129. [Google Scholar] [CrossRef]
- Paglia, G.; Antonini, L.; Cervoni, L.; Ragno, R.; Sabatino, M.; Minacori, M.; Rubini, E.; Altieri, F. A Comparative Analysis of Punicalagin Interaction with PDIA1 and PDIA3 by Biochemical and Computational Approaches. Biomedicines 2021, 9, 1533. [Google Scholar] [CrossRef]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Bilches Medinas, D.; Malik, S.; Yıldız-Bölükbaşı, E.; Borgonovo, J.; Saaranen, M.J.; Urra, H.; Pulgar, E.; Afzal, M.; Contreras, D.; Wright, M.T.; et al. Mutation in Protein Disulfide Isomerase A3 Causes Neurodevelopmental Defects by Disturbing Endoplasmic Reticulum Proteostasis. EMBO J. 2022, 41, e105531. [Google Scholar] [CrossRef]
- Hussmann, M.; Janke, K.; Kranz, P.; Neumann, F.; Mersch, E.; Baumann, M.; Goepelt, K.; Brockmeier, U.; Metzen, E. Depletion of the Thiol Oxidoreductase ERp57 in Tumor Cells Inhibits Proliferation and Increases Sensitivity to Ionizing Radiation and Chemotherapeutics. Oncotarget 2015, 6, 39247–39261. [Google Scholar] [CrossRef]
- Shi, W.; Han, H.; Zou, J.; Zhang, Y.; Li, H.; Zhou, H.; Cui, G. Identification of Dihydrotanshinone I as an ERp57 Inhibitor with Anti-Breast Cancer Properties via the UPR Pathway. Biochem. Pharmacol. 2021, 190, 114637. [Google Scholar] [CrossRef]
- McKinnon, C.; Nandhabalan, M.; Murray, S.A.; Plaha, P. Glioblastoma: Clinical Presentation, Diagnosis, and Management. BMJ 2021, 374, n1560. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. In Cell Cycle Control: Mechanisms and Protocols; Humana Press: New York, NY, USA, 2014; pp. 29–40. [Google Scholar] [CrossRef]
- Nagelkerke, A.; Span, P.N. Staining Against Phospho-H2AX (γ-H2AX) as a Marker for DNA Damage and Genomic Instability in Cancer Tissues and Cells. In Tumor Microenvironment: Study Protocols; Springer: Cham, Switzerland, 2016; pp. 1–10. [Google Scholar] [CrossRef]
- Zahid, S.; Seif El Dahan, M.; Iehl, F.; Fernandez-Varela, P.; Le Du, M.-H.; Ropars, V.; Charbonnier, J.B. The Multifaceted Roles of Ku70/80. Int. J. Mol. Sci. 2021, 22, 4134. [Google Scholar] [CrossRef] [PubMed]
- Hirose, Y.; Berger, M.S.; Pieper, R.O. p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res. 2001, 61, 1957–1963. [Google Scholar]
- Van Meir, E.G.; Kikuchi, T.; Tada, M.; Li, H.; Diserens, A.C.; Wojcik, B.E.; Huang, H.J.; Friedmann, T.; de Tribolet, N.; Cavenee, W.K. Analysis of the p53 gene and its expression in human glioblastoma cells. Cancer Res. 1994, 54, 649–652. [Google Scholar]
- Ellgaard, L.; Frickel, E.-M. Calnexin, Calreticulin, and ERp57: Teammates in Glycoprotein Folding. Cell Biochem. Biophys. 2003, 39, 223–248. [Google Scholar] [CrossRef]
- Russell, S.J.; Ruddock, L.W.; Salo, K.E.H.; Oliver, J.D.; Roebuck, Q.P.; Llewellyn, D.H.; Roderick, H.L.; Koivunen, P.; Myllyharju, J.; High, S. The Primary Substrate Binding Site in the B′ Domain of ERp57 Is Adapted for Endoplasmic Reticulum Lectin Association. J. Biol. Chem. 2004, 279, 18861–18869. [Google Scholar] [CrossRef]
- Coppari, S.; Altieri, F.; Ferraro, A.; Chichiarelli, S.; Eufemi, M.; Turano, C. Nuclear Localization and DNA Interaction of Protein Disulfide Isomerase ERp57 in Mammalian Cells. J. Cell Biochem. 2002, 85, 325–333. [Google Scholar] [CrossRef]
- Eufemi, M.; Coppari, S.; Altieri, F.; Grillo, C.; Ferraro, A.; Turano, C. ERp57 Is Present in STAT3–DNA Complexes. Biochem. Biophys. Res. Commun. 2004, 323, 1306–1312. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, J.-X.; Nie, Z.-Y.; Wen, Y.; Jia, X.-J.; Zhang, L.-N.; Duan, H.-J.; Shi, Y.-H. Upregulation of ERp57 Promotes Clear Cell Renal Cell Carcinoma Progression by Initiating a STAT3/ILF3 Feedback Loop. J. Exp. Clin. Cancer Res. 2019, 38, 439. [Google Scholar] [CrossRef]
- Li, S.; Zhao, X.; Chang, S.; Li, Y.; Guo, M.; Guan, Y. ERp57 small Interfering RNA Silencing Can Enhance the Sensitivity of Drug resistant Human Ovarian Cancer Cells to Paclitaxel. Int. J. Oncol. 2018, 54, 249–260. [Google Scholar] [CrossRef]
- Young, H.S.; McGowan, L.M.; Jepson, K.A.; Adams, J.C. Impairment of Cell Adhesion and Migration by Inhibition of Protein Disulphide Isomerases in Three Breast Cancer Cell Lines. Biosci. Rep. 2020, 40, BSR20193271. [Google Scholar] [CrossRef]
- Kurpińska, A.; Suraj-Prażmowska, J.; Stojak, M.; Jarosz, J.; Mateuszuk, Ł.; Niedzielska-Andres, E.; Smolik, M.; Wietrzyk, J.; Kalvins, I.; Walczak, M.; et al. Comparison of Anti-Cancer Effects of Novel Protein Disulphide Isomerase (PDI) Inhibitors in Breast Cancer Cells Characterized by High and Low PDIA17 Expression. Cancer Cell Int. 2022, 22, 218. [Google Scholar] [CrossRef]
- Kondo, R.; Ishino, K.; Wada, R.; Takata, H.; Peng, W.; Kudo, M.; Kure, S.; Kaneya, Y.; Taniai, N.; Yoshida, H.; et al. Downregulation of Protein Disulfide isomerase A3 Expression Inhibits Cell Proliferation and Induces Apoptosis through STAT3 Signaling in Hepatocellular Carcinoma. Int. J. Oncol. 2019, 54, 1409–1421. [Google Scholar] [CrossRef]
- Song, D.; Guo, M.; Wu, K.; Hao, J.; Nie, Y.; Fan, D. Silencing of ER-Resident Oxidoreductase PDIA3 Inhibits Malignant Biological Behaviors of Multidrug-Resistant Gastric Cancer. Acta Biochim. Biophys. Sin. 2021, 53, 1216–1226. [Google Scholar] [CrossRef]
- Grillo, C.; D’Ambrosio, C.; Scaloni, A.; Maceroni, M.; Merluzzi, S.; Turano, C.; Altieri, F. Cooperative activity of Ref-1/APE and ERp57 in reductive activation of transcription factors. Free Radic. Biol. Med. 2006, 41, 1113–1123. [Google Scholar] [CrossRef]
- Chichiarelli, S.; Ferraro, A.; Altieri, F.; Eufemi, M.; Coppari, S.; Grillo, C.; Arcangeli, V.; Turano, C. The Stress Protein ERp57/GRP58 Binds Specific DNA Sequences in HeLa Cells. J. Cell Physiol. 2007, 210, 343–351. [Google Scholar] [CrossRef]
- Barroso, S.I.; Aguilera, A. Detection of DNA Double-Strand Breaks by γ-H2AX Immunodetection. In Homologous Recombination: Methods and Protocols; Humana: New York, NY, USA, 2021; pp. 1–8. [Google Scholar] [CrossRef]
- Wilson, M.D.; Durocher, D. Reading Chromatin Signatures after DNA Double-Strand Breaks. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160280. [Google Scholar] [CrossRef]
- Cocchiola, R.; Rubini, E.; Altieri, F.; Chichiarelli, S.; Paglia, G.; Romaniello, D.; Carissimi, S.; Giorgi, A.; Giamogante, F.; Macone, A.; et al. STAT3 Post-Translational Modifications Drive Cellular Signaling Pathways in Prostate Cancer Cells. Int. J. Mol. Sci. 2019, 20, 1815. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paglia, G.; Minacori, M.; Meschiari, G.; Fiorini, S.; Chichiarelli, S.; Eufemi, M.; Altieri, F. Protein Disulfide Isomerase A3 (PDIA3): A Pharmacological Target in Glioblastoma? Int. J. Mol. Sci. 2023, 24, 13279. https://doi.org/10.3390/ijms241713279
Paglia G, Minacori M, Meschiari G, Fiorini S, Chichiarelli S, Eufemi M, Altieri F. Protein Disulfide Isomerase A3 (PDIA3): A Pharmacological Target in Glioblastoma? International Journal of Molecular Sciences. 2023; 24(17):13279. https://doi.org/10.3390/ijms241713279
Chicago/Turabian StylePaglia, Giuliano, Marco Minacori, Giorgia Meschiari, Sara Fiorini, Silvia Chichiarelli, Margherita Eufemi, and Fabio Altieri. 2023. "Protein Disulfide Isomerase A3 (PDIA3): A Pharmacological Target in Glioblastoma?" International Journal of Molecular Sciences 24, no. 17: 13279. https://doi.org/10.3390/ijms241713279