
Preservation of Biomarkers Associated with Alzheimer’s Disease (Amyloid Peptides 1-38, 1-40, 1-42, Tau Protein, Beclin 1) in the Blood of Neonates after Perinatal Asphyxia
 , ,
, ,
Abstract
:1. Introduction
2. Results
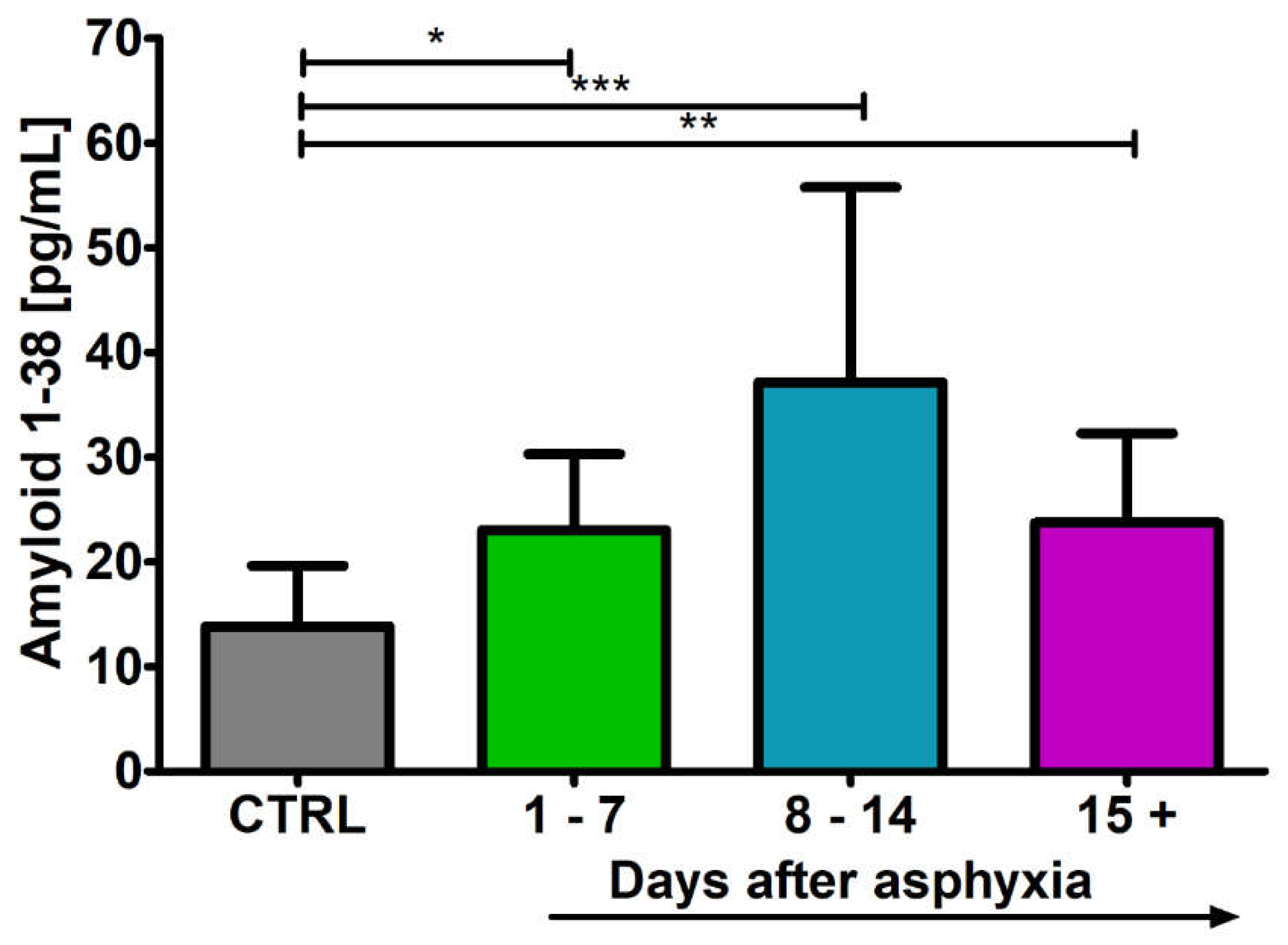
2.1. Level of the β-Amyloid Peptide 1-38 in Blood
2.2. Level of the β-Amyloid Peptide 1-40 in Blood
2.3. Level of the β-Amyloid Peptide 1-42 in Blood
2.4. Level of the Tau Protein in Blood
2.5. Level of the Beclin 1 in Blood
3. Discussion
4. Materials and Methods
- Newborns (preterm and full-term) > 31 weeks of gestational age.
- Acidosis with pH < 7.0 (in umbilical cord or blood sample obtained throughout the 60 min post-asphyxia).
- Or base deficit > −12.
- Or Apgar score of 0–5 at 10 min after birth or during 10 min of resuscitation.
- The presence of multiple organ dysfunctions.
- Clinical manifestations of encephalopathy: aberrant oculomotor or pupillary activity, feeble or elusive suck, episodic breathing/apnea, or seizures.
- Neurological deficits cannot be related to alternative disease.
4.1. Research Groups
4.2. Elisa Studies
4.3. Statistical Analysis of the Results
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mathew, J.L.; Kaur, N.; Dsouza, J.M. Therapeutic hypothermia in neonatal hypoxic encephalopathy: A systematic review and meta-analysis. J. Glob. Health 2022, 12, 04030. [Google Scholar] [CrossRef]
- Parmentier, C.E.J.; de Vries, L.S.; Groenendaal, F. Magnetic resonance imaging in (Near-)term infants with hypoxic-ischemic encephalopathy. Diagnostics 2022, 12, 645. [Google Scholar] [CrossRef] [PubMed]
- Uzianbaeva, L.; Yan, Y.; Joshi, T.; Yin, N.; Hsu, C.D.; Hernandez-Andrade, E.; Mehrmohammadi, M. Methods for monitoring risk of hypoxic damage in fetal and neonatal brains: A review. Fetal Diagn. Ther. 2022, 49, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, L.; Zhang, X.; Xie, W.; Li, L.; Yang, D.; Heng, X.; Du, Y.; Doody, R.S.; Le, W. Prenatal hypoxia may aggravate the cognitive impairment and Alzheimer’s disease neuropathology in APPSwe/PS1A246E transgenic mice. Neurobiol. Aging 2013, 34, 663–678. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Liang, L.; Xu, H.; Xu, J.; Yao, L.; Li, Y.; Tan, Y.; Li, X.; Huang, Q.; Yang, Z.; et al. Short term exposure to bilirubin induces encephalopathy similar to Alzheimer’s disease in late life. J. Alzheimers Dis. 2020, 73, 277–295. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Lechner, C.; Emerson, P.; Northington, F.J.; Martin, L.J. Accumulation of PSA-NCAM marks nascent neurodegeneration in the dorsal hippocampus after neonatal hypoxic-ischemic brain injury in mice. Br. J. Pharmacol. 2021, 41, 1039–1057. [Google Scholar] [CrossRef]
- Lv, H.; Wang, Q.; Wu, S.; Yang, L.; Ren, P.; Yang, Y.; Gao, J.; Li, L. Neonatal hypoxic ischemic encephalopathy related biomarkers in serum and cerebrospinal fluid. Clin. Chim. Acta. 2015, 450, 282–927. [Google Scholar] [CrossRef] [PubMed]
- Bano, S.; Chaudhary, V.; Garga, U.C. Neonatal Hypoxic-ischemic Encephalopathy: A Radiological Review. J. Pediatr. Neurosci. 2017, 12, 1–6. [Google Scholar] [CrossRef]
- Graham, E.M.; Everett, A.D.; Delpech, J.C.; Northington, F.J. Blood biomarkers for evaluation of perinatal encephalopathy: State of the art. Curr. Opin. Pediatr. 2018, 30, 199–203. [Google Scholar] [CrossRef]
- Satriano, A.; Pluchinotta, F.; Gazzolo, F.; Serpero, L.; Gazzolo, D. The potentials and limitations of neuro-biomarkers as predictors of outcome in neonates with birth asphyxia. Early Hum. Dev. 2017, 105, 63–67. [Google Scholar] [CrossRef]
- Tarkowska, A.; Furmaga-Jabłońska, W.; Bogucki, J.; Kocki, J.; Pluta, R. Hypothermia after perinatal asphyxia does not affect genes responsible for amyloid production in neonatal peripheral lymphocytes. J. Clin. Med. 2022, 11, 3263. [Google Scholar] [CrossRef]
- Tarkowska, A.; Furmaga-Jabłońska, W.; Bogucki, J.; Kocki, J.; Pluta, R. Alzheimer’s disease associated presenilin 1 and 2 genes dysregulation in neonatal lymphocytes following perinatal asphyxia. Int. J. Mol. Sci. 2021, 22, 5140. [Google Scholar] [CrossRef] [PubMed]
- Denihan, N.M.; Kirwan, J.A.; Walsh, B.H.; Dunn, W.B.; Broadhurst, D.I.; Boylan, G.B.; Murray, D.M. Untargeted metabolomic analysis and pathway discovery in perinatal asphyxia and hypoxic-ischaemic encephalopathy. J. Cereb. Blood Flow Metab. 2017, 39, 147–162. [Google Scholar] [CrossRef] [PubMed]
- Chavez-Valdez, R.; Miller, S.; Spahic, H.; Vaidya, D.; Parkinson, C.; Dietrick, B.; Brooks, S.; Gerner, G.J.; Tekes, A.; Graham, E.M.; et al. Therapeutic hypothermia modulates the relationships between indicators of severity of neonatal hypoxic ischemic encephalopathy and serum biomarkers. Front. Neurol. 2021, 12, 748150. [Google Scholar] [CrossRef]
- Toorell, H.; Zetterberg, H.; Blennow, K.; Sävman, K.; Hagberg, H. Increase of neuronal injury markers tau and neurofilament light proteins in umbilical blood after intrapartum asphyxia. J. Matern. Fetal Neonatal Med. 2018, 31, 2468–2472. [Google Scholar] [CrossRef]
- Takahashi, K.; Hasegawa, S.; Maeba, S.; Fukunaga, S.; Motoyama, M.; Hamano, H.; Ichiyama, T. Serum tau protein level serves as a predictive factor for neurological prognosis in neonatal asphyxia. Brain Dev. 2014, 36, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Kis, J.; Januszewski, S.; Jabłonski, M.; Czuczwar, S.J. Cross-Talk between Amyloid, Tau Protein and Free Radicals in Post-Ischemic Brain Neurodegeneration in the Form of Alzheimer’s Disease Proteinopathy. Antioxidants 2022, 11, 146. [Google Scholar] [CrossRef]
- Schiefecker, A.J.; Putzer, G.; Braun, P.; Martini, J.; Strapazzon, G.; Antunes, A.P.; Mulino, M.; Pinggera, D.; Glodny, B.; Brugger, H.; et al. Total Tau Protein as Investigated by Cerebral Microdialysis Increases in Hypothermic Cardiac Arrest: A Pig Study. Ther. Hypothermia Temp. Manag. 2021, 11, 28–34. [Google Scholar] [CrossRef]
- Li, R.; Lee, J.K.; Govindan, R.B.; Graham, E.M.; Everett, A.D.; Perin, J.; Vezina, G.; Tekes, A.; Chen, M.W.; Northington, F.; et al. Plasma Biomarkers of Evolving Encephalopathy and Brain Injury in Neonates with Hypoxic-Ischemic Encephalopathy. J. Pediatr. 2023, 252, 146–153.e2. [Google Scholar] [CrossRef]
- Lv, H.Y.; Wu, S.J.; Gu, X.L.; Wang, Q.L.; Ren, P.S.; Ma, Y.; Peng, L.Y.; Jin, L.H.; Li, L.X. Predictive Value of Neurodevelopmental Outcome and Serum Tau Protein Level in Neonates with Hypoxic Ischemic Encephalopathy. Clin. Lab. 2017, 63, 1153–1162. [Google Scholar] [CrossRef] [PubMed]
- Massaro, A.N.; Wu, Y.W.; Bammler, T.K.; Comstock, B.; Mathur, A.; McKinstry, R.C.; Chang, T.; Mayock, D.E.; Mulkey, S.B.; Van Meurs, K.; et al. Plasma biomarkers of brain injury in neonatal hypoxic-ischemic encephalopathy. J. Pediatr. 2018, 194, 67–75.e1. [Google Scholar] [CrossRef]
- Li, S.; Wang, W.; Wang, C.; Tang, Y.Y. Possible involvement of NO/NOS signaling in hippocampal amyloid-beta production induced by transient focal cerebral ischemia in aged rats. Neurosci. Lett. 2010, 470, 106–110. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Zhang, D.; Zeng, Y.; Huang, T.Y.; Xu, H.; Zhao, Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol. Neurodegener. 2020, 15, 40. [Google Scholar] [CrossRef]
- Pluta, R.; Ułamek-Kozioł, M.; Januszewski, S.; Czuczwar, S.J. Participation of amyloid and tau protein in neuronal death and neurodegeneration after brain ischemia. Int. J. Mol. Sci. 2020, 21, 4599. [Google Scholar] [CrossRef] [PubMed]
- Pluta, R.; Ouyang, L.; Januszewski, S.; Li, Y.; Czuczwar, S.J. Participation of Amyloid and Tau Protein in Post-Ischemic Neurodegeneration of the Hippocampus of a Nature Identical to Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2460. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.B.; Ni, Y.S.; Liu, N.; Wei, W.; Liu, Y.; Yang, J.M.; Ma, L.; Bai, R.; Zhang, J.; Yu, J.Q. Neuroprotective effects of oxymatrine on hypoxic-ischemic brain damage in neonatal rats by activating the Wnt/β-catenin pathway. Biomed Pharmacother. 2023, 159, 114266. [Google Scholar] [CrossRef] [PubMed]
- Mehta, S.L.; Kim, T.; Chelluboina, B.; Vemuganti, R. Tau and GSK-3β are Critical Contributors to α-Synuclein-Mediated Post-Stroke Brain Damage. Neuromolecular Med. 2023, 25, 94–101. [Google Scholar] [CrossRef]
- Rana, A.K.; Kumar, R.; Shukla, D.N.; Singh, D. Lithium co-administration with rutin improves post-stroke neurological outcomes via suppressing Gsk-3β activity in a rat model. Free. Radic. Biol. Med. 2023, 207, 107–119. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VARIABLES | N | CONTROL | ASPHYXIA | ||
|---|---|---|---|---|---|
| Mean ± SD | Median | Mean ± SD | Median | ||
| Age (Days) | 10 | 15 ± 7 | 15 | 10 ± 7 | 7 |
| Gestational Age (Weeks) | 10 | 39 ± 1 | 39 | 39 ± 2 | 39 |
| Apgar Score (1 min) | 10 | 10 ± 0 | 10 | 2 ± 1.5 | 1.50 |
| WBC (/µL) | 10 | 13,640 ± 2651 | 13,870 | 25,298 ± 9300 | 24,300 |
| RBC (×1000/µL) | 10 | 4654 ± 539 | 4855 | 4498 ± 761 | 4715 |
| Hct (%) | 10 | 44 ± 5 | 46 | 48 ± 10 | 49 |
| PLT (×1000/µL) | 10 | 446 ± 165 | 455 | 250 ± 64 | 260 |
| Lymphocytes (/µL) | 10 | 7180 ± 907 | 7520 | 5609 ± 2247 | 4640 |
| pH | 10 | 7.41 ± 0.02 | 7.41 | 7.23 ± 0.25 | 7.23 |
| BE (mmol/L) | 10 | 0.80 ± 1.7 | −0.10 | −12.77 ± 9.9 | −11.20 |
| Birth Weight (g) | 10 | 3572 ± 664 | 3675 | 3572 ± 664 | 3620 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarkowska, A.; Furmaga-Jabłońska, W.; Bogucki, J.; Kocki, J.; Pluta, R. Preservation of Biomarkers Associated with Alzheimer’s Disease (Amyloid Peptides 1-38, 1-40, 1-42, Tau Protein, Beclin 1) in the Blood of Neonates after Perinatal Asphyxia. Int. J. Mol. Sci. 2023, 24, 13292. https://doi.org/10.3390/ijms241713292
Tarkowska A, Furmaga-Jabłońska W, Bogucki J, Kocki J, Pluta R. Preservation of Biomarkers Associated with Alzheimer’s Disease (Amyloid Peptides 1-38, 1-40, 1-42, Tau Protein, Beclin 1) in the Blood of Neonates after Perinatal Asphyxia. International Journal of Molecular Sciences. 2023; 24(17):13292. https://doi.org/10.3390/ijms241713292
Chicago/Turabian StyleTarkowska, Agata, Wanda Furmaga-Jabłońska, Jacek Bogucki, Janusz Kocki, and Ryszard Pluta. 2023. "Preservation of Biomarkers Associated with Alzheimer’s Disease (Amyloid Peptides 1-38, 1-40, 1-42, Tau Protein, Beclin 1) in the Blood of Neonates after Perinatal Asphyxia" International Journal of Molecular Sciences 24, no. 17: 13292. https://doi.org/10.3390/ijms241713292