
The Mitochondrial m.3243A>G Mutation on the Dish, Lessons from In Vitro Models


Abstract
:1. Mitochondria
2. Mitochondrial Disease
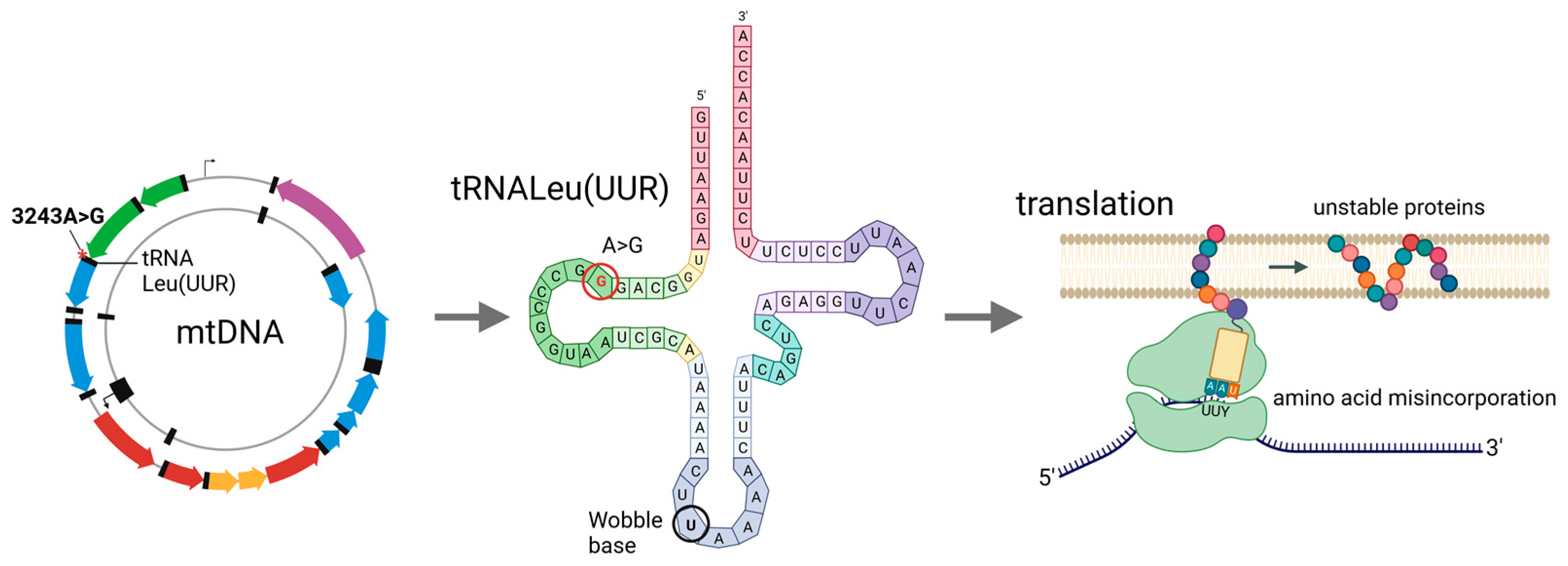
3. m.3243A>G Mutation
4. Molecular and Functional Consequences of the m.3243A>G Mutation
5. Cell Models of m.3243A>G
5.1. Fibroblast Studies
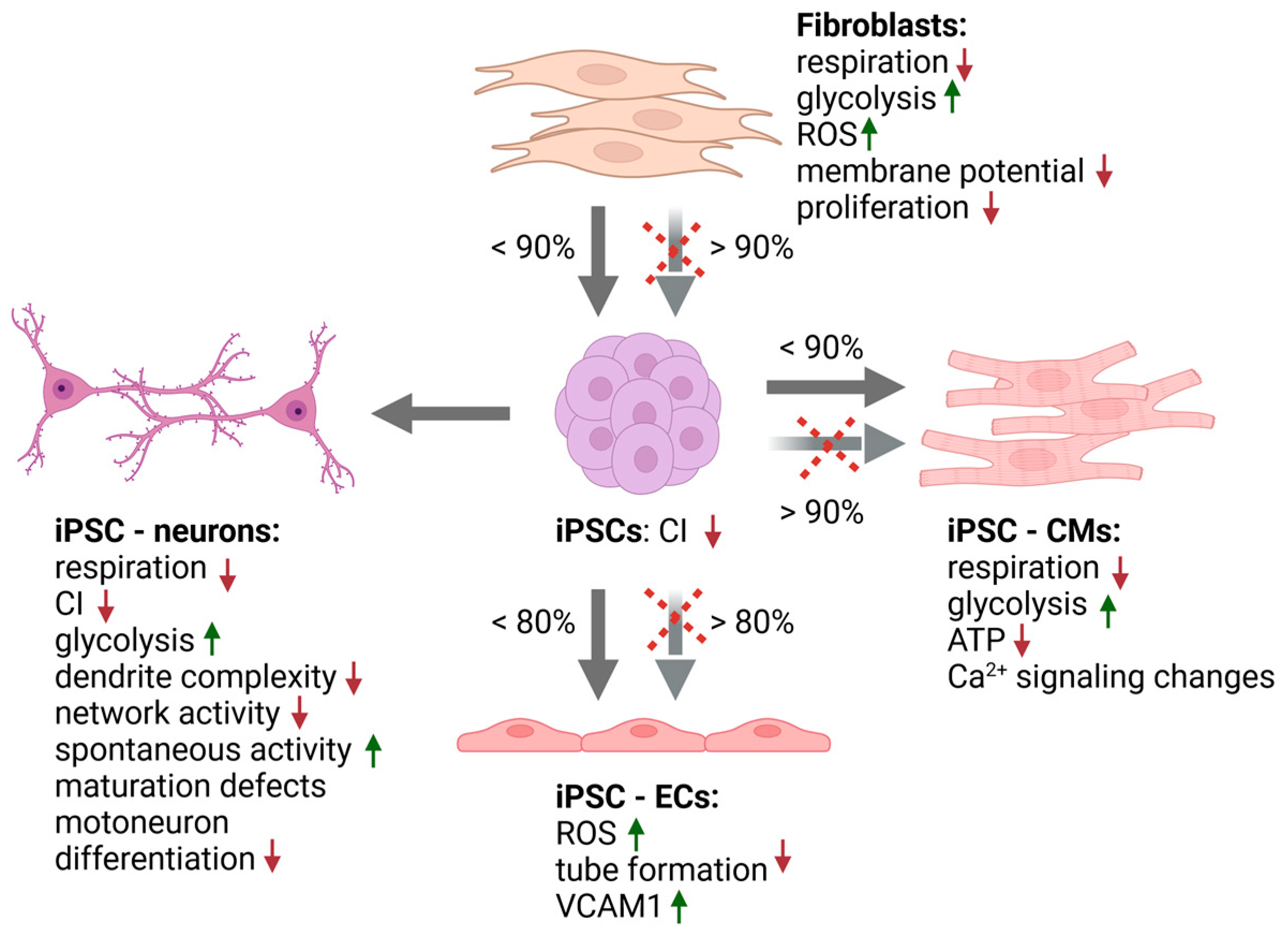
5.2. Stem Cell Based Models
5.2.1. Pluripotent Stem Cells
5.2.2. Neural Cells
5.2.3. Cardiac Cells
5.2.4. Other iPSC- Derived Cell Types
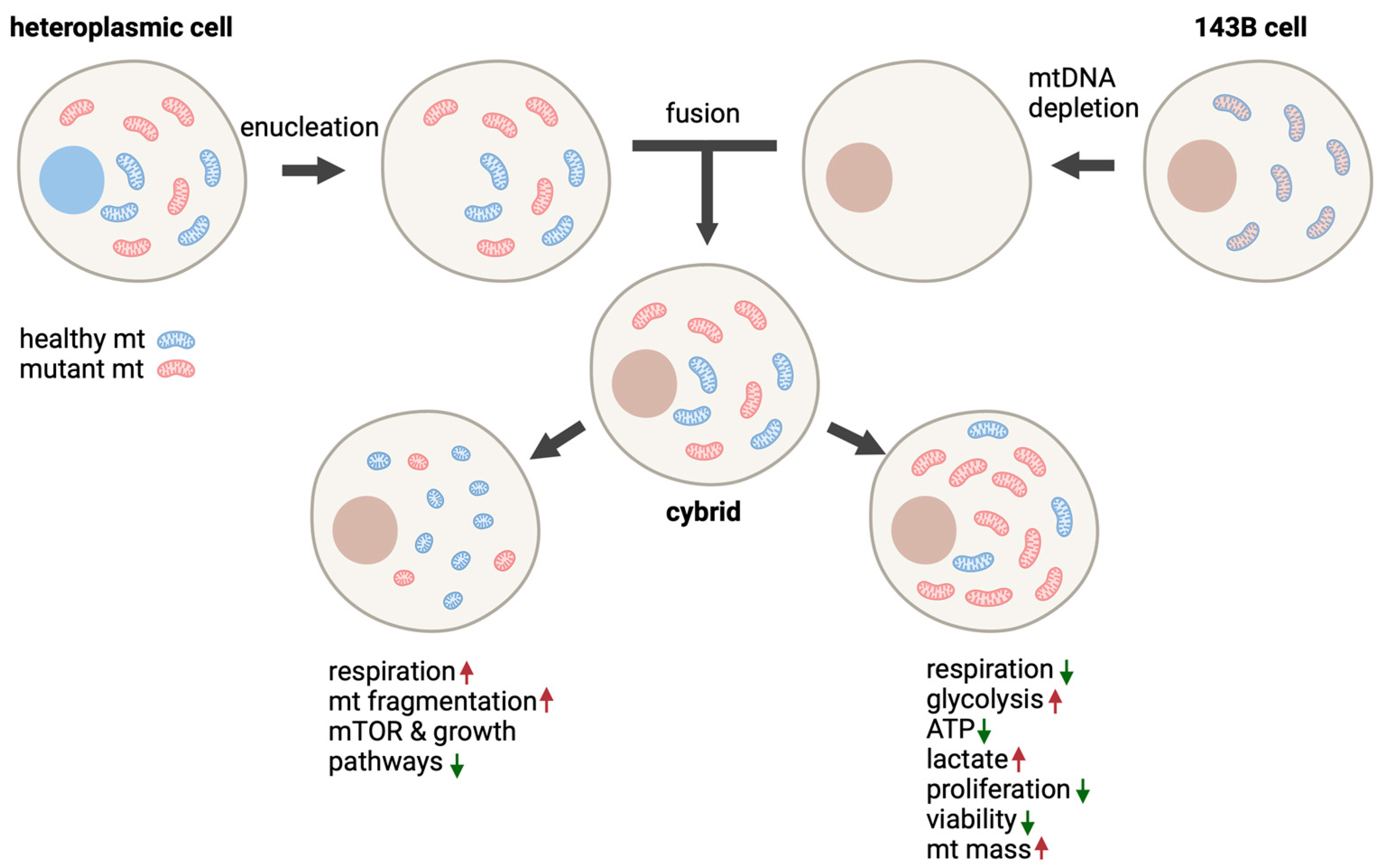
5.3. Cybrid Models
5.3.1. 143B Human Osteosarcoma Cells
5.3.2. SH-SY5Y Human Neuroblastoma Cybrids
5.3.3. Other Cybrid Models of m.3243A>G
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nunnari, J.; Suomalainen, A. Mitochondria: In Sickness and in Health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Kühlbrandt, W. Structure and Function of Mitochondrial Membrane Protein Complexes. BMC Biol. 2015, 13, 89. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Gorman, G.S.; Schaefer, A.M.; Ng, Y.; Gomez, N.; Blakely, E.L.; Alston, C.L.; Feeney, C.; Horvath, R.; Yu-Wai-Man, P.; Chinnery, P.F.; et al. Prevalence of Nuclear and Mitochondrial DNA Mutations Related to Adult Mitochondrial Disease. Ann. Neurol. 2015, 77, 753–759. [Google Scholar] [CrossRef]
- Alston, C.L.; Rocha, M.C.; Lax, N.Z.; Turnbull, D.M.; Taylor, R.W. The Genetics and Pathology of Mitochondrial Disease. J. Pathol. 2017, 241, 236–250. [Google Scholar] [CrossRef]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef]
- Pickett, S.J.; Grady, J.P.; Ng, Y.S.; Gorman, G.S.; Schaefer, A.M.; Wilson, I.J.; Cordell, H.J.; Turnbull, D.M.; Taylor, R.W.; McFarland, R. Phenotypic Heterogeneity in m.3243A>G Mitochondrial Disease: The Role of Nuclear Factors. Ann. Clin. Transl. Neurol. 2018, 5, 333–345. [Google Scholar] [CrossRef]
- Manwaring, N.; Jones, M.M.; Wang, J.J.; Rochtchina, E.; Howard, C.; Mitchell, P.; Sue, C.M. Population Prevalence of the MELAS A3243G Mutation. Mitochondrion 2007, 7, 230–233. [Google Scholar] [CrossRef]
- Majamaa, K.; Moilanen, J.S.; Uimonen, S.; Remes, A.M.; Salmela, P.I.; Kärppä, M.; Majamaa-Voltti, K.A.; Rusanen, H.; Sorri, M.; Peuhkurinen, K.J.; et al. Epidemiology of A3243G, the Mutation for Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes: Prevalence of the Mutation in an Adult Population. Am. J. Hum. Genet. 1998, 63, 447–454. [Google Scholar] [CrossRef]
- Li, D.; Liang, C.; Zhang, T.; Marley, J.L.; Zou, W.; Lian, M.; Ji, D. Pathogenic Mitochondrial DNA 3243A>G Mutation: From Genetics to Phenotype. Front. Genet. 2022, 13, 951185. [Google Scholar] [CrossRef]
- Shen, X.; Du, A. The Non-Syndromic Clinical Spectrums of MtDNA 3243A>G Mutation. Neurosciences 2021, 26, 128–133. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the TRNA(Leu)(UUR) Gene Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS Syndrome: Clinical Manifestations, Pathogenesis, and Treatment Options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Nesbitt, V.; Pitceathly, R.D.S.; Turnbull, D.M.; Taylor, R.W.; Sweeney, M.G.; Mudanohwo, E.E.; Rahman, S.; Hanna, M.G.; McFarland, R. The UK MRC Mitochondrial Disease Patient Cohort Study: Clinical Phenotypes Associated with the m.3243A>G Mutation—Implications for Diagnosis and Management. J. Neurol. Neurosurg. Psychiatry 2013, 84, 936–938. [Google Scholar] [CrossRef]
- Boggan, R.M.; Lim, A.; Taylor, R.W.; McFarland, R.; Pickett, S.J. Resolving Complexity in Mitochondrial Disease: Towards Precision Medicine. Mol. Genet. Metab. 2019, 128, 19–29. [Google Scholar] [CrossRef]
- Haast, R.A.M.; Ivanov, D.; IJsselstein, R.J.T.; Sallevelt, S.C.E.H.; Jansen, J.F.A.; Smeets, H.J.M.; de Coo, I.F.M.; Formisano, E.; Uludağ, K. Anatomic & Metabolic Brain Markers of the m.3243A>G Mutation: A Multi-Parametric 7T MRI Study. NeuroImage Clin. 2018, 18, 231–244. [Google Scholar] [CrossRef]
- Rahman, S.; Poulton, J.; Marchington, D.; Suomalainen, A. Decrease of 3243 A→G MtDNA Mutation from Blood in MELAS Syndrome: A Longitudinal Study. Am. J. Hum. Genet. 2001, 68, 238–240. [Google Scholar] [CrossRef]
- Grady, J.P.; Pickett, S.J.; Ng, Y.S.; Alston, C.L.; Blakely, E.L.; Hardy, S.A.; Feeney, C.L.; Bright, A.A.; Schaefer, A.M.; Gorman, G.S.; et al. MtDNA Heteroplasmy Level and Copy Number Indicate Disease Burden in m.3243A>G Mitochondrial Disease. EMBO Mol. Med. 2018, 10, e8262. [Google Scholar] [CrossRef]
- Ng, Y.S.; Lax, N.Z.; Blain, A.P.; Erskine, D.; Baker, M.R.; Polvikoski, T.; Thomas, R.H.; Morris, C.M.; Lai, M.; Whittaker, R.G.; et al. Forecasting Stroke-like Episodes and Outcomes in Mitochondrial Disease. Brain 2021, 145, 542–554. [Google Scholar] [CrossRef]
- van den Ouweland, J.M.W.; Lemkes, H.H.P.J.; Ruitenbeek, W.; Sandkuijl, L.A.; de Vijlder, M.F.; Struyvenberg, P.A.A.; van de Kamp, J.J.P.; Maassen, J.A. Mutation in Mitochondrial TRNALeu(UUR) Gene in a Large Pedigree with Maternally Transmitted Type II Diabetes Mellitus and Deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Motlagh Scholle, L.; Zierz, S.; Mawrin, C.; Wickenhauser, C.; Lehmann Urban, D. Heteroplasmy and Copy Number in the Common m.3243A>G Mutation—A Post-Mortem Genotype–Phenotype Analysis. Genes 2020, 11, 212. [Google Scholar] [CrossRef] [PubMed]
- Chomyn, A.; Enriquez, J.A.; Micol, V.; Fernandez-Silva, P.; Attardi, G. The Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episode Syndrome-Associated Human Mitochondrial TRNALeu(UUR) Mutation Causes Aminoacylation Deficiency and Concomitant Reduced Association of MRNA with Ribosomes. J. Biol. Chem. 2000, 275, 19198–19209. [Google Scholar] [CrossRef]
- Kirino, Y.; Suzuki, T. Human Mitochondrial Diseases Associated with TRNA Wobble Modification Deficiency. RNA Biol. 2005, 2, 41–44. [Google Scholar] [CrossRef]
- Wittenhagen, L.M.; Kelley, S.O. Dimerization of a Pathogenic Human Mitochondrial TRNA. Nat. Struct. Biol. 2002, 9, 586–590. [Google Scholar] [CrossRef]
- Levinger, L.; Oestreich, I.; Florentz, C.; Mörl, M. A Pathogenesis-Associated Mutation in Human Mitochondrial TRNALeu(UUR) Leads to Reduced 3′-End Processing and CCA Addition. J. Mol. Biol. 2004, 337, 535–544. [Google Scholar] [CrossRef]
- Hess, J.F.; Parisi, M.A.; Bennett, J.L.; Clayton, D.A. Impairment of Mitochondrial Transcription Termination by a Point Mutation Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1991, 351, 236–239. [Google Scholar] [CrossRef]
- Guo, J.; Chen, X.; Liu, Z.; Sun, H.; Zhou, Y.; Dai, Y.; Ma, Y.; He, L.; Qian, X.; Wang, J.; et al. DdCBE Mediates Efficient and Inheritable Modifications in Mouse Mitochondrial Genome. Mol. Ther. Nucleic Acids 2022, 27, 73–80. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.-C.; Seo, H.; Kim, J.-S. Mitochondrial DNA Editing in Mice with DddA-TALE Fusion Deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef]
- Ylikallio, E.; Suomalainen, A. Mechanisms of Mitochondrial Diseases. Ann. Med. 2012, 44, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive Increase in MtDNA 3243A>G Heteroplasmy Causes Abrupt Transcriptional Reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of Nuclear Epigenome by Mitochondrial DNA Heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef] [PubMed]
- Chichagova, V.; Hallam, D.; Collin, J.; Buskin, A.; Saretzki, G.; Armstrong, L.; Yu-Wai-Man, P.; Lako, M.; Steel, D.H. Human IPSC Disease Modelling Reveals Functional and Structural Defects in Retinal Pigment Epithelial Cells Harbouring the m.3243A > G Mitochondrial DNA Mutation. Sci. Rep. 2017, 7, 12320. [Google Scholar] [CrossRef] [PubMed]
- Hämäläinen, R.H.; Manninen, T.; Koivumäki, H.; Kislin, M.; Otonkoski, T.; Suomalainen, A. Tissue- and Cell-Type-Specific Manifestations of Heteroplasmic MtDNA 3243A>G Mutation in Human Induced Pluripotent Stem Cell-Derived Disease Model. Proc. Natl. Acad. Sci. USA 2013, 110, E3622–E3630. [Google Scholar] [CrossRef]
- Malena, A.; Pantic, B.; Borgia, D.; Sgarbi, G.; Solaini, G.; Holt, I.J.; Spinazzola, A.; Perissinotto, E.; Sandri, M.; Baracca, A.; et al. Mitochondrial Quality Control: Cell-Type-Dependent Responses to Pathological Mutant Mitochondrial DNA. Autophagy 2016, 12, 2098–2112. [Google Scholar] [CrossRef]
- Yokota, M.; Hatakeyama, H.; Okabe, S.; Ono, Y.; Goto, Y. Mitochondrial Respiratory Dysfunction Caused by a Heteroplasmic Mitochondrial DNA Mutation Blocks Cellular Reprogramming. Hum. Mol. Genet. 2015, 24, 4698–4709. [Google Scholar] [CrossRef]
- Matsubara, M.; Kanda, H.; Imamura, H.; Inoue, M.; Noguchi, M.; Hosoda, K.; Kakizuka, A.; Nakao, K. Analysis of Mitochondrial Function in Human Induced Pluripotent Stem Cells from Patients with Mitochondrial Diabetes Due to the A3243G Mutation. Sci. Rep. 2018, 8, 949. [Google Scholar] [CrossRef]
- Ryytty, S.; Modi, S.R.; Naumenko, N.; Shakirzyanova, A.; Rahman, M.O.; Vaara, M.; Suomalainen, A.; Tavi, P.; Hämäläinen, R.H. Varied Responses to a High m.3243A>G Mutation Load and Respiratory Chain Dysfunction in Patient-Derived Cardiomyocytes. Cells 2022, 11, 2593. [Google Scholar] [CrossRef]
- Sreedhar, A.; Aguilera-Aguirre, L.; Singh, K.K. Mitochondria in Skin Health, Aging, and Disease. Cell Death Dis. 2020, 11, 444. [Google Scholar] [CrossRef]
- Chung, C.-Y.; Singh, K.; Kotiadis, V.N.; Valdebenito, G.E.; Ahn, J.H.; Topley, E.; Tan, J.; Andrews, W.D.; Bilanges, B.; Pitceathly, R.D.S.; et al. Constitutive Activation of the PI3K-Akt-MTORC1 Pathway Sustains the m.3243 A > G MtDNA Mutation. Nat. Commun. 2021, 12, 6409. [Google Scholar] [CrossRef]
- Cotán, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Ávila, M.; Rodríguez-Hernández, A.; Gómez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary Coenzyme Q10 Deficiency Triggers Mitochondria Degradation by Mitophagy in MELAS Fibroblasts. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 2669–2687. [Google Scholar] [CrossRef]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Ávila, M.; de la Mata, M.; Pavón, A.D.; de Lavera, I.; Alcocer-Gómez, E.; Galán, F.; et al. Critical Role of AMP-Activated Protein Kinase in the Balance between Mitophagy and Mitochondrial Biogenesis in MELAS Disease. Biochim. Biophys. Acta 2015, 1852, 2535–2553. [Google Scholar] [CrossRef]
- Lin, D.-S.; Huang, Y.-W.; Ho, C.-S.; Huang, T.-S.; Lee, T.-H.; Wu, T.-Y.; Huang, Z.-D.; Wang, T.-J. Impact of Mitochondrial A3243G Heteroplasmy on Mitochondrial Bioenergetics and Dynamics of Directly Reprogrammed MELAS Neurons. Cells 2022, 12, 15. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Cook, A.N.; Evans, J.M.; Roellinger, S.; Secreto, F.; Emmanuele, V.; Oglesbee, D.; Mootha, V.K.; Hirano, M.; Schon, E.A.; et al. Natural Underlying MtDNA Heteroplasmy as a Potential Source of Intra-Person HiPSC Variability. EMBO J. 2016, 35, 1979–1990. [Google Scholar] [CrossRef] [PubMed]
- Klein Gunnewiek, T.M.; Van Hugte, E.J.H.; Frega, M.; Guardia, G.S.; Foreman, K.; Panneman, D.; Mossink, B.; Linda, K.; Keller, J.M.; Schubert, D.; et al. m.3243A > G-Induced Mitochondrial Dysfunction Impairs Human Neuronal Development and Reduces Neuronal Network Activity and Synchronicity. Cell Rep. 2020, 31, 107538. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Ma, H.; Juanes, R.C.; Tachibana, M.; Sparman, M.; Woodward, J.; Ramsey, C.; Xu, J.; Kang, E.-J.; Amato, P.; et al. Rapid Mitochondrial DNA Segregation in Primate Preimplantation Embryos Precedes Somatic and Germline Bottleneck. Cell Rep. 2012, 1, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Shitara, H.; Horii, T.; Nagao, Y.; Imai, H.; Abe, K.; Hara, T.; Hayashi, J.-I.; Yonekawa, H. The Mitochondrial Bottleneck Occurs without Reduction of MtDNA Content in Female Mouse Germ Cells. Nat. Genet. 2007, 39, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Burr, S.P.; Chinnery, P.F. The Mitochondrial DNA Genetic Bottleneck: Inheritance and Beyond. Essays Biochem. 2018, 62, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Cherry, A.B.C.; Gagne, K.E.; McLoughlin, E.M.; Baccei, A.; Gorman, B.; Hartung, O.; Miller, J.D.; Zhang, J.; Zon, R.L.; Ince, T.A.; et al. Induced Pluripotent Stem Cells with a Mitochondrial DNA Deletion. Stem Cells 2013, 31, 1287–1297. [Google Scholar] [CrossRef]
- Kodaira, M.; Hatakeyama, H.; Yuasa, S.; Seki, T.; Egashira, T.; Tohyama, S.; Kuroda, Y.; Tanaka, A.; Okata, S.; Hashimoto, H.; et al. Impaired Respiratory Function in MELAS-Induced Pluripotent Stem Cells with High Heteroplasmy Levels. FEBS Open Bio 2015, 5, 219–225. [Google Scholar] [CrossRef]
- Yokota, M.; Hatakeyama, H.; Ono, Y.; Kanazawa, M.; Goto, Y. Mitochondrial Respiratory Dysfunction Disturbs Neuronal and Cardiac Lineage Commitment of Human IPSCs. Cell Death Dis. 2018, 8, e2551. [Google Scholar] [CrossRef]
- Pek, N.M.Q.; Phua, Q.H.; Ho, B.X.; Pang, J.K.S.; Hor, J.-H.; An, O.; Yang, H.H.; Yu, Y.; Fan, Y.; Ng, S.-Y.; et al. Mitochondrial 3243A > G Mutation Confers Pro-Atherogenic and pro-Inflammatory Properties in MELAS IPS Derived Endothelial Cells. Cell Death Dis. 2019, 10, 802. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Yin, J.; Huo, W.; Chaum, E. Modeling of Mitochondrial Bioenergetics and Autophagy Impairment in MELAS-Mutant IPSC-Derived Retinal Pigment Epithelial Cells. Stem Cell Res. Ther. 2022, 13, 260. [Google Scholar] [CrossRef]
- Winanto; Khong, Z.J.; Soh, B.-S.; Fan, Y.; Ng, S.-Y. Organoid Cultures of MELAS Neural Cells Reveal Hyperactive Notch Signaling That Impacts Neurodevelopment. Cell Death Dis. 2020, 11, 182. [Google Scholar] [CrossRef]
- Dong, Z.; Huo, J.; Liang, A.; Chen, J.; Chen, G.; Liu, D. Gamma-Secretase Inhibitor (DAPT), a Potential Therapeutic Target Drug, Caused Neurotoxicity in Planarian Regeneration by Inhibiting Notch Signaling Pathway. Sci. Total Environ. 2021, 781, 146735. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, J.; Lyu, Z.; Chen, Y.; Ji, X.; Cao, H.; Jin, M.; Zhu, J.; Yang, J.; Ling, R.; et al. Positive Feedback Loop between Mitochondrial Fission and Notch Signaling Promotes Survivin-Mediated Survival of TNBC Cells. Cell Death Dis. 2018, 9, 1050. [Google Scholar] [CrossRef]
- Klein Gunnewiek, T.M.; Verboven, A.H.A.; Pelgrim, I.; Hogeweg, M.; Schoenmaker, C.; Renkema, H.; Beyrath, J.; Smeitink, J.; de Vries, B.B.A.; ’t Hoen, P.-B.A.C.; et al. Sonlicromanol Improves Neuronal Network Dysfunction and Transcriptome Changes Linked to m.3243A>G Heteroplasmy in IPSC-Derived Neurons. Stem Cell Rep. 2021, 16, 2197–2212. [Google Scholar] [CrossRef]
- Majamaa-Voltti, K.; Peuhkurinen, K.; Kortelainen, M.-L.; Hassinen, I.E.; Majamaa, K. Cardiac Abnormalities in Patients with Mitochondrial DNA Mutation 3243A>G. BMC Cardiovasc. Disord. 2002, 2, 12. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. The Heart in m.3243A>G Carriers. Herz 2020, 45, 356–361. [Google Scholar] [CrossRef]
- Anan, R.; Nakagawa, M.; Miyata, M.; Higuchi, I.; Nakao, S.; Suehara, M.; Osame, M.; Tanaka, H. Cardiac Involvement in Mitochondrial Diseases. A Study on 17 Patients with Documented Mitochondrial DNA Defects. Circulation 1995, 91, 955–961. [Google Scholar] [CrossRef]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Bobryshev, Y.V.; Orekhov, A.N. Mitochondrial Mutations Are Associated with Atherosclerotic Lesions in the Human Aorta. Clin. Dev. Immunol. 2012, 2012, 832464. [Google Scholar] [CrossRef]
- Sue, C.M.; Mitchell, P.; Crimmins, D.S.; Moshegov, C.; Byrne, E.; Morris, J.G. Pigmentary Retinopathy Associated with the Mitochondrial DNA 3243 Point Mutation. Neurology 1997, 49, 1013–1017. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, H.M.; Carl, S.M.; Swerdlow, R.H. Cytoplasmic Hybrid (Cybrid) Cell Lines as a Practical Model for Mitochondriopathies. Redox Biol. 2014, 2, 619–631. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The Hallmarks of Cancer Metabolism: Still Emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef]
- King, M.P.; Koga, Y.; Davidson, M.; Schon, E.A. Defects in Mitochondrial Protein Synthesis and Respiratory Chain Activity Segregate with the TRNA(Leu(UUR)) Mutation Associated with Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes. Mol. Cell. Biol. 1992, 12, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Schon, E.A.; Koga, Y.; Davidson, M.; Moraes, C.T.; King, M.P. The Mitochondrial TRNA(Leu)(UUR)) Mutation in MELAS: A Model for Pathogenesis. Biochim. Biophys. Acta 1992, 1101, 206–209. [Google Scholar] [PubMed]
- de Andrade, P.B.M.; Rubi, B.; Frigerio, F.; van den Ouweland, J.M.W.; Maassen, J.A.; Maechler, P. Diabetes-Associated Mitochondrial DNA Mutation A3243G Impairs Cellular Metabolic Pathways Necessary for Beta Cell Function. Diabetologia 2006, 49, 1816–1826. [Google Scholar] [CrossRef]
- McMillan, R.P.; Stewart, S.; Budnick, J.A.; Caswell, C.C.; Hulver, M.W.; Mukherjee, K.; Srivastava, S. Quantitative Variation in m.3243A > G Mutation Produce Discrete Changes in Energy Metabolism. Sci. Rep. 2019, 9, 5752. [Google Scholar] [CrossRef]
- DiFrancesco, J.C.; Cooper, J.M.; Lam, A.; Hart, P.E.; Tremolizzo, L.; Ferrarese, C.; Schapira, A.H. MELAS Mitochondrial DNA Mutation A3243G Reduces Glutamate Transport in Cybrids Cell Lines. Exp. Neurol. 2008, 212, 152–156. [Google Scholar] [CrossRef]
- Meseguer, S.; Panadero, J.; Navarro-González, C.; Villarroya, M.; Boutoual, R.; Comi, G.P.; Armengod, M.-E. The MELAS Mutation m.3243A>G Promotes Reactivation of Fetal Cardiac Genes and an Epithelial-Mesenchymal Transition-like Program via Dysregulation of MiRNAs. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2018, 1864, 3022–3037. [Google Scholar] [CrossRef]
- Burgin, H.J.; Lopez Sanchez, M.I.G.; Smith, C.M.; Trounce, I.A.; McKenzie, M. Pioglitazone and Deoxyribonucleoside Combination Treatment Increases Mitochondrial Respiratory Capacity in m.3243A>G MELAS Cybrid Cells. Int. J. Mol. Sci. 2020, 21, 2139. [Google Scholar] [CrossRef] [PubMed]
- Capriglia, F.; Rizzo, F.; Petrosillo, G.; Morea, V.; d’Amati, G.; Cantatore, P.; Roberti, M.; Loguercio Polosa, P.; Bruni, F. Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype. Life 2021, 11, 674. [Google Scholar] [CrossRef]
- Perli, E.; Fiorillo, A.; Giordano, C.; Pisano, A.; Montanari, A.; Grazioli, P.; Campese, A.F.; Di Micco, P.; Tuppen, H.A.; Genovese, I.; et al. Short Peptides from Leucyl-TRNA Synthetase Rescue Disease-Causing Mitochondrial TRNA Point Mutations. Hum. Mol. Genet. 2016, 25, 903–915. [Google Scholar] [CrossRef] [PubMed]
- Jahangir Tafrechi, R.S.; Svensson, P.J.; Janssen, G.M.C.; Szuhai, K.; Maassen, J.A.; Raap, A.K. Distinct Nuclear Gene Expression Profiles in Cells with MtDNA Depletion and Homoplasmic A3243G Mutation. Mutat. Res. 2005, 578, 43–52. [Google Scholar] [CrossRef]
- Aras, S.; Purandare, N.; Gladyck, S.; Somayajulu-Nitu, M.; Zhang, K.; Wallace, D.C.; Grossman, L.I. Mitochondrial Nuclear Retrograde Regulator 1 (MNRR1) Rescues the Cellular Phenotype of MELAS by Inducing Homeostatic Mechanisms. Proc. Natl. Acad. Sci. USA 2020, 117, 32056–32065. [Google Scholar] [CrossRef] [PubMed]
- Shu, L.; Hu, C.; Xu, M.; Yu, J.; He, H.; Lin, J.; Sha, H.; Lu, B.; Engelender, S.; Guan, M.; et al. ATAD3B Is a Mitophagy Receptor Mediating Clearance of Oxidative Stress-Induced Damaged Mitochondrial DNA. EMBO J. 2021, 40, e106283. [Google Scholar] [CrossRef]
- Meseguer, S.; Martínez-Zamora, A.; García-Arumí, E.; Andreu, A.L.; Armengod, M.-E. The ROS-Sensitive MicroRNA-9/9* Controls the Expression of Mitochondrial TRNA-Modifying Enzymes and Is Involved in the Molecular Mechanism of MELAS Syndrome. Hum. Mol. Genet. 2015, 24, 167–184. [Google Scholar] [CrossRef]
- Meseguer, S.; Navarro-González, C.; Panadero, J.; Villarroya, M.; Boutoual, R.; Sánchez-Alcázar, J.A.; Armengod, M.-E. The MELAS Mutation m.3243A>G Alters the Expression of Mitochondrial TRNA Fragments. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1433–1449. [Google Scholar] [CrossRef]
- Belal, S.; Goudenège, D.; Bocca, C.; Dumont, F.; Chao De La Barca, J.M.; Desquiret-Dumas, V.; Gueguen, N.; Geffroy, G.; Benyahia, R.; Kane, S.; et al. Glutamate-Induced Deregulation of Krebs Cycle in Mitochondrial Encephalopathy Lactic Acidosis Syndrome Stroke-Like Episodes (MELAS) Syndrome Is Alleviated by Ketone Body Exposure. Biomedicines 2022, 10, 1665. [Google Scholar] [CrossRef]
- Desquiret-Dumas, V.; Gueguen, N.; Barth, M.; Chevrollier, A.; Hancock, S.; Wallace, D.C.; Amati-Bonneau, P.; Henrion, D.; Bonneau, D.; Reynier, P.; et al. Metabolically Induced Heteroplasmy Shifting and L-Arginine Treatment Reduce the Energetic Defect in a Neuronal-like Model of MELAS. Biochim. Biophys. Acta 2012, 1822, 1019–1029. [Google Scholar] [CrossRef]
- Frey, S.; Geffroy, G.; Desquiret-Dumas, V.; Gueguen, N.; Bris, C.; Belal, S.; Amati-Bonneau, P.; Chevrollier, A.; Barth, M.; Henrion, D.; et al. The Addition of Ketone Bodies Alleviates Mitochondrial Dysfunction by Restoring Complex I Assembly in a MELAS Cellular Model. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Besse, A.; Wu, P.; Bruni, F.; Donti, T.; Graham, B.H.; Craigen, W.J.; McFarland, R.; Moretti, P.; Lalani, S.; Scott, K.L.; et al. The GABA Transaminase, ABAT, Is Essential for Mitochondrial Nucleoside Metabolism. Cell Metab. 2015, 21, 417–427. [Google Scholar] [CrossRef]
- Davidson, M.M.; Walker, W.F.; Hernandez-Rosa, E. The m.3243A>G MtDNA Mutation Is Pathogenic in an in Vitro Model of the Human Blood Brain Barrier. Mitochondrion 2009, 9, 463–470. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Cell Type/Phenotype | Fibroblasts | iPSCs | Neurons | Cardiac Cells |
|---|---|---|---|---|
| Metabolism | Decreased respiration. Increased glycolytic activity [40]. | Decreased respiration in some patients [33,37,44]. | Decreased respiration in some studies. Increased glycolytic activity [45]. | Decreased respiration. Increased glycolytic activity [38,46]. |
| Mitochondrial effects | Decreased membrane potential [40,41]. CI and CIV deficiency [35,40,41,42,43]. | Membrane potential not affected. CI deficiency [33,44]. | CI deficiency [34] | Membrane potential not affected. CI deficiency [38]. |
| Cellular energy | Decreased ATP [40,41]. | Not affected [37]. | n.d | Decreased ATP [38]. |
| Functional defects | Reduced growth rate [40]. | n.d | Increased spontaneous activity, decreased synchronous activity, decreased excitatory synapses, and dendritic complexity [45]. | Changes in calcium signaling [38]. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ryytty, S.; Hämäläinen, R.H. The Mitochondrial m.3243A>G Mutation on the Dish, Lessons from In Vitro Models. Int. J. Mol. Sci. 2023, 24, 13478. https://doi.org/10.3390/ijms241713478
Ryytty S, Hämäläinen RH. The Mitochondrial m.3243A>G Mutation on the Dish, Lessons from In Vitro Models. International Journal of Molecular Sciences. 2023; 24(17):13478. https://doi.org/10.3390/ijms241713478
Chicago/Turabian StyleRyytty, Sanna, and Riikka H. Hämäläinen. 2023. "The Mitochondrial m.3243A>G Mutation on the Dish, Lessons from In Vitro Models" International Journal of Molecular Sciences 24, no. 17: 13478. https://doi.org/10.3390/ijms241713478
APA StyleRyytty, S., & Hämäläinen, R. H. (2023). The Mitochondrial m.3243A>G Mutation on the Dish, Lessons from In Vitro Models. International Journal of Molecular Sciences, 24(17), 13478. https://doi.org/10.3390/ijms241713478