The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome



1. Introduction
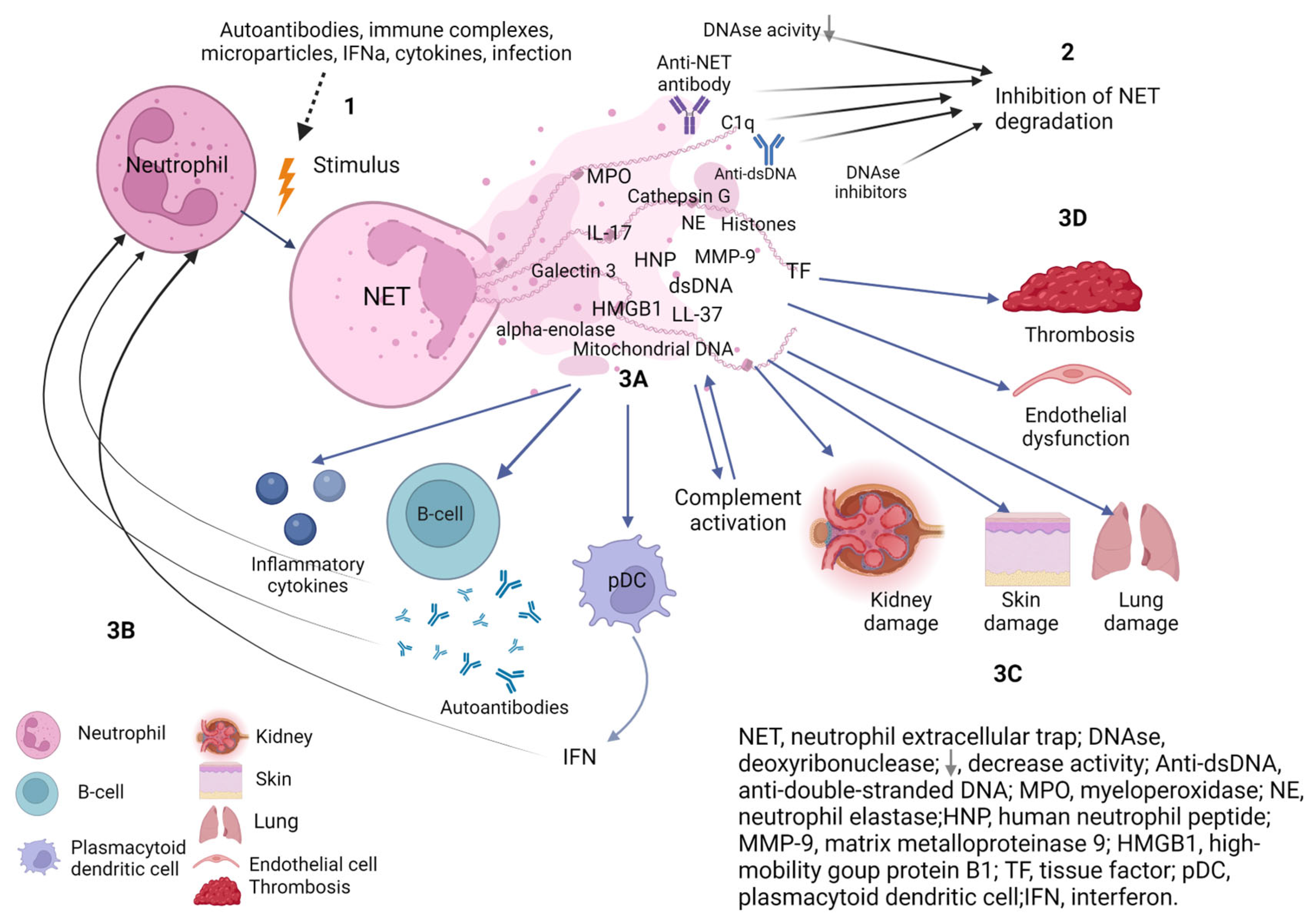
2. NETosis in SLE
2.1. NETs Formation in SLE
2.1.1. Spontaneous and Stimulated Formation of NETs by Neutrophils of SLE Patients
2.1.2. Peptidyl Arginine Deiminase 4 (PAD4) in SLE
2.1.3. NOX-2 NADPH-Oxidase in SLE
2.1.4. Mitochondria and NETs
Mitochondrial ROS (mtROS)
Mitochondrial DNA (mtDNA)
2.2. NETs Degradation in SLE
2.3. NETs in SLE
2.3.1. NETs and B-Cells
2.3.2. NETs and IFN
2.3.3. NETs and Complement System
2.3.4. NETs and Tissue Damage
NETs and Endothelial Dysfunction
NETs in the Affected Tissues
2.4. NETosis Markers in Peripheral Blood in SLE
2.4.1. The MPO-DNA Complex
2.4.2. Cell-Free DNA (cfDNA)
2.5. Blockade of NETs as a Therapeutic Strategy in SLE
2.6. NETs and Thrombosis in SLE
3. NETs in Thrombosis
4. NETs in APS
4.1. Spontaneous and Induced NETosis in APS
4.2. NETs Degradation in APS
4.3. NETosis Markers in Peripheral Blood in APS
4.4. Mechanism of NETs Effect on the Hemostasis System in APS
4.5. Blockade of NETs as a Therapeutic Strategy in APS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nasonov, E.L.; Reshetnyak, T.M.; Solovyev, S.K.; Popkova, T.V. Systemic lupus erythematosus and antiphospholipid syndrome: Past, present, future. Ter. Arkhiv 2023, 95, 365–374. [Google Scholar] [CrossRef]
- Reshetnyak, T.M.; Cheldieva, F.A.; Nurbayeva, K.S.; Lila, A.M.; Nasonov, E.L. Antiphospholipid syndrome: Diagnosis, development mechanism, therapy issues. Thromb. Hemost. Rheol. 2020, 4, 4–21. (In Russian) [Google Scholar] [CrossRef]
- Gerhardsson, J.; Sundelin, B.; Zickert, A.; Padyukov, L.; Svenungsson, E.; Gunnarsson, I. Histological antiphospholipid-associated nephropathy versus lupus nephritis in patients with systemic lupus erythematosus: An observational cross-sectional study with longitudinal follow-up. Arthritis Res. Ther. 2015, 17, 109. [Google Scholar] [CrossRef] [PubMed]
- Montero-Olvera, P.R.; Berebichez-Fridman, R.; Velázquez-Álvarez, L.; Ríos-Morales, J.R.; Rodríguez-Guiza, M.A. Late diagnosis of systemic lupus erythematosus and antiphospholipid syndrome in an older woman with psychosis: A case report and review of the literature. Clin. Case Rep. 2017, 5, 1819–1825. [Google Scholar] [CrossRef]
- Meroni, P.L.; Tsokos, G.C. Editorial: Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 199. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, L.L.; Bisoendial, R.J. B-Cells and BAFF in Primary Antiphospholipid Syndrome, Targets for Therapy? J. Clin. Med. 2022, 12, 18. [Google Scholar] [CrossRef]
- Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. [Google Scholar] [CrossRef]
- Pérez, D.; Stojanovich, L.; Naranjo, L.; Stanisavljevic, N.; Bogdanovic, G.; Serrano, M.; Serrano, A. Presence of Immune Complexes of IgG/IgM Bound to B2-glycoprotein I Is Associated With Non-criteria Clinical Manifestations in Patients With Antiphospholipid Syndrome. Front. Immunol. 2018, 9, 2644. [Google Scholar] [CrossRef]
- Naranjo, L.; Stojanovich, L.; Djokovic, A.; Andreoli, L.; Tincani, A.; Maślińska, M.; Sciascia, S.; Infantino, M.; Garcinuño, S.; Kostyra-Grabczak, K.; et al. Circulating immune-complexes of IgG/IgM bound to B2-glycoprotein-I associated with complement consumption and thrombocytopenia in antiphospholipid syndrome. Front. Immunol. 2022, 13, 957201. [Google Scholar] [CrossRef]
- Bonegio, R.G.; Lin, J.D.; Beaudette-Zlatanova, B.; York, M.R.; Menn-Josephy, H.; Yasuda, K. Lupus-Associated Immune Complexes Activate Human Neutrophils in an FcγRIIA-Dependent but TLR-Independent Response. J. Immunol. 2019, 202, 675–683. [Google Scholar] [CrossRef]
- Menachem, A.; Chapman, J.; Katzav, A. Significant Changes in the Levels of Secreted Cytokines in Brains of Experimental Antiphospholipid Syndrome Mice. Autoimmune Dis. 2012, 2012, 404815. [Google Scholar] [CrossRef] [PubMed]
- Ohl, K.; Tenbrock, K. Inflammatory Cytokines in Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2011, 2011, 432595. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, S.; Braunstein, E.M.; Brodsky, R.A. Antiphospholipid syndrome: Complement activation, complement gene mutations, and therapeutic implications. J. Thromb. Haemost. 2021, 19, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.; Alexander, R.V.; Zack, D.J. A Review of Complement Activation in SLE. Curr. Rheumatol. Rep. 2021, 23, 16. [Google Scholar] [CrossRef]
- Grenn, R.C.; Yalavarthi, S.; Gandhi, A.A.; Kazzaz, N.M.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Cabral, A.R.; McCune, W.J.; Bockenstedt, P.L.; Knight, J.S. Endothelial progenitor dysfunction associates with a type I interferon signature in primary antiphospholipid syndrome. Ann. Rheum. Dis. 2017, 76, 450–457. [Google Scholar] [CrossRef]
- Palli, E.; Kravvariti, E.; Tektonidou, M.G. Type I Interferon Signature in Primary Antiphospholipid Syndrome: Clinical and Laboratory Associations. Front. Immunol. 2019, 10, 487. [Google Scholar] [CrossRef]
- Sim, T.M.; Ong, S.J.; Mak, A.; Tay, S.H. Type I Interferons in Systemic Lupus Erythematosus: A Journey from Bench to Bedside. Int. J. Mol. Sci. 2022, 23, 2505. [Google Scholar] [CrossRef]
- Wirestam, L.; Arve, S.; Linge, P.; Bengtsson, A.A. Neutrophils—Important Communicators in Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Front. Immunol. 2019, 10, 2734. [Google Scholar] [CrossRef]
- Quintero-González, D.C.; Muñoz-Urbano, M.; Vásquez, G. Mitochondria as a key player in systemic lupus erythematosus. Autoimmunity 2022, 55, 497–505. [Google Scholar] [CrossRef]
- Wang, M.; Ishikawa, T.; Lai, Y.; Nallapothula, D.; Singh, R.R. Diverse Roles of NETosis in the Pathogenesis of Lupus. Front. Immunol. 2022, 13, 895216. [Google Scholar] [CrossRef]
- Yalavarthi, S.; Gould, T.J.; Rao, A.N.; Mazza, L.F.; Morris, A.E.; Núñez-Álvarez, C.; Hernández-Ramírez, D.; Bockenstedt, P.L.; Liaw, P.C.; Cabral, A.R.; et al. Release of Neutrophil Extracellular Traps by Neutrophils Stimulated with Antiphospholipid Antibodies: A Newly Identified Mechanism of Thrombosis in the Antiphospholipid Syndrome. Arthritis Rheumatol. 2015, 67, 2990–3003. [Google Scholar] [CrossRef]
- Vorobjeva, N.V.; Chernyak, B.V. NETosis: Molecular Mechanisms, Role in Physiology and Pathology. Biochemistry 2020, 85, 1178–1190. [Google Scholar] [CrossRef] [PubMed]
- Schultz, B.M.; Acevedo, O.A.; Kalergis, A.M.; Bueno, S.M. Role of Extracellular Trap Release during Bacterial and Viral Infection. Front. Microbiol. 2022, 13, 798853. [Google Scholar] [CrossRef] [PubMed]
- Mutua, V.; Gershwin, L.J. A Review of Neutrophil Extracellular Traps (NETs) in Disease: Potential Anti-NETs Therapeutics. Clin. Rev. Allergy Immunol. 2020, 61, 194–211. [Google Scholar] [CrossRef]
- Kaplan, M.J.; Radic, M. Neutrophil Extracellular Traps: Double-Edged Swords of Innate Immunity. J. Immunol. 2012, 189, 2689–2695. [Google Scholar] [CrossRef] [PubMed]
- Ulfig, A.; Leichert, L.I. The effects of neutrophil-generated hypochlorous acid and other hypohalous acids on host and pathogens. Cell. Mol. Life Sci. 2020, 78, 385–414. [Google Scholar] [CrossRef]
- Huang, J.; Hong, W.; Wan, M.; Zheng, L. Molecular mechanisms and therapeutic target of NETosis in diseases. Medcomm 2022, 3, e162. [Google Scholar] [CrossRef]
- Villanueva, E.; Yalavarthi, S.; Berthier, C.C.; Hodgin, J.B.; Khandpur, R.; Lin, A.M.; Rubin, C.J.; Zhao, W.; Olsen, S.H.; Klinker, M.; et al. Netting Neutrophils Induce Endothelial Damage, Infiltrate Tissues, and Expose Immunostimulatory Molecules in Systemic Lupus Erythematosus. J. Immunol. 2011, 187, 538–552. [Google Scholar] [CrossRef]
- Leffler, J.; Martin, M.; Gullstrand, B.; Tydén, H.; Lood, C.; Truedsson, L.; Bengtsson, A.A.; Blom, A.M. Neutrophil Extracellular Traps That Are Not Degraded in Systemic Lupus Erythematosus Activate Complement Exacerbating the Disease. J. Immunol. 2012, 188, 3522–3531. [Google Scholar] [CrossRef]
- Lande, R.; Ganguly, D.; Facchinetti, V.; Frasca, L.; Conrad, C.; Gregorio, J.; Meller, S.; Chamilos, G.; Sebasigari, R.; Riccieri, V.; et al. Neutrophils Activate Plasmacytoid Dendritic Cells by Releasing Self-DNA–Peptide Complexes in Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra19. [Google Scholar] [CrossRef]
- Kienhöfer, D.; Hahn, J.; Stoof, J.; Csepregi, J.Z.; Reinwald, C.; Urbonaviciute, V.; Johnsson, C.; Maueröder, C.; Podolska, M.J.; Biermann, M.H.; et al. Experimental lupus is aggravated in mouse strains with impaired induction of neutrophil extracellular traps. JCI Insight 2017, 2, e92920. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Romo, G.S.; Caielli, S.; Vega, B.; Connolly, J.; Allantaz, F.; Xu, Z.; Punaro, M.; Baisch, J.; Guiducci, C.; Coffman, R.L.; et al. Netting Neutrophils Are Major Inducers of Type I IFN Production in Pediatric Systemic Lupus Erythematosus. Sci. Transl. Med. 2011, 3, 73ra20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-Y.; Wang, C.-T.; Chen, C.-Y.; Kuo, P.-Y.; Wang, C.-R.; Shiau, A.-L.; Chang, C.-H.; Wu, C.-L. Galectin-3 Mediates NETosis and Acts as an Autoantigen in Systemic Lupus Erythematosus-Associated Diffuse Alveolar Haemorrhage. Int. J. Mol. Sci. 2023, 24, 9493. [Google Scholar] [CrossRef]
- Kraaij, T.; Kamerling, S.W.; de Rooij, E.N.; van Daele, P.L.; Bredewold, O.W.; Bakker, J.A.; Bajema, I.M.; Scherer, H.U.; Toes, R.E.; Huizinga, T.J.; et al. The NET-effect of combining rituximab with belimumab in severe systemic lupus erythematosus. J. Autoimmun. 2018, 91, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Frangou, E.; Chrysanthopoulou, A.; Mitsios, A.; Kambas, K.; Arelaki, S.; Angelidou, I.; Arampatzioglou, A.; Gakiopoulou, H.; Bertsias, G.K.; Verginis, P.; et al. REDD1/autophagy pathway promotes thromboinflammation and fibrosis in human systemic lupus erythematosus (SLE) through NETs decorated with tissue factor (TF) and interleukin-17A (IL-17A). Ann. Rheum. Dis. 2019, 78, 238–248. [Google Scholar] [CrossRef]
- Knight, J.S.; Zhao, W.; Luo, W.; Subramanian, V.; O’dell, A.A.; Yalavarthi, S.; Hodgin, J.B.; Eitzman, D.T.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition is immunomodulatory and vasculoprotective in murine lupus. J. Clin. Investig. 2013, 123, 2981–2993. [Google Scholar] [CrossRef]
- Knight, J.S.; Subramanian, V.; O’Dell, A.A.; Yalavarthi, S.; Zhao, W.; Smith, C.K.; Hodgin, J.B.; Thompson, P.R.; Kaplan, M.J. Peptidylarginine deiminase inhibition disrupts NET formation and protects against kidney, skin and vascular disease in lupus-prone MRL/lpr mice. Ann. Rheum. Dis. 2015, 74, 2199–2206. [Google Scholar] [CrossRef]
- Van Avondt, K.; Fritsch-Stork, R.; Derksen, R.H.W.M.; Meyaard, L. Ligation of Signal Inhibitory Receptor on Leukocytes-1 Suppresses the Release of Neutrophil Extracellular Traps in Systemic Lupus Erythematosus. PLoS ONE 2013, 8, e78459. [Google Scholar] [CrossRef]
- Gestermann, N.; Di Domizio, J.; Lande, R.; Demaria, O.; Frasca, L.; Feldmeyer, L.; Di Lucca, J.; Gilliet, M. Netting Neutrophils Activate Autoreactive B Cells in Lupus. J. Immunol. 2018, 200, 3364–3371. [Google Scholar] [CrossRef]
- Sule, G.; Abuaita, B.H.; Steffes, P.A.; Fernandes, A.T.; Estes, S.K.; Dobry, C.; Pandian, D.; Gudjonsson, J.E.; Kahlenberg, J.M.; O’riordan, M.X.; et al. Endoplasmic reticulum stress sensor IRE1α propels neutrophil hyperactivity in lupus. J. Clin. Investig. 2021, 131, e137866. [Google Scholar] [CrossRef]
- Carmona-Rivera, C.; Zhao, W.; Yalavarthi, S.; Kaplan, M.J. Neutrophil extracellular traps induce endothelial dysfunction in systemic lupus erythematosus through the activation of matrix metalloproteinase-2. Ann. Rheum. Dis. 2015, 74, 1417–1424. [Google Scholar] [CrossRef] [PubMed]
- van der Linden, M.; van den Hoogen, L.L.; Westerlaken, G.H.A.; Fritsch-Stork, R.D.E.; van Roon, J.A.G.; Radstake, T.R.D.J.; Meyaard, L. Neutrophil extracellular trap release is associated with antinuclear antibodies in systemic lupus erythematosus and anti-phospholipid syndrome. Rheumatology 2018, 57, 1228–1234. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Shida, H.; Tomaru, U.; Yoshida, M.; Nishio, S.; Atsumi, T.; Ishizu, A. Enhanced Formation and Disordered Regulation of NETs in Myeloperoxidase-ANCA–Associated Microscopic Polyangiitis. J. Am. Soc. Nephrol. 2014, 25, 990–997. [Google Scholar] [CrossRef] [PubMed]
- van Dam, L.S.; Kraaij, T.; Kamerling, S.W.A.; Bakker, J.A.; Scherer, U.H.; Rabelink, T.J.; van Kooten, C.; Teng, Y.K.O. Intrinsically Distinct Role of Neutrophil Extracellular Trap Formation in Antineutrophil Cytoplasmic Antibody–Associated Vasculitis Compared to Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 2047–2058. [Google Scholar] [CrossRef] [PubMed]
- El-Ghoneimy, D.H.; Hesham, M.; Hasan, R.; Tarif, M.; Gouda, S. The behavior of neutrophil extracellular traps and NADPH oxidative activity in pediatric systemic lupus erythematosus: Relation to disease activity and lupus nephritis. Clin. Rheumatol. 2019, 38, 2585–2593. [Google Scholar] [CrossRef]
- Bruschi, M.; Bonanni, A.; Petretto, A.; Vaglio, A.; Pratesi, F.; Santucci, L.; Migliorini, P.; Bertelli, R.; Galetti, M.; Belletti, S.; et al. Neutrophil Extracellular Traps Profiles in Patients with Incident Systemic Lupus Erythematosus and Lupus Nephritis. J. Rheumatol. 2020, 47, 377–386. [Google Scholar] [CrossRef]
- Thiam, H.R.; Wong, S.L.; Wagner, D.D.; Waterman, C.M. Cellular Mechanisms of NETosis. Annu. Rev. Cell Dev. Biol. 2020, 36, 191–218. [Google Scholar] [CrossRef]
- Liu, Y.; Lightfoot, Y.L.; Seto, N.; Carmona-Rivera, C.; Moore, E.; Goel, R.; O’neil, L.; Mistry, P.; Hoffmann, V.; Mondal, S.; et al. Peptidylarginine deiminases 2 and 4 modulate innate and adaptive immune responses in TLR-7–dependent lupus. JCI Insight 2018, 3, e124729. [Google Scholar] [CrossRef]
- Hanata, N.; Shoda, H.; Hatano, H.; Nagafuchi, Y.; Komai, T.; Okamura, T.; Suzuki, A.; Gunarta, I.K.; Yoshioka, K.; Yamamoto, K.; et al. Peptidylarginine Deiminase 4 Promotes the Renal Infiltration of Neutrophils and Exacerbates the TLR7 Agonist-Induced Lupus Mice. Front. Immunol. 2020, 11, 1095. [Google Scholar] [CrossRef]
- Saisorn, W.; Saithong, S.; Phuengmaung, P.; Udompornpitak, K.; Bhunyakarnjanarat, T.; Visitchanakun, P.; Chareonsappakit, A.; Pisitkun, P.; Chiewchengchol, D.; Leelahavanichkul, A. Acute Kidney Injury Induced Lupus Exacerbation Through the Enhanced Neutrophil Extracellular Traps (and Apoptosis) in Fcgr2b Deficient Lupus Mice With Renal Ischemia Reperfusion Injury. Front. Immunol. 2021, 12, 669162. [Google Scholar] [CrossRef]
- Gordon, R.A.; Herter, J.M.; Rosetti, F.; Campbell, A.M.; Nishi, H.; Kashgarian, M.; Bastacky, S.I.; Marinov, A.; Nickerson, K.M.; Mayadas, T.N.; et al. Lupus and proliferative nephritis are PAD4 independent in murine models. JCI Insight 2017, 2, e92926. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.M.; Kashgarian, M.; Shlomchik, M.J. NADPH Oxidase Inhibits the Pathogenesis of Systemic Lupus Erythematosus. Sci. Transl. Med. 2012, 4, 157ra141. [Google Scholar] [CrossRef]
- Hultqvist, M.; Olsson, L.M.; Gelderman, K.A.; Holmdahl, R. The protective role of ROS in autoimmune disease. Trends Immunol. 2009, 30, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.; Blanco, L.P.; Purmalek, M.M.; Carmona-Rivera, C.; De Ravin, S.S.; Smith, C.K.; Malech, H.L.; Ledbetter, J.A.; Elkon, K.B.; Kaplan, M.J. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat. Med. 2016, 22, 146–153. [Google Scholar] [CrossRef] [PubMed]
- A Fortner, K.; Blanco, L.P.; Buskiewicz, I.; Huang, N.; Gibson, P.C.; Cook, D.L.; Pedersen, H.L.; Yuen, P.S.T.; Murphy, M.P.; Perl, A.; et al. Targeting mitochondrial oxidative stress with MitoQ reduces NET formation and kidney disease in lupus-prone MRL-lpr mice. Lupus Sci. Med. 2020, 7, e000387. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef]
- Demkow, U. Molecular Mechanisms of Neutrophil Extracellular Trap (NETs) Degradation. Int. J. Mol. Sci. 2023, 24, 4896. [Google Scholar] [CrossRef]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Al Abrawi, S.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Skiljevic, D.; Jeremic, I.; Nikolic, M.; Andrejevic, S.; Sefik-Bukilica, M.; Stojimirovic, B.; Bonaci-Nikolic, B. Serum DNase I activity in systemic lupus erythematosus: Correlation with immunoserological markers, the disease activity and organ involvement. Clin. Chem. Lab. Med. 2013, 51, 1083–1091. [Google Scholar] [CrossRef]
- Hartl, J.; Serpas, L.; Wang, Y.; Rashidfarrokhi, A.; Perez, O.A.; Sally, B.; Sisirak, V.; Soni, C.; Khodadadi-Jamayran, A.; Tsirigos, A.; et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J. Exp. Med. 2021, 218, e20201138. [Google Scholar] [CrossRef]
- Pruchniak, M.P.; Ostafin, M.; Wachowska, M.; Jakubaszek, M.; Kwiatkowska, B.; Olesinska, M.; Zycinska, K.; Demkow, U. Neutrophil extracellular traps generation and degradation in patients with granulomatosis with polyangiitis and systemic lupus erythematosus. Autoimmunity 2019, 52, 126–135. [Google Scholar] [CrossRef]
- Kenny, E.F.; Raupach, B.; Abu Abed, U.; Brinkmann, V.; Zychlinsky, A. Dnase1-deficient mice spontaneously develop a systemic lupus erythematosus-like disease. Eur. J. Immunol. 2019, 49, 590–599. [Google Scholar] [CrossRef]
- Sisirak, V.; Sally, B.; D’agati, V.; Martinez-Ortiz, W.; Özçakar, Z.B.; David, J.; Rashidfarrokhi, A.; Yeste, A.; Panea, C.; Chida, A.S.; et al. Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 2016, 166, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Hakkim, A.; Fürnrohr, B.G.; Amann, K.; Laube, B.; Abed, U.A.; Brinkmann, V.; Herrmann, M.; Voll, R.E.; Zychlinsky, A. Impairment of neutrophil extracellular trap degradation is associated with lupus nephritis. Proc. Natl. Acad. Sci. USA 2010, 107, 9813–9818. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Gullstrand, B.; Jönsen, A.; Nilsson, J.; Martin, M.; Blom, A.M.; Bengtsson, A.A. Degradation of neutrophil extracellular traps co-varies with disease activity in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2013, 15, R84. [Google Scholar] [CrossRef] [PubMed]
- Leffler, J.; Ciacma, K.; Gullstrand, B.; Bengtsson, A.A.; Martin, M.; Blom, A.M. A subset of patients with systemic lupus erythematosus fails to degrade DNA from multiple clinically relevant sources. Arthritis Res. Ther. 2015, 17, 205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Lu, X.; Shu, X.; Tian, X.; Yang, H.; Yang, W.; Zhang, Y.; Wang, G. Elevated Plasma cfDNA May be Associated with Active Lupus Nephritis and Partially Attributed to Abnormal Regulation of Neutrophil Extracellular Traps (NETs) in Patients with Systemic Lupus Erythematosus. Intern. Med. 2014, 53, 2763–2771. [Google Scholar] [CrossRef]
- Rother, N.; Pieterse, E.; Lubbers, J.; Hilbrands, L.; van der Vlag, J. Acetylated Histones in Apoptotic Microparticles Drive the Formation of Neutrophil Extracellular Traps in Active Lupus Nephritis. Front. Immunol. 2017, 8, 1136. [Google Scholar] [CrossRef]
- Bruschi, M.; Petretto, A.; Santucci, L.; Vaglio, A.; Pratesi, F.; Migliorini, P.; Bertelli, R.; Lavarello, C.; Bartolucci, M.; Candiano, G.; et al. Neutrophil Extracellular Traps protein composition is specific for patients with Lupus nephritis and includes methyl-oxidized αenolase (methionine sulfoxide 93). Sci. Rep. 2019, 9, 7934. [Google Scholar] [CrossRef]
- Bruschi, M.; Sinico, R.A.; Moroni, G.; Pratesi, F.; Migliorini, P.; Galetti, M.; Murtas, C.; Tincani, A.; Madaio, M.; Radice, A.; et al. Glomerular Autoimmune Multicomponents of Human Lupus Nephritis In Vivo: α-enolase and annexin AI. J. Am. Soc. Nephrol. 2014, 25, 2483–2498. [Google Scholar] [CrossRef]
- Bruschi, M.; Moroni, G.; Sinico, R.A.; Franceschini, F.; Fredi, M.; Vaglio, A.; Cavagna, L.; Petretto, A.; Pratesi, F.; Migliorini, P.; et al. Neutrophil Extracellular Traps in the Autoimmunity Context. Front. Med. 2021, 8, 614829. [Google Scholar] [CrossRef] [PubMed]
- Reshetnyak, T.; Nurbaeva, K.; Ptashnik, I.; Kudriaeva, A.; Belogurov, A., Jr.; Lila, A.; Nasonov, E. Markers of NETosis in Patients with Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Int. J. Mol. Sci. 2023, 24, 9210. [Google Scholar] [CrossRef] [PubMed]
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320. [Google Scholar] [CrossRef] [PubMed]
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.; Berden, J.H.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379. [Google Scholar] [CrossRef]
- Lin, H.; Liu, J.; Li, N.; Zhang, B.; Nguyen, V.D.; Yao, P.; Feng, J.; Liu, Q.; Chen, Y.; Li, G.; et al. NETosis promotes chronic inflammation and fibrosis in systemic lupus erythematosus and COVID-19. Clin. Immunol. 2023, 254, 109687. [Google Scholar] [CrossRef]
- Donkel, S.J.; Wolters, F.J.; Ikram, M.A.; de Maat, M.P.M. Circulating Myeloperoxidase (MPO)-DNA complexes as marker for Neutrophil Extracellular Traps (NETs) levels and the association with cardiovascular risk factors in the general population. PLoS ONE 2021, 16, e0253698. [Google Scholar] [CrossRef]
- Moore, S.; Juo, H.-H.; Nielsen, C.T.; Tyden, H.; Bengtsson, A.A.; Lood, C. Role of Neutrophil Extracellular Traps Regarding Patients at Risk of Increased Disease Activity and Cardiovascular Comorbidity in Systemic Lupus Erythematosus. J. Rheumatol. 2020, 47, 1652–1660. [Google Scholar] [CrossRef]
- Hanata, N.; Ota, M.; Tsuchida, Y.; Nagafuchi, Y.; Okamura, T.; Shoda, H.; Fujio, K. Serum extracellular traps associate with the activation of myeloid cells in SLE patients with the low level of anti-DNA antibodies. Sci. Rep. 2022, 12, 18397. [Google Scholar] [CrossRef]
- Duvvuri, B.; Lood, C. Cell-Free DNA as a Biomarker in Autoimmune Rheumatic Diseases. Front. Immunol. 2019, 10, 502. [Google Scholar] [CrossRef]
- Atamaniuk, J.; Hsiao, Y.-Y.; Mustak, M.; Bernhard, D.; Erlacher, L.; Fodinger, M.; Tiran, B.; Stuhlmeier, K.M. Analysing cell-free plasma DNA and SLE disease activity. Eur. J. Clin. Investig. 2011, 41, 579–583. [Google Scholar] [CrossRef]
- Tug, S.; Helmig, S.; Menke, J.; Zahn, D.; Kubiak, T.; Schwarting, A.; Simon, P. Correlation between cell free DNA levels and medical evaluation of disease progression in systemic lupus erythematosus patients. Cell. Immunol. 2014, 292, 32–39. [Google Scholar] [CrossRef]
- Hendy, O.M.; Abdel-Motalib, T.; El Shafie, M.A.; Khalaf, F.A.; Kotb, S.E.; Khalil, A.; Ali, S.R. Circulating cell free DNA as a predictor of systemic lupus erythematosus severity and monitoring of therapy. Egypt. J. Med. Hum. Genet. 2016, 17, 79–85. [Google Scholar] [CrossRef]
- Abdelal, I.T.; Zakaria, M.A.; Sharaf, D.M.; Elakad, G.M. Levels of plasma cell-free DNA and its correlation with disease activity in rheumatoid arthritis and systemic lupus erythematosus patients. Egypt. Rheumatol. 2016, 38, 295–300. [Google Scholar] [CrossRef]
- Xu, Y.; Song, Y.; Chang, J.; Zhou, X.; Qi, Q.; Tian, X.; Li, M.; Zeng, X.; Xu, M.; Zhang, W.; et al. High levels of circulating cell-free DNA are a biomarker of active SLE. Eur. J. Clin. Investig. 2018, 48, e13015. [Google Scholar] [CrossRef] [PubMed]
- Furumoto, Y.; Smith, C.K.; Blanco, L.; Zhao, W.; Brooks, S.R.; Thacker, S.G.; Zarzour, A.; Sciumè, G.; Tsai, W.L.; Trier, A.M.; et al. Tofacitinib Ameliorates Murine Lupus and Its Associated Vascular Dysfunction. Arthritis Rheumatol. 2017, 69, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Al-Homood, I.A. Thrombosis in Systemic Lupus Erythematosus: A Review Article. ISRN Rheumatol. 2012, 2012, 428269. [Google Scholar] [CrossRef] [PubMed]
- Manzano, E.M.; Fernandez-Bello, I.; Sanz, R.J.; Marhuenda, Á.R.; Acuña, P.; Román, M.T.A.; Arias-Salgado, E.G.; Barcenilla, S.G.; Canales, M.A.; Jimenez-Yuste, V.; et al. Thrombin Generation Related to Netosis in Patients with Systemic Lupus Erythematosus. Blood 2020, 136 (Suppl. S1), 10–11. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, Z.; Liu, Z. Impact of Neutrophil Extracellular Traps on Thrombosis Formation: New Findings and Future Perspective. Front. Cell. Infect. Microbiol. 2022, 12, 910908. [Google Scholar] [CrossRef] [PubMed]
- Sira, J.; Eyre, L. Physiology of haemostasis. Anaesth. Intensive Care Med. 2016, 17, 79–82. [Google Scholar] [CrossRef]
- Stark, K.; Massberg, S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat. Rev. Cardiol. 2021, 18, 666–682. [Google Scholar] [CrossRef]
- Wagner, D.D.; Heger, L.A. Thromboinflammation: From Atherosclerosis to COVID-19. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1103–1112. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885. [Google Scholar] [CrossRef] [PubMed]
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; DE Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Martinod, K.; Demers, M.; Fuchs, T.A.; Wong, S.L.; Brill, A.; Gallant, M.; Hu, J.; Wang, Y.; Wagner, D.D. Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. USA 2013, 110, 8674–8679. [Google Scholar] [CrossRef] [PubMed]
- Nakazawa, D.; Tomaru, U.; Yamamoto, C.; Jodo, S.; Ishizu, A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front. Immunol. 2012, 3, 333. [Google Scholar] [CrossRef]
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870. [Google Scholar] [CrossRef] [PubMed]
- de Boer, O.J.; Li, X.; Teeling, P.; Mackaay, C.; Ploegmakers, H.J.; van der Loos, C.M.; Daemen, M.J.; de Winter, R.J.; van der Wal, A.C. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb. Haemost. 2013, 109, 290–297. [Google Scholar] [CrossRef]
- Döring, Y.; Soehnlein, O.; Weber, C. Neutrophil Extracellular Traps in Atherosclerosis and Atherothrombosis. Circ. Res. 2017, 120, 736–743. [Google Scholar] [CrossRef]
- Megens, R.T.A.; Vijayan, S.; Lievens, D.; Döring, Y.; van Zandvoort, M.A.M.J.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598. [Google Scholar] [CrossRef]
- Nappi, F.; Bellomo, F.; Singh, S.S.A. Worsening Thrombotic Complication of Atherosclerotic Plaques Due to Neutrophils Extracellular Traps: A Systematic Review. Biomedicines 2023, 11, 113. [Google Scholar] [CrossRef]
- Laridan, E.; Denorme, F.; Desender, L.; François, O.; Andersson, T.; Deckmyn, H.; Vanhoorelbeke, K.; De Meyer, S.F. Neutrophil extracellular traps in ischemic stroke thrombi. Ann. Neurol. 2017, 82, 223–232. [Google Scholar] [CrossRef]
- Noubouossie, D.F.; Whelihan, M.F.; Yu, Y.-B.; Sparkenbaugh, E.; Pawlinski, R.; Monroe, D.M.; Key, N.S. In vitro activation of coagulation by human neutrophil DNA and histone proteins but not neutrophil extracellular traps. Blood 2017, 129, 1021–1029. [Google Scholar] [CrossRef]
- Semeraro, F.; Ammollo, C.T.; Morrissey, J.H.; Dale, G.L.; Friese, P.; Esmon, N.L.; Esmon, C.T. Extracellular histones promote thrombin generation through platelet-dependent mechanisms: Involvement of platelet TLR2 and TLR4. Blood 2011, 118, 1952–1961. [Google Scholar] [CrossRef]
- Elaskalani, O.; Abdol Razak, N.B.; Metharom, P. Neutrophil extracellular traps induce aggregation of washed human platelets independently of extracellular DNA and histones. Cell Commun. Signal. 2018, 16, 24. [Google Scholar] [CrossRef] [PubMed]
- Sambrano, G.R.; Huang, W.; Faruqi, T.; Mahrus, S.; Craik, C.; Coughlin, S.R. Cathepsin G Activates Protease-activated Receptor-4 in Human Platelets. J. Biol. Chem. 2000, 275, 6819–6823. [Google Scholar] [CrossRef] [PubMed]
- Seif, K.; Alidzanovic, L.; Tischler, B.; Ibrahim, N.; Zagrapan, B.; Rauscher, S.; Salzmann, M.; Hell, L.; Mauracher, L.-M.; Budde, U.; et al. Neutrophil-Mediated Proteolysis of Thrombospondin-1 Promotes Platelet Adhesion and String Formation. Thromb. Haemost. 2018, 118, 2074–2085. [Google Scholar] [CrossRef] [PubMed]
- de Los Reyes-García, A.M.; Aroca, A.; Arroyo-Rodríguez, A.B.; García-Barbera, N.; Vicente, V.; González-Conejero, R.; Martinez, C. Neutrophil extracellular trap components increase the expression of coagulation factors. Biomed. Rep. 2019, 10, 195–201. [Google Scholar] [CrossRef]
- Oehmcke, S.; Mörgelin, M.; Herwald, H. Activation of the Human Contact System on Neutrophil Extracellular Traps. J. Innate Immun. 2009, 1, 225–230. [Google Scholar] [CrossRef]
- Gould, T.J.; Vu, T.T.; Swystun, L.L.; Dwivedi, D.J.; Mai, S.H.; Weitz, J.I.; Liaw, P.C. Neutrophil extracellular traps promote thrombin generation through platelet-dependent and platelet-independent mechanisms. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1977–1984. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, L.; Braun, O.; Westman, J.; Madhi, R.; Herwald, H.; Mörgelin, M.; Thorlacius, H. Neutrophil extracellular trap-microparticle complexes enhance thrombin generation via the intrinsic pathway of coagulation in mice. Sci. Rep. 2018, 8, 4020. [Google Scholar] [CrossRef]
- Kambas, K.; Chrysanthopoulou, A.; Vassilopoulos, D.; Apostolidou, E.; Skendros, P.; Girod, A.; Arelaki, S.; Froudarakis, M.; Nakopoulou, L.; Giatromanolaki, A.; et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann. Rheum. Dis. 2014, 73, 1854–1863. [Google Scholar] [CrossRef]
- Stakos, D.A.; Kambas, K.; Konstantinidis, T.; Mitroulis, I.; Apostolidou, E.; Arelaki, S.; Tsironidou, V.; Giatromanolaki, A.; Skendros, P.; Konstantinides, S.; et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur. Heart J. 2015, 36, 1405–1414. [Google Scholar] [CrossRef]
- Folco, E.J.; Mawson, T.L.; Vromman, A.; Bernardes-Souza, B.; Franck, G.; Persson, O.; Nakamura, M.; Newton, G.; Luscinskas, F.W.; Libby, P. Neutrophil Extracellular Traps Induce Endothelial Cell Activation and Tissue Factor Production Through Interleukin-1α and Cathepsin G. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Petersen, L.C.; Bjørn, S.E.; Nordfang, O. Effect of Leukocyte Proteinases on Tissue Factor Pathway Inhibitor. Thromb. Haemost. 1992, 67, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Jordan, R.E.; Nelson, R.M.; Kilpatrick, J.; Newgren, J.O.; Esmon, P.C.; Fournel, M.A. Inactivation of human antithrombin by neutrophil elastase. Kinetics of the heparin-dependent reaction. J. Biol. Chem. 1989, 264, 10493–10500. [Google Scholar] [CrossRef] [PubMed]
- Ammollo, C.T.; Semeraro, F.; Xu, J.; Esmon, N.L.; Esmon, C.T. Extracellular histones increase plasma thrombin generation by impairing thrombomodulin-dependent protein C activation. J. Thromb. Haemost. 2011, 9, 1795–1803. [Google Scholar] [CrossRef]
- Meng, H.; Yalavarthi, S.; Kanthi, Y.; Mazza, L.F.; Elfline, M.A.; Luke, C.E.; Pinsky, D.J.; Henke, P.K.; Knight, J.S. In Vivo Role of Neutrophil Extracellular Traps in Antiphospholipid Antibody-Mediated Venous Thrombosis. Arthritis Rheumatol. 2017, 69, 655–667. [Google Scholar] [CrossRef]
- Zha, C.; Zhang, W.; Gao, F.; Xu, J.; Jia, R.; Cai, J.; Liu, Y. Anti-β2GPI/β2GPI induces neutrophil extracellular traps formation to promote thrombogenesis via the TLR4/MyD88/MAPKs axis activation. Neuropharmacology 2018, 138, 140–150. [Google Scholar] [CrossRef]
- Grossi, C.; Capitani, N.; Benagiano, M.; Baldari, C.T.; Della Bella, C.; Macor, P.; Tedesco, F.; Borghi, M.O.; Maugeri, N.; D’elios, M.M.; et al. Beta 2 glycoprotein I and neutrophil extracellular traps: Potential bridge between innate and adaptive immunity in anti-phospholipid syndrome. Front. Immunol. 2023, 13, 1076167. [Google Scholar] [CrossRef]
- You, Y.; Liu, Y.; Li, F.; Mu, F.; Zha, C. Anti-β2GPI/β2GPI induces human neutrophils to generate NETs by relying on ROS. Cell Biochem. Funct. 2019, 37, 56–61. [Google Scholar] [CrossRef]
- Lu, Y.; Dong, Y.; Zhang, Y.; Shen, D.; Wang, X.; Ge, R.; Zhang, M.; Xia, Y.; Wang, X. Antiphospholipid antibody-activated NETs exacerbate trophoblast and endothelial cell injury in obstetric antiphospholipid syndrome. J. Cell. Mol. Med. 2020, 24, 6690–6703. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zuo, Y.; Zhang, S.; Makris, U.E.; Karp, D.R.; Li, Z. Additional risk factors associated with thrombosis and pregnancy morbidity in a unique cohort of antiphospholipid antibody-positive patients. Chin. Med. J. 2022, 135, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.A.; Estes, S.K.; Gandhi, A.A.; Yalavarthi, S.; Hoy, C.K.; Shi, H.; Zuo, Y.; Erkan, D.; Knight, J.S. Defibrotide Inhibits Antiphospholipid Antibody–Mediated Neutrophil Extracellular Trap Formation and Venous Thrombosis. Arthritis Rheumatol. 2022, 74, 902–907. [Google Scholar] [CrossRef]
- Mauracher, L.-M.; Krall, M.; Roiß, J.; Hell, L.; Koder, S.; Hofbauer, T.M.; Gebhart, J.; Hayden, H.; Brostjan, C.; Ay, C.; et al. Neutrophil subpopulations and their activation potential in patients with antiphospholipid syndrome and healthy individuals. Rheumatology 2020, 60, 1687–1699. [Google Scholar] [CrossRef] [PubMed]
- van den Hoogen, L.L.; Fritsch-Stork, R.D.E.; van Roon, J.A.G.; Radstake, T.R.D.J. Low density granulocytes are increased in the antiphospholipid syndrome and are associated with anti-β2GPI antibodies: Comment on the article by Yalavarthi et al. Arthritis Rheumatol. 2016, 68, 1320–1321. [Google Scholar] [CrossRef]
- Leffler, J.; Stojanovich, L.; Shoenfeld, Y.; Bogdanovic, G.; Hesselstrand, R.; Blom, A.M. Degradation of neutrophil extracellular traps is decreased in patients with antiphospholipid syndrome. Clin. Exp. Rheumatol. 2014, 32, 66–70. [Google Scholar] [PubMed]
- Zuo, Y.; Yalavarthi, S.; Gockman, K.; Madison, J.A.; Gudjonsson, J.E.; Kahlenberg, J.M.; McCune, W.J.; Bockenstedt, P.L.; Karp, D.R.; Knight, J.S. Anti–Neutrophil Extracellular Trap Antibodies and Impaired Neutrophil Extracellular Trap Degradation in Antiphospholipid Syndrome. Arthritis Rheumatol. 2020, 72, 2130–2135. [Google Scholar] [CrossRef]
- Zuo, Y.; Navaz, S.; Tsodikov, A.; Kmetova, K.; Kluge, L.; Ambati, A.; Hoy, C.K.; Yalavarthi, S.; de Andrade, D.; Tektonidou, M.G.; et al. Anti–Neutrophil Extracellular Trap Antibodies in Antiphospholipid Antibody–Positive Patients: Results From the Antiphospholipid Syndrome Alliance for Clinical Trials and InternatiOnal Networking Clinical Database and Repository. Arthritis Rheumatol. 2023, 75, 1407–1414. [Google Scholar] [CrossRef]
- Hell, L.; Lurger, K.; Mauracher, L.-M.; Grilz, E.; Reumiller, C.M.; Schmidt, G.J.; Ercan, H.; Koder, S.; Assinger, A.; Basilio, J.; et al. Altered platelet proteome in lupus anticoagulant (LA)-positive patients—Protein disulfide isomerase and NETosis as new players in LA-related thrombosis. Exp. Mol. Med. 2020, 52, 66–78. [Google Scholar] [CrossRef]
- Mazetto, B.d.M.; Hounkpe, B.W.; da Silva Saraiva, S.; Vieira-Damiani, G.; dos Santos, A.P.R.; Jacinto, B.C.; Vaz, C.d.O.; Mesquita, G.T.V.; Annichino-Bizzacchi, J.M.; De Paula, E.V.; et al. Association between neutrophil extracellular traps (NETs) and thrombosis in antiphospholipid syndrome. Thromb. Res. 2022, 214, 132–137. [Google Scholar] [CrossRef]
- Foret, T.; Dufrost, V.; du Mont, L.S.; Costa, P.; Lakomy, C.; Lagrange, J.; Lacolley, P.; Regnault, V.; Zuily, S.; Wahl, D. A new pro-thrombotic mechanism of neutrophil extracellular traps in antiphospholipid syndrome: Impact on activated protein C resistance. Rheumatology 2022, 61, 2993–2998. [Google Scholar] [CrossRef]
- Salet, D.M.; Bekkering, S.; Middeldorp, S.; van den Hoogen, L.L. Targeting thromboinflammation in antiphospholipid syndrome. J. Thromb. Haemost. 2023, 21, 744–757. [Google Scholar] [CrossRef] [PubMed]
- Sule, G.; Kelley, W.J.; Gockman, K.; Yalavarthi, S.; Vreede, A.P.; Banka, A.L.; Bockenstedt, P.L.; Eniola-Adefeso, O.; Knight, J.S. Increased Adhesive Potential of Antiphospholipid Syndrome Neutrophils Mediated by β2 Integrin Mac-1. Arthritis Rheumatol. 2020, 72, 114–124. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.S.; Meng, H.; Coit, P.; Yalavarthi, S.; Sule, G.; Gandhi, A.A.; Grenn, R.C.; Mazza, L.F.; Ali, R.A.; Renauer, P.; et al. Activated signature of antiphospholipid syndrome neutrophils reveals potential therapeutic target. JCI Insight 2017, 2, e93897. [Google Scholar] [CrossRef] [PubMed]
- Nayak, L.; Sweet, D.R.; Thomas, A.; Lapping, S.D.; Kalikasingh, K.; Madera, A.; Vinayachandran, V.; Padmanabhan, R.; Vasudevan, N.T.; Myers, J.T.; et al. A targetable pathway in neutrophils mitigates both arterial and venous thrombosis. Sci. Transl. Med. 2022, 14, eabj7465. [Google Scholar] [CrossRef] [PubMed]
- Stachowicz, A.; Zabczyk, M.; Natorska, J.; Suski, M.; Olszanecki, R.; Korbut, R.; Wiśniewski, J.R.; Undas, A. Differences in plasma fibrin clot composition in patients with thrombotic antiphospholipid syndrome compared with venous thromboembolism. Sci. Rep. 2018, 8, 17301. [Google Scholar] [CrossRef]
- Ali, R.A.; Gandhi, A.A.; Meng, H.; Yalavarthi, S.; Vreede, A.P.; Estes, S.K.; Palmer, O.R.; Bockenstedt, P.L.; Pinsky, D.J.; Greve, J.M.; et al. Adenosine receptor agonism protects against NETosis and thrombosis in antiphospholipid syndrome. Nat Commum. 2019, 10, 1916. [Google Scholar] [CrossRef]
- Manfredi, A.A.; Rovere-Querini, P.; D’angelo, A.; Maugeri, N. Low molecular weight heparins prevent the induction of autophagy of activated neutrophils and the formation of neutrophil extracellular traps. Pharmacol. Res. 2017, 123, 146–156. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Authors | NET Components /Intact NET | Results | Impact on Hemostasis |
|---|---|---|---|
| Fuchs et al. [92] | Intact NET | Adhesion, activation and aggregation of platelets | -Platelet activation -NETs provide a scaffold for platelets and RBCs adhesion. |
| Elaskalani et al. [104] | Intact NET | Platelet activation and aggregation | Platelet activation |
| Sambrano et al. [105] | Cathepsin G | PAR4-dependent platelet activation | Platelet activation |
| Seif. et al. [106] | Neutrophil elastase, cathepsin G | Induction of thrombospondin-1 proteolysis | Platelet activation |
| Noubouossie et al. [102] | DNA, histones, intact NET | DNA and histones activated thrombin generation | Activation of coagulation by DNA and histones but not intact NETs |
| Semeraro et al. [103] | Histones | -Adhesion, activation and aggregation of platelets -Stimulation of platelet-dependent thrombin generation | -Platelet activation -Activation of coagulation |
| Gould et al. [109] | Intact NETs | DNA-dependent thrombin generation | Activation of coagulation |
| Wang et al. [110] | Intact NET | Factor XII activation and thrombin formation | Activation of coagulation |
| Oehmcke et al. [108] | Intact NET | Activation of factor XII | Activation of coagulation |
| Kambas et al. [111] | Intact NET | TF expressing NETs | Activation of coagulation |
| Stakos et al. [112] | Intact NET | TF expressing NETs | Activation of coagulation |
| Folco et al. [113] | Intact NET | NETs induce endothelial TF expression | Activation of coagulation |
| Petersen et al. [114] | Neutrophil elastase and cathepsin G | Inactivation of tissue factor pathway inhibitor (TFPI). | Inhibition of the natural anticoagulants |
| Jordan et al. [115] | Neutrophil elastase | Inhibition of antithrombin III | Inhibition of the natural anticoagulants |
| Ammollo et al. [116] | Histones | Disruption of thrombomodulin-dependent protein C activation | Inhibition of the natural anticoagulants |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reshetnyak, T.; Nurbaeva, K. The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. Int. J. Mol. Sci. 2023, 24, 13581. https://doi.org/10.3390/ijms241713581
Reshetnyak T, Nurbaeva K. The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. International Journal of Molecular Sciences. 2023; 24(17):13581. https://doi.org/10.3390/ijms241713581
Chicago/Turabian StyleReshetnyak, Tatiana, and Kamila Nurbaeva. 2023. "The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome" International Journal of Molecular Sciences 24, no. 17: 13581. https://doi.org/10.3390/ijms241713581
APA StyleReshetnyak, T., & Nurbaeva, K. (2023). The Role of Neutrophil Extracellular Traps (NETs) in the Pathogenesis of Systemic Lupus Erythematosus and Antiphospholipid Syndrome. International Journal of Molecular Sciences, 24(17), 13581. https://doi.org/10.3390/ijms241713581