
Antibody Cross-Reactivity in Auto-Immune Diseases


Abstract
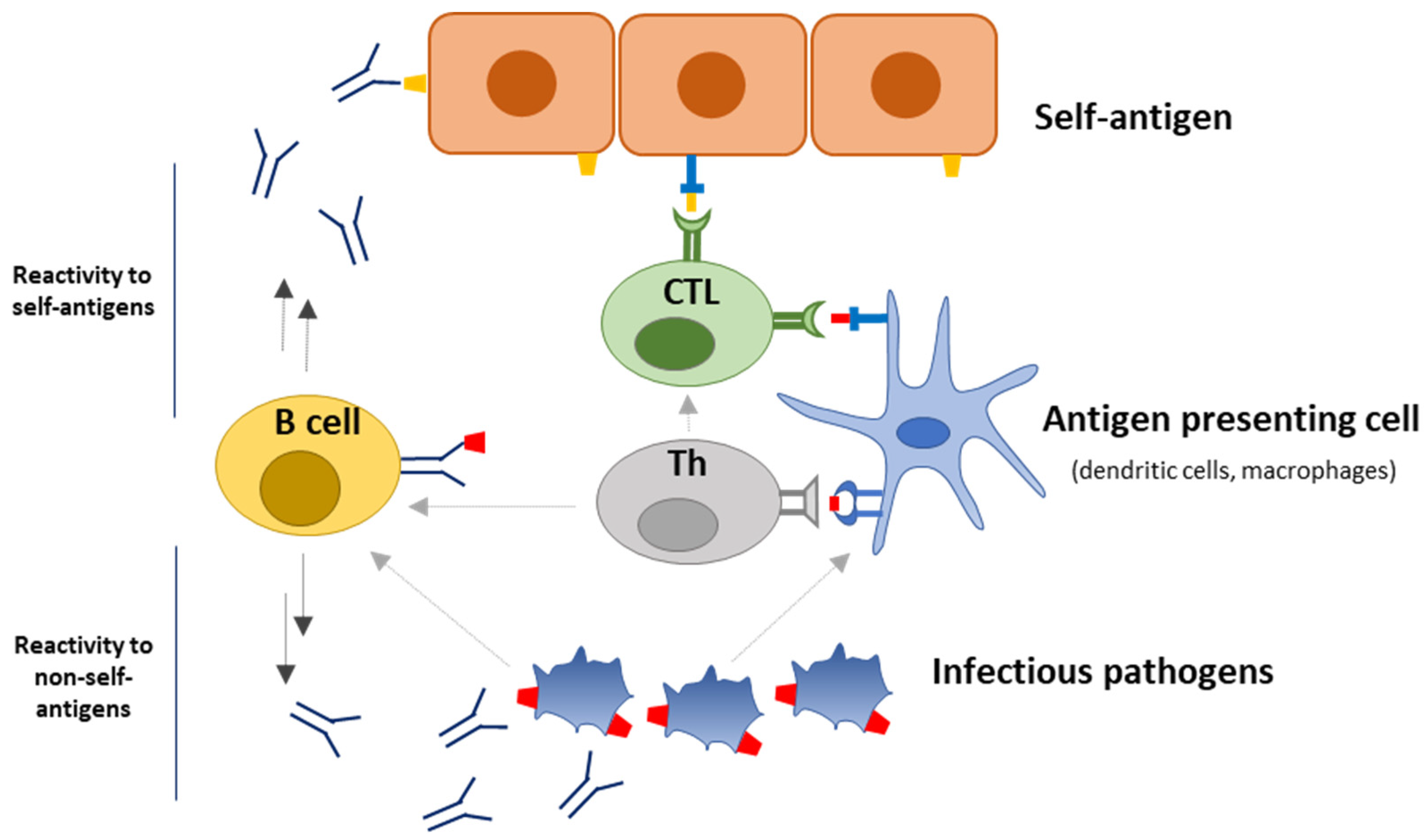
:1. Introduction
2. Autoimmune Diseases
3. Factors and Mechanisms Associated with Autoimmune Diseases

{kind=link}
{kind=link}
| Mechanisms | Reference |
|---|---|
| Bystander activation: implies activation of cells without antigen recognition. During an immune response to non-self pathogens, bystander activation of autoreactive T cells via inflammatory markers, e.g., cytokines, can trigger autoimmunity. | [31,68] |
| Defective self-tolerance: self-tolerance is maintained through central and peripheral tolerance mechanisms, which entails neutralization of autoreactive cells in the bone marrow/ thymus or in the periphery, corresponding to central and peripheral tolerance, respectively. Breach of these leads to autoimmunity. | [50,51] |
| Epitope spreading: implies diversification of epitope specificity from the initial dominant epitope-specific immune response to subdominant and eventually cryptic epitopes of the same protein (intramolecular spreading) or other proteins (intermolecular spreading). | [52,53] |
| Molecular mimicry: implies cross-reactivity between infectious pathogenic antigens and self-antigens, which favor activation of autoreactive T or B cells. | [7,8] |
4. Antibody Specificity and Cross-Reactivity
5. Cross-Reactivity in Autoimmune Diseases
5.1. Rheumatoid Arthritis
5.2. Systemic Lupus Erythematosus
5.3. Multiple Sclerosis
5.4. Type 1 Diabetes
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murphy, K.; Weaver, C.; Berg, L.J. Janeway’s Immunobiology, 10th ed.; W. W. Norton & Company: New York, NY, USA, 2022. [Google Scholar]
- Wang, L.; Wang, F.S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef] [PubMed]
- Rosenblum, M.D.; Remedios, K.A.; Abbas, A.K. Mechanisms of human autoimmunity. J. Clin. Investig. 2015, 125, 2228–2233. [Google Scholar] [CrossRef] [PubMed]
- Touil, H.; Mounts, K.; De Jager, P.H. Differential impact of environmental factors on systemic and localized autoimmunity. Front. Immunol. 2023, 14, 1147447. [Google Scholar] [CrossRef]
- Sundaresan, B.; Shirafkan, F.; Ripperger, K.; Rattay, K. The Role of Viral Infections in the Onset of Autoimmune Diseases. Viruses 2023, 15, 782. [Google Scholar] [CrossRef]
- Ramos, P.R.; Shedlock, A.M.; Langefeld, C.D. Genetics of autoimmune diseases: Insights from population genetics. J. Hum. Genet. 2015, 60, 657–664. [Google Scholar] [CrossRef]
- Cusick, M.F.; Libbey, J.E.; Fujinami, R.S. Molecular mimicry as a mechanism of autoimmune disease. Clin. Rev. Allergy Immunol. 2012, 42, 102–111. [Google Scholar] [CrossRef]
- Rojas, M.; Restrepo-Jimenez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [CrossRef]
- Lerner, A.; Jeremias, P.; Matthias, T. The world incidence and prevalence of autoimmune diseases is increasing. Int. J. Celiac Dis. 2015, 3, 55–155. [Google Scholar] [CrossRef]
- DiMeglio, L.A.; Evans-Molina, C.; Oram, R.A. Type 1 diabetes. Lancet 2018, 391, 2449–2462. [Google Scholar] [CrossRef]
- Filippi, M.; Bar-Or, A.; Piehl, F.; Preziosa, P.; Solaria, A.; Vukusic, S.; Rocca, M.A. Multiple sclerosis. Nat. Rev. 2018, 4, 43. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef]
- Rahman, A.; Isenberg, D.A. Systemic lupus erythematosus. N. Engl. J. Med. 2008, 358, 929–939. [Google Scholar] [CrossRef]
- Xiao, Z.X.; Miller, J.S.; Zheng, S.G. An updated advance of autoantibodies in autoimmune diseases. Autoimmun Rev. 2021, 20, 102743. [Google Scholar] [CrossRef]
- Marrack, P.; Kotzin, B.L. Autoimmune disease: Why and where it occurs. Nat. Med. 2001, 7, 899–905. [Google Scholar] [CrossRef]
- Goodnow, C.C.; Sprent, J.; de St Groth, B.F.; Vinuesa, C.G. Cellular and genetic mechanisms of self tolerance and autoimmunity. Nature 2005, 435, 590–597. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H.; Frederiksen, J.L. Epstein-Barr virus and multiple sclerosis. Front. Immunol. 2020, 11, 587078. [Google Scholar] [CrossRef]
- Op de Beeck, A.; Eizirik, D.L. Viral infections in type 1 diabetes mellitus–why the beta cells? Nat. Rev. Endocrinol. 2016, 12, 263–273. [Google Scholar] [CrossRef]
- McGonagle, D.; Conaghan, P.G.; O’Connor, P.; Gibbon, W.; Green, M.; Wakefield, R.; Ridgway, J.; Emery, P. The relationship between synovitis and bone changes in early untreated rheumatoid arthritis: A controlled magnetic resonance imaging study. Arthritis Rheum. 1999, 42, 1706–1711. [Google Scholar] [CrossRef]
- Snir, O.; Widhe, M.; Hermansson, M.; von Spee, C.; Lindberg, J.; Hensen, S.; Lundberg, K.; Engstöm, A.; Venables, P.J.W.; Toes, R.E.M.; et al. Antibodies to several citrullinated antigens are enriched in the joints of rheumatoid arthritis patients. Arthritis Rheum. 2010, 62, 44–52. [Google Scholar] [CrossRef]
- Cojocaru, M.I.; Socoliuc, G.; Sapira, V.; Cojocaru, M. Primary Sjogren’s syndrome or multiple sclerosis? Our experience concerning the dilemma of clinically isolated syndrome. Rom. J. Intern. Med. 2011, 49, 301–318. [Google Scholar]
- Anaya, J.M. The diagnosis and clinical significance of polyautoimmunity. Autoimmun. Rev. 2014, 13, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Provost, T.T.; Talal, N.; Harley, J.B.; Reichlin, M.; Alexander, E. The relationship between anti-Ro (SS-A) antibody-positive Sjogren’s syndrome and anti-Ro (SS-A) antibody-positive lupus erythematosus. Arch. Dermatol. 1988, 124, 63–71. [Google Scholar] [CrossRef] [PubMed]
- Amador-Patarroyo, M.J.; Arbelaez, J.G.; Mantilla, R.D.; Rodriguez-Rodriguez, A.; Cárdenas-Roldán, J.; Pineda-Tamayo, R.; Guarin, M.R.; Kleine, L.L.; Villarraga, A.; Anaya, J.M. Sjogren’s syndrome at the crossroad of polyautoimmunity. J. Autoimmun. 2012, 39, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Cojocaru, M.; Cojocaru, I.M.; Silosi, I. Multiple autoimmune syndrome. Maedica 2010, 5, 132–134. [Google Scholar]
- Sinha, A.A.; Lopez, M.A.; McDevitt, H.O. Autoimmune diseases: The failure of self-tolerance. Science 1990, 248, 1380–1388. [Google Scholar] [CrossRef]
- Mueller, D.L.; Jenkins, M.K. Autoimmunity: When self-tolerance breaks down. Curr. Biol. 1997, 7, R255–R257. [Google Scholar] [CrossRef]
- Vojdani, A.; Pollard, K.M.; Campbell, A.W. Environmental triggers and autoimmunity. Autoimmune Dis. 2014, 2014, 798029. [Google Scholar] [CrossRef]
- Houen, G.; Trier, N.H. Epstein-Barr virus and systemic autoimmune diseases. Front. Immunol. 2020, 11, 587380. [Google Scholar] [CrossRef] [PubMed]
- Smatti, M.K.; Cyprian, F.S.; Nasrallah, G.K.; Al Thani, A.A.; Almishal, R.O.; Yassine, H.M. Viruses and autoimmunity: A review on the potential interaction and molecular mechanisms. Viruses 2019, 11, 762. [Google Scholar] [CrossRef]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 115. [Google Scholar] [CrossRef]
- Rodriguez, Y.; Rojas, M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramirez-Santana, C.; Monsalve, D.M.; Gershwin, M.E.; Anaya, J.M. Guillain-Barre syndrome, transverse myelitis and infectious diseases. Cell. Mol. Immunol. 2018, 15, 547–562. [Google Scholar] [CrossRef]
- Shahrizaila, N.; Yuki, N. Guillain-barre syndrome animal model: The first proof of molecular mimicry in human autoimmune disorder. J. Biomed. Biotechnol. 2011, 2011, 829129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L. A common mechanism links Epstein-Barr virus infections and autoimmune diseases. J. Med. Virol. 2023, 95, e28363. [Google Scholar] [CrossRef] [PubMed]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus in systemic autoimmune diseases. Clin. Dev. Immunol. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Draborg, A.H.; Duus, K.; Houen, G. Epstein-Barr virus and systemic lupus erythematosus. Clin. Dev. Immunol. 2012, 2012, 1–10. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390, 211–240. [Google Scholar]
- Poole, B.D.; Scofield, R.H.; Harley, J.B.; James, J.A. Epstein-Barr virus and molecular mimicry in systemic lupus erythematosus. Autoimmunity 2006, 39, 63–70. [Google Scholar] [CrossRef]
- McClain, M.T.; Heinlen, L.D.; Dennis, G.J.; Roebuck, J.; Harley, J.B.; James, J.A. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat. Med. 2005, 11, 85–89. [Google Scholar] [CrossRef]
- Christen, U. Molecular mimicry. In Autoantibodies, 3rd ed.; Shoenfeld, Y., Meroni, P.L., Gershwin, M.E., Eds.; Elsevier: San Diego, CA, USA, 2014; pp. 35–42. [Google Scholar]
- Guarneri, F.; Guarneri, C.; Benvenga, S. Helicobacter pylori and autoimmune pancreatitis: Role of carbonic anhydrase via molecular mimicry? J. Cell Mol. Med. 2005, 9, 741–744. [Google Scholar] [CrossRef]
- Albert, L.J.; Inman, R.D. Molecular mimicry and autoimmunity. N. Engl. J. Med. 1999, 341, 2068–2074. [Google Scholar] [CrossRef]
- Andersen, M.H.; Schrama, D.; Straten, P.T.; Becker, J.C. Cytotoxic T cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Takada, K.; Jameson, S.C. Self-class I MHC molecules support survival of naïve CD8 T cells, but depress their functional sensitivity through regulation of CD8 expression levels. J. Exp. Med. 2009, 206, 2253–2269. [Google Scholar] [CrossRef]
- Zhang, X.M.; Liu, C.Y.; Shao, Z.H. Advances in the role of Helper T cells in autoimmune diseases. Chin. Med. J. 2020, 133, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Xu, P.; Zhou, Q.; Xu, J. The function of T follicular Helper cells in the autoimmune liver diseases. J. Immunol. Res. 2020, 2020, 5679254. [Google Scholar] [CrossRef]
- Fasano, R.; Malerba, E.; Prete, M.; Solimando, A.G.; Buonavoglia, A.; Silvestris, N.; Leone, P.; Racanelli, V. Impact of Antigen Presentation Mechanisms on Immune Response in Autoimmune Hepatitis. Front. Immunol. 2022, 12, 814155. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Iadarola, M.J.; Keller, J.M.; Warner, B.M. Autoantibodies targeting intracellular and extracellular proteins in autoimmune diseases. Front. Immunol. 2021, 12, 548469. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, R.J.; Vanhoorelbeke, K.; Leypoldt, F.; Kaya, Z.; Bieber, K.; McLachlan, S.M.; Komorowski, L.; Luo, J.; Cabral-Marques, O.; Hammers, C.M.; et al. Mechanisms of Autoantibody Induced Pathology. Front. Immunol. 2017, 8, 603. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and Pheripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Theofilopoulos, A.; Kono, D.; Baccala, R. The multiple pathways to autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef]
- Venkatesha, S.H.; Durai, M.; Moudgil, K.D. Chapter 4-Epitope Spreading in Autoimmune Diseases. Infect. Autoimmun. (Second Ed.) 2015, 45–68. [Google Scholar]
- Cornaby, C.; Gibbons, L.; Mayhew, V.; Sloan, C.S.; Welling, A.; Poole, B.D. B cell epitope spreading: Mechanisms and contribution to autoimmune diseases. Immunol. Lett. 2015, 163, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M.; Karpati, S.; Prussik, R.; Klaus-Kovtun, V.; Stanley, J.R. Autoantibodies against the amino-terminal cadherin-like binding domain of pemphigus vulgaris antigen are pathogenic. J. Clin. Investig. 1992, 90, 919–926. [Google Scholar] [CrossRef] [PubMed]
- Charytan, D.M.; Le, D.D.; Grossman, J.M.; Holthaus, K.A.; Kalluri, R. Antiglomerular basement membrane autoantibodies are nonpathogenic in Wegener’s granulomatosis. Am. J. Med. 2003, 115, 414–415. [Google Scholar] [CrossRef]
- Sillevis Smitt, P.A.; Manley, G.T.; Posner, J.B. Immunization with the paraneoplastic encephalomyelitis antigen HuD does not cause neurologic disease in mice. Neurology 1995, 45, 1873–1878. [Google Scholar]
- Waldman, M.; Madaio, M.P. Pathogenic autoantibodies in lupus nephritis. Lupus 2005, 14, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Bizzaro, N. Autoantibodies as predictors of disease: The clinical and experimental evidence. Autoimmun. Rev. 2007, 6, 325–333. [Google Scholar] [CrossRef] [PubMed]
- von Muhlen, C.A. Autoantibodies in the diagnosis of systemic rheumatic diseases. Semin. Arthritis Rheum. 1995, 24, 323–358. [Google Scholar] [CrossRef]
- Sato, S.; Kuwana, M.; Fujita, T.; Suzuki, Y. Anti-CADM-140/MDA5 autoantibody titer correlates with disease activity and predicts disease outcome in patients with dermatomyositis and rapidly progressive interstitial lung disease. Mod. Rheumatol. 2013, 23, 496–502. [Google Scholar] [CrossRef]
- Arbuckle, M.R.; McClain, M.T.; Rubertone, M.V.; Scofield, R.H.; Dennis, G.J.; James, J.A.; Harley, J.B. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med. 2003, 349, 1526–1533. [Google Scholar] [CrossRef]
- Kokkonen, H.; Mullazehi, M.; Berglin, E.; Hallmans, G.; Wadell, G.; Rönnelid, J.; Ranrapää-Dahlqvist, S. Antibodies of IgG, IgA and IgM isotypes against cyclic citrullinated peptide precede the development of rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, 1–10. [Google Scholar] [CrossRef]
- Ma, H.; Murphy, C.; Loscher, C.E.; O’Kennedy, R. Autoantibodies-Enemies, and/or protential allies? Front. Immunol. 2022, 13, 953726. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, X.; Song, W.; Fang, Y.; Yu, L.; Liu, S.; Churilov, L.P.; Zhang, F. The roles and applications of autoantibodies in progression, diagnosis, treatment and prognosis of human malignant tumours. Autoimmun. Rev. 2017, 16, 1270–1281. [Google Scholar] [CrossRef]
- Gronwall, C.; Silverman, G.J. Natural IgM: Beneficial autoantibodies for the control of inflammatory and autoimmune disease. J. Clin. Immunol. 2014, 34, 12–21. [Google Scholar] [CrossRef]
- Schwartz-Albiez, R.; Monteiro, R.C.; Rodriguez, M.; Binder, C.J.; Schoenfeld, Y. Natural antibodies, intravenous immunoglobulin and their role in autoimmunity, cancer and inflammation. Clin. Exp. Immunol. 2009, 158 (Suppl. 1), 43–50. [Google Scholar] [CrossRef] [PubMed]
- Lobo, P.I. Role of Natural IgM Autoantibodies (IgM-NAA) and IgM Anti-Leukocyte Antibodies (IgM-ALA) in Regulating Inflammation. Curr. Top Microbiol. Immunol. 2017, 408, 89–117. [Google Scholar] [PubMed]
- Shim, C.H.; Cho, S.; Shin, Y.M.; Choi, J.M. Emerging role of bystander T cell activation in autoimmune diseases. BMB Rep. 2022, 55, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Marrack, P.; Scott-Browne, J.P.; Dai, S.; Gapin, L.; Kappler, J.W. Evolutionarily conserved amino acids that control TCR-MHC interaction. Annu. Rev. Immunol. 2008, 26, 171–203. [Google Scholar] [CrossRef]
- Bridgeman, J.S.; Sewell, A.K.; Miles, J.J.; Price, D.A.; Cole, D.K. Structural and biophysical determinants of αβ T-cell antigen recognition. Immunology 2012, 135, 9–18. [Google Scholar] [CrossRef]
- Natarajan, K.; Jiang, J.; May, N.A.; Mage, M.G.; Boyd, L.F.; McShan, A.C.; Sgourakis, N.G.; Bax, A.; Margulies, D.H. The role of molecular flexibility in antigen presentation and T Cell Receptor-mediated signaling. Front. Immunol. 2018, 9, 1657. [Google Scholar] [CrossRef]
- Szeto, C.; Lobos, C.A.; Nguyen, A.T.; Gras, S. TCR Recognition of Peptide-MHC-I: Rule Makers and Breakers. Int. J. Mol. Sci. 2020, 22, 68. [Google Scholar] [CrossRef]
- Wu, Y.; Zhang, N.; Hashimoto, K.; Xia, C.; Dijkstra, J.M. Structural Comparison Between MHC Classes I and II; in Evolution, a Class-II-Like Molecule Probably Came First. Front. Immunol. 2021, 12, 621153. [Google Scholar] [CrossRef]
- Takaba, H.; Takayanagi, H. The mechanisms of T cell selection in the thymus. Trends Immunol. 2017, 38, 805–816. [Google Scholar] [CrossRef]
- Kondo, K.; Ohigashi, I.; Takahama, Y. Thymus machinery for T-cell selection. Int. Immunol. 2019, 31, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Jiang, N.; Malone, M.; Chizari, S. Antigen-specific and cross-reactive T cells in protection and disease. Immunol. Rev. 2023, 316, 120–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and co-inhibitory pathways in autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M. Co-signal molecules in T-cell activation: Historical overview and perspective. Adv. Exp. Med. Biol. 2019, 1189, 3–23. [Google Scholar]
- Barnas, J.L.; Looney, R.J.; Anolik, J.H. B cell targeted therapies in autoimmune disease. Curr. Opin. Immunol. 2019, 61, 92–99. [Google Scholar] [CrossRef]
- Merino-Vico, A.; Frazzei, G.; van Hamburg, J.P.; Tas, S.W. Targeting B cells and plasma cells in autoimmune diseases: From established treatments to novel therapeutic approaches. Eur. J. Immunol. 2023, 53, e2149675. [Google Scholar] [CrossRef]
- Damian, R.T. Molecular mimicry: Antigen sharing by parasite and host and its consequences. Am. Nat. 1964, 98, 129–149. [Google Scholar] [CrossRef]
- Zabriskie, J.B.; Freimer, E.H. An immunological relationship between the group. A streptococcus and mammalian muscle. J. Exp. Med. 1966, 124, 661–678. [Google Scholar] [CrossRef]
- Kaplan, M.H.; Meyeserian, M. An immunological cross-reaction between group-A streptococcal cells and human heart tissue. Lancet 1962, 1, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Trier, N.H.; Valdarnini, N.; Fanelli, I.; Rovero, P.; Hansen, P.R.; Schafer-Nielsen, C.; Ciplys, E.; Slibinskas, R.; Pociot, F.; Friis, T.; et al. Peptide Antibody Reactivity to Homologous Regions in Glutamate Decarboxylase Isoforms and Coxsackievirus B4 P2C. Int. J. Mol. Sci. 2022, 23, 4424. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Kinloch, A.; Fisher, B.A.; Wegner, N.; Wait, R.; Charles, P.; Mikuls, T.R.; Venables, P.J. Antibodies to citrullinated alpha-enolase peptide 1 are specific for rheumatoid arthritis and cross-react with bacterial enolase. Arthritis Rheum. 2008, 58, 3009–3019. [Google Scholar] [CrossRef]
- Birkenfeld, P.; Haratz, N.; Klein, G.; Sulitzeanu, D. Cross-reactivity between the EBNA-1 p107 peptide, collagen, and keratin: Implications for the pathogenesis of rheumatoid arthritis. Clin. Immunol. Immunopathol. 1990, 54, 14–25. [Google Scholar] [CrossRef]
- James, J.A.; Harley, J.B. Linear epitope mapping of an Sm B/B′ polypeptide. J. Immunol. 1992, 148, 2074–2079. [Google Scholar] [CrossRef] [PubMed]
- Yuki, N. Ganglioside mimicry and peripheral nerve disease. Muscle Nerve 2007, 35, 691–711. [Google Scholar] [CrossRef]
- Lang, H.L.; Jacobsen, H.; Ikemizu, S.; Andersson, C.; Harlos, K.; Madsen, L.; Hjorth, P.; Sondergaard, L.; Svejgaard, A.; Wucherpfennig, K.; et al. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat. Immunol. 2002, 3, 940–943. [Google Scholar] [CrossRef] [PubMed]
- Soderberg, C.; Larsson, S.; Rozell, B.L.; Sumitran-Karuppan, S.; Ljungman, P.; Moller, E. Cytomegalovirus-induced CD13-specific autoimmunity-a possible cause of chronic graft-vs-host disease. Transplantation 1996, 61, 600–609. [Google Scholar] [CrossRef]
- Schulz, U.; Randalls, B.; Counsell, C. Anti-hu syndrome: A rare presentation and a very difficult decision. Pract. Neurol. 2007, 7, 336–341. [Google Scholar] [CrossRef]
- Douglas, C.A.; Ellershaw, J. Anti-Hu antibodies may indicate a positive response to chemotherapy in paraneoplastic syndrome secondary to small cell lung cancer. Palliat. Med. 2003, 17, 638–639. [Google Scholar] [CrossRef]
- Peterson, L.K.; Fujijami, R.S. Molecular mimicry. In Autoantibodies; Shoenfeld, M.E., Gershwin, M.E., Eds.; Elsevier: Burlington, NJ, USA, 2007. [Google Scholar]
- Tam, C.C.; O’Brien, S.J.; Petersen, I.; Islam, A.; Hayward, A.; Rodrigues, L.C. GuillainBarre syndrome and preceding infection with campylobacter, influenza and Epstein-Barr virus in the general practice research database. PLoS ONE 2007, 2, e344. [Google Scholar] [CrossRef] [PubMed]
- MacCalum, R.M.; Martin, A.C.; Thornton, J.M. Antibody-antigen interactions: Contact analysis and binding site topography. J. Mol. Biol. 1996, 262, 732–745. [Google Scholar] [CrossRef] [PubMed]
- Chitarra, V.; Alzari, P.M.; Bentley, G.A.; Bhat, T.N.; Eiselé, J.L.; Houdusse, A.; Lescar, J.; Souchon, H.; Poljak, R.J. Three-dimensional structure of a heteroclitic antigen-antibody cross-reaction complex. Proc. Natl. Acad. Sci. USA 1993, 90, 7711–7715. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Stanfield, R.L. Structural aspects of antibodies and antibody-antigen complexes. Ciba Found. Symp. 1991, 159, 13–28. [Google Scholar] [PubMed]
- Ofran, Y.; Schelssinger, A.; Rost, B. Automated identification of complementarity determining regions (CDRs) reveals peculiar characteristics of CDRs and B cell epitopes. J. Immunol. 2008, 181, 6230–6235. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Stanfield, R.L. Antibody-antigen interactions: New structures and new conformational changes. Curr. Opin. Struct. Biol. 1994, 4, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Reverberi, R.; Reverberi, L. Factors affecting the antigen-antibody reaction. Blood Transfus. 2007, 5, 227–240. [Google Scholar]
- Uysal, H.; Bockermann, R.; Nandakumar, K.S.; Sehnert, B.; Bajtner, E.; Engström, A.; Serre, G.; Burkhardt, H.; Thunnissen, M.G.M.; Holmdahl, R. Structure and pathogenicity of antibodies specific for citrullinated collagen type II in experimental arthritis. J. Exp. Med. 2009, 206, 449–462. [Google Scholar] [CrossRef]
- Mian, I.S.; Bradwell, A.R.; Olson, A.J. Structure, function and properties of antibody binding sites. J. Mol. Biol. 1991, 217, 133–151. [Google Scholar] [CrossRef]
- Tian, Y.; Ramesh, C.V.; Ma, X.; Naqvi, S.; Patel, T.; Cenizal, T.; Diaz, K.; Crea, K.; Arnold, E.; Arnold, G.F.; et al. Structure-affinity relationships in the gp41 ELDKWA epitope for the HIV-1 neutralizing monoclonal antibody 2F5: Effects of side-chain and backbone modifications and conformational constraints. J. Pept. Res. 2002, 59, 264–276. [Google Scholar] [CrossRef]
- Trier, N.H.; Hansen, P.R.; Vadelser, C.A.; Somnier, F.E.; Houen, G. Identification of continuous epitopes of HuD antibodies related to paraneoplastic diseases/small cell lung cancer. J. Neuroimmunol. 2012, 243, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.M.; Henry, A.H.; Rees, A.R. Antibody-antigen interactions. Curr. Opin. Struct. Biol. 1994, 4, 123–129. [Google Scholar] [CrossRef]
- Frank, S.A. Specificity and cross-reactivity. In Immunology and Evolution of Infectious Disease; Princeton University Press: Princeton, NJ, USA, 2002. [Google Scholar]
- Agca, S.; Houen, G.; Trier, N.H. Characterization of continuous B-cell epitopes in the N-terminus of glutamate decarboxylase67 using monoclonal antibodies. J. Pept. Sci. 2014, 20, 928. [Google Scholar] [CrossRef]
- Trier, N.H.; Holm, B.E.; Slot, O.; Locht, H.; Lindegaard, H.; Svendsen, A.; Houen, G. Physical characteristics of a citrullinated pro-filaggrin epitope recognized by anti-citrullinated protein antibodies in rheumatoid arthritis sera. PLoS ONE 2016, 11, e0168542. [Google Scholar] [CrossRef]
- Welner, S.; Trier, N.H.; Houen, G.; Hansen, P.R. Identification and mapping of a linear epitope of centromere protein F using monoclonal antibodies. J. Pept. Sci. 2013, 19, 95. [Google Scholar] [CrossRef] [PubMed]
- Trier, N.H.; Leth, M.L.; Hansen, P.R.; Houen, G. Cross-reactivity of a human IgG1 anticitrullinated fibrinogen monoclonal antibody to a citrullinated pro-filaggrin peptide. Protein Sci. 2012, 21, 1929. [Google Scholar] [CrossRef]
- Tiwana, H.; Wilson, C.; Alvarez, A.; Abuknesha, R.; Bansal, S.; Ebringer, A. Cross-reactivity between rheumatoid arthritis-associated motif EQKRAA and structurally related sequences found in proteus mirabillis. Infect. Immun. 1999, 67, 2769–2775. [Google Scholar] [CrossRef]
- Yadav, P.; Tran, H.; Ebegbe, R.; Gottlieb, P.; Wei, H.; Lewis, R.H.; Mumbey-Wafula, A.; Kaplan, A.; Kholdarova, E.; Spatz, L. Antibodies Elicited in Response to EBNA-1 May Cross-React with DsDNA. PLoS ONE 2011, 6, e14488. [Google Scholar] [CrossRef]
- Yadav, P.; Carr, M.T.; Yu, R.; Mumbey-Wafula, A.; Spatz, L.A. Mapping an Epitope in EBNA-1 That Is Recognized by Monoclonal Antibodies to EBNA-1 That Cross-React with DsDNA. Immun. Inflamm. Dis. 2016, 4, 362–375. [Google Scholar] [CrossRef]
- Tejada-Simon, M.V.; Zang, Y.C.; Hong, J.; Rivera, V.M.; Zhang, J.Z. Cross-reactivity with myelin basic protein and human herpesvirus-6 in multiple sclerosis. Ann. Neurol. 2003, 53, 189–197. [Google Scholar] [CrossRef]
- Cheng, W.; Ma, Y.; Gong, F.; Hu, C.; Qian, L.; Huang, Q.; Yu, Q.; Zhang, J.; Chen, S.; Liu, Z.; et al. Cross-reactivity of autoreactive T cells with MBP and viral antigens in patients with MS. Front. Biosci. (Landmark Ed.) 2012, 17, 1648–1658. [Google Scholar] [CrossRef]
- Chunder, R.; Weier, A.; Mäurer, H.; Luber, N.; Enders, M.; Luber, G.; Heider, T.; Spitzer, A.; Tacke, S.; Becker-Gotot, J.; et al. Antibody cross-reactivity between casein and myelin-associated glycoprotein results in central nervous system demyelination. Proc. Natl. Acad. Sci. USA 2022, 119, e2117034119. [Google Scholar] [CrossRef] [PubMed]
- Amrutkar, S.D.; Trier, N.H.; Hansen, P.R.; Houen, G. Fine mapping of a monoclonal antibody to the N-Methyl D-aspartate receptor reveals a short linear epitope. Biopolymers 2012, 98, 567. [Google Scholar] [CrossRef] [PubMed]
- Valdarnini, N.; Holm, B.; Hansen, P.; Rovero, P.; Houen, G.; Trier, N. Fine mapping of glutamate decarbocylase 65 epitopes reveals dependency of hydrophobic amino acids for specific interactions. Int. J. Mol. Sci. 2019, 20, 2909. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, I.Ø.; Trier, N.H.; Friis, T.; Houen, G. Characterisation of continuous monoclonal antibody epitopes in the N-terminus of Ro60. Biopolymers 2016, 106, 62. [Google Scholar] [CrossRef]
- Trier, N.H.; Dam, C.E.; Olsen, D.T.; Hansen, P.R.; Houen, G. Contribution of peptide backbone to citrulline-dependent antibody reactivity. PLoS ONE 2015, 10, e0144707. [Google Scholar] [CrossRef] [PubMed]
- Petersen, N.H.; Hansen, P.R.; Houen, G. Fast and efficient characterization of an anti-gliadin monoclonal antibody related to celiac disease using resin-bound peptides. J. Immunol. Methods 2011, 365, 174. [Google Scholar] [CrossRef] [PubMed]
- Barreira, S.C.; Fonseca, J.E. The impact of conventional and biological disease modifying antirheumatic drugs on bone biology. Rheumatoid arthritis as a case study. Clin. Rev. Allergy Immunol. 2016, 51, 100–109. [Google Scholar] [CrossRef]
- Cambridge, G.; Leandro, M.J.; Lahey, L.J.; Fairhead, T.; Robinson, W.H.; Sokolove, J. B cell depletion with rituximab in patients with rheumatoid arthritis: Multiplex bead array reveals the kinetics of IgG and IgA antibodies to citrullinated antigens. J. Autoimmun. 2016, 70, 22–30. [Google Scholar] [CrossRef]
- Arleevskaya, M.; Takha, E.; Petrov, S.; Kazarian, G.; Renaudineau, Y.; Brooks, W.; Laionova, R.; Korovina, M.; Valeeva, A.; Shuralev, E.; et al. Interplay of environmental, individual and genetic factors in rheumatoid arthritis provocation. Int. J. Mol. Sci. 2022, 23, 8140. [Google Scholar] [CrossRef]
- Carlberg, C. Nutrogenomics of Vitamin D. Nutrients 2019, 11, 676. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagaski, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Tuomi, T.; Heliovaara, M.; Palosuo, T.; Aho, K. Smoking, lung function, and rheumatoid factors. Ann. Rheum. Dis. 1990, 49, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Begovich, A.B.; Carlton, V.E.; Honigberg, L.A.; Schrodi, S.J.; Chokkalingam, A.P.; Alexander, H.C.; Ardlie, K.G.; Huang, Q.; Smith, A.M.; Spoerke, J.M.; et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phos-phatase (PTPN22) is associated with rheumatoid arthritis. Am. J. Hum. Genet. 2004, 75, 330–337. [Google Scholar] [CrossRef]
- Song, G.G.; Bae, S.C.; Le, Y.H. Association between vitamin D intake and the risk of rheumatoid arthritis: A meta-analysis. Clin. Rheumatol. 2012, 31, 1733–1739. [Google Scholar] [CrossRef]
- Stastny, P. Mixed lymphocyte cultures in rheumatoid arthritis. J. Clin. Investig. 1976, 57, 1148–1157. [Google Scholar] [CrossRef]
- Stastny, P. Association of the B-Cell alloantigen DRw4 with rheumatoid arthritis. N. Engl. J. Med. 1978, 298, 869–871. [Google Scholar] [CrossRef]
- Scally, S.W.; Petersen, J.; Law, S.C.; Dudek, N.L.; Nel, H.J.; Loh, K.L.; Wijeyewickrema, L.C.; Eckle, S.B.; van Heemst, J.; Pike, R.N.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569–2582. [Google Scholar] [CrossRef]
- Rashid, T.; Jayakumar, K.S.; Binder, A.; Ellis, S.; Cunningham, P.; Ebringer, A. Rheumatoid arthritis patients have elevated antibodies to cross-reactive and non cross-reactive antigens from Proteus microbes. Clin. Exp. Rheumatol. 2007, 25, 259–267. [Google Scholar]
- Lundberg, K.; Wegner, N.; Yucel-Lindberg, T.; Venables, P. Periodintitis in RA—The citrullinated enolase connection. Nat. Rev. Rheumatol. 2010, 6, 727–730. [Google Scholar] [CrossRef]
- Kawahito, Y.; Ichinose, S.; Sano, H.; Tsubouchi, Y.; Kohno, M.; Yoshikawa, T.; Tokunaga, D.; Hojo, T.; Harasawa, R.; Nakano, T.; et al. Mycoplasma fermentans glycolipid-antigen as a pathogen of rheumatoid arthritis. Biochem. Biophys. Res. Commun. 2008, 369, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Ebringer, A.; Rashid, T. Rheumatoid arthritis is an autoimmune disease triggered by Proteus urinary tract infection. Clin. Dev. Immunol. 2006, 13, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Senior, B.W.; Anderson, G.A.; Morley, K.D.; Kerr, M.A. Evidence that patients with rheumatoid arthritis have asymptomatic “non-significant” Proteus mirabilis bacteriuria more frequently than healthy controls. J. Infect. 1999, 38, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Christopoulos, G.; Christopoulou, V.; Routsias, J.G.; Babionitakis, A.; Antoniadis, C.; Vaiopoulos, G. Greek rheumatoid arthritis patients have elevated levels of antibodies against antigens from Proteus mirabilis. Clin. Rheumatol. 2017, 36, 527–553. [Google Scholar] [CrossRef]
- de Smit, M.; Westra, J.; Vissink, A.; Doornbos-van der Meer, B.; Brouwer, E.; van Winkelhoff, A.J. Periodontitis in established rheumatoid arthritis patients: A crosssectional clinical, microbiological and serological study. Arthritis Res. Ther. 2012, 14, R222. [Google Scholar] [CrossRef]
- de Pablo, P.; Dietrich, T.; McAlindon, T.E. Association of periodontal disease and tooth loss with rheumatoid arthritis in the US population. J. Rheumatol. 2008, 35, 70–76. [Google Scholar]
- Dissick, A.; Redman, R.S.; Jones, M.; Rangan, B.V.; Reimold, A.; Griffiths, G.R.; Mikuls, T.R.; Amdur, R.L.; Richards, J.S.; Kerr, G.S. Association of periodontitis with rheumatoid arthritis: A pilot study. J. Periodontol. 2010, 81, 223–230. [Google Scholar] [CrossRef]
- Kinloch, A.J.; Alzabin, S.; Brintnell, W.; Wilson, E.; Barra, L.; Wegner, N.; Bell, D.A.; Cairns, E.; Venables, P.J. Immunization with Porphyromonas gingivalis enolase induces autoimmunity to mammalian alpha-enolase and arthritis in DR4-IE transgenic mice. Arthritis Rheum. 2011, 63, 3818–3823. [Google Scholar] [CrossRef]
- Cantley, M.D.; Haynes, D.R.; Marino, V.; Bartold, P.M. Pre-existing periodontitis exacerbates experimental arthritis in a mouse model. J. Clin. Periodontol. 2011, 38, 532–541. [Google Scholar] [CrossRef]
- Bartold, P.M.; Marino, V.; Cantley, M.; Haynes, D.R. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J. Clin. Periodontol. 2010, 37, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Maresz, K.J.; Hellvard, A.; Sroka, A.; Adamowicz, K.; Bielecka, E.; Koziel, J.; Gawron, K.; Mizgalska, D.; Marcinska, K.A.; Benedyk, M.; et al. Porphyromonas gingivalis facilitates the development and progression of destructive arthritis through its unique bacterial peptidylarginine deiminase (PAD). PLoS Pathog. 2013, 9, e1003627. [Google Scholar] [CrossRef] [PubMed]
- Trier, N.H.; Holm, B.E.; Heiden, J.; Slot, O.; Locht, H.; Jensen, B.; Lindegaard, H.; Svendsen, A.; Nielsen, C.T.; Jacobsen, S.; et al. The use of synthetic peptides for detection of anti-citrullinated protein antibodies in rheumatoid arthritis. J. Immunol. Methods 2018, 454, 6–14. [Google Scholar] [CrossRef]
- Trier, N.H.; Holm, B.E.; Heiden, J.; Slot, O.; Locht, H.; Lindegaard, H.; Svendsen, A.; Nielsen, C.T.; Jacobsen, S.; Theander, E.; et al. Antibodies to a strain-specific citrullinated Epstein-Barr virus peptide diagnoses rheumatoid arthritis. Sci. Rep. 2018, 8, 3684. [Google Scholar] [CrossRef] [PubMed]
- Alspaugh, M.A.; Jensen, F.C.; Rabin, H.; Tan, E.M. Lymphocytes transformed by Epstein-Barr virus. Induction of nuclear antigen reactive with antibody in rheumatoid arthritis. J. Exp. Med. 1978, 147, 1018–1027. [Google Scholar] [CrossRef]
- Westergaard, M.W.; Draborg, A.H.; Troelsen, L.; Jacobsen, S.; Houen, G. Isotypes of Epstein-Barr virus antibodies in rheumatoid arthritis: Association with rheumatoid factors and citrulline-dependent antibodies. Biomed. Res. Int. 2015, 2015, 472174. [Google Scholar] [CrossRef]
- Blaschke, S.; Schwarz, G.; Moneke, D.; Binder, L.; Muller, G.; Reuss-Borst, M. Epstein-Barr virus infection in peripheral blood mononuclear cells, synovial fluid cells, and synovial membranes of patients with rheumatoid arthritis. J. Rheumatol. 2000, 27, 866–873. [Google Scholar]
- Ferrell, P.B.; Aitcheson, C.T.; Pearson, G.R.; Tan, E.M. Seroepidemiological study of relationships between Epstein-Barr virus and rheumatoid arthritis. J. Clin. Investig. 1981, 67, 681–687. [Google Scholar] [CrossRef]
- Balandraud, N.; Meynard, J.B.; Auger, I.; Sovran, H.; Mugnier, B.; Reviron, D.; Roudier, J.; Roudier, C. EpsteinBarr virus load in the peripheral blood of patients with rheumatoid arthritis: Accurate quantification using real-time polymerase chain reaction. Arthritis Rheum. 2003, 48, 1223–1228. [Google Scholar] [CrossRef]
- Saal, J.G.; Krimmel, M.; Steidle, M.; Gerneth, F.; Wagner, S.; Fritz, P.; Koch, S.; Zacher, J.; Sell, S.; Einsele, H.; et al. Synovial Epstein-Barr virus infection increases the risk of rheumatoid arthritis in individuals with the shared HLA-DR4 epitope. Arthritis Rheum. 1999, 42, 1485–1496. [Google Scholar] [CrossRef]
- Takeda, T.; Mizugaki, Y.; Matsubara, L.; Imai, S.; Koike, T.; Takada, K. Lytic Epstein-Barr virus infection in the synovial tissue of patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Takei, M.; Mitamura, K.; Fujiwara, S.; Horie, T.; Ryu, J.; Osaka, S.; Yoshino, S.; Sawada, S. Detection of Epstein-Barr virus-encoded small RNA 1 and latent membrane protein 1 in synovial lining cells from rheumatoid arthritis patients. Int. Immunol. 1997, 9, 739–743. [Google Scholar] [CrossRef]
- Trier, N.H.; Izarzugaza, J.; Chailyan, A.; Marcatili, P.; Houen, G. Human MHC-II with Shared Epitope Motifs Are Optimal Epstein-Barr Virus Glycoprotein 42 Ligands-Relation to Rheumatoid Arthritis. Int. J. Mol. Sci. 2018, 19, 317. [Google Scholar] [CrossRef]
- Fox, R.; Sportsman, R.; Rhodes, G.; Luka, J.; Pearson, G.; Vaughan, J. Rheumatoid arthritis synovial membrane contains a 62,000-molecular-weight protein that shares an antigenic epitope with the Epstein-Barr virus encoded associated nuclear antigen. J. Clin. Investig. 1986, 77, 1539–1547. [Google Scholar] [CrossRef]
- Kouri, T.; Petersen, J.; Rhodes, G.; Aho, K.; Palosuo, T.; Heliovaara, M.; Isomaki, H.; von Essen, R.; Vaughan, J.H. Antibodies to synthetic peptides from Epstein-Barr nuclear antigen-1 in sera of patients with early rheumatoid arthritis and in preillness sera. J. Rheumatol. 1990, 17, 1442–1449. [Google Scholar] [PubMed]
- Lunemann, J.D.; Frey, O.; Eidner, T.; Baier, M.; Roberts, S.; Sashihara, J.; Volkmer, R.; Cohen, J.I.; Hein, G.; Kamradt, T.; et al. Increased frequency of EBV-specific effector memory CD8+ T cells correlates with higher viral load in rheumatoid arthritis. J. Immunol. 2008, 181, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Scotet, E.; David-Ameline, J.; Peyrat, M.A.; Moreau-Aubry, A.; Pinczon, D.; Lim, A.; Even, J.; Semana, G.; Berthelot, J.M.; Breathnach, R.; et al. T cell response to Epstein-Barr virus transactivators in chronic rheumatoid arthritis. J. Exp. Med. 1996, 184, 1791–1800. [Google Scholar] [CrossRef]
- Warde, N. Experimental arthritis: EBV induces arthritis in mice. Nat. Rev. Rheumatol. 2011, 683, 176. [Google Scholar]
- Kuwana, Y.; Takei, M.; Yajima, M.; Imadome, K.; Inomata, H.; Shiozaki, M.; Ikumi, N.; Nozaki, T.; Shiraiwa, H.; Kitamura, N.; et al. Epstein-Barr virus induces erosive arthritis in humanized mice. PLoS ONE 2011, 6, e26630. [Google Scholar] [CrossRef]
- Schellekens, G.A.; de Jong, B.A.; van den Hoogen, F.H.; van de Putte, L.B.; van Venrooij, W.J. Citrulline is an essential constituent of antigenic determinants recognized rheumatoid arthritis-specific autoantibodies. J. Clin. Investig. 1998, 101, 273–281. [Google Scholar] [CrossRef]
- Helmick, C.G.; Felson, D.T.; Lawrence, R.C.; Gabriel, S.; Hirsch, R.; Kwoh, C.K.; Liang, M.H.; Kremers, H.M.; Mayes, M.D.; Merkel, P.A.; et al. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States Part I. Arthritis Rheum. 2008, 58, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Danchenko, N.; Satia, J.A.; Anthony, M.S. Epidemiology of systemic lupus erythematosus: A comparison of worldwide disease burden. Lupus 2006, 15, 308–318. [Google Scholar] [CrossRef]
- Yu, C.; Gershwin, M.E.; Chang, C. Diagnostic criteria for systemic lupus erythematosus: A critical review. J. Autoimmun. 2014, 48, 10–13. [Google Scholar] [CrossRef]
- Cunha, J.S.; Gilek-Seibert, K. Systemic lupus erythematosus: A review of the clinical approach to diagnosis and update on current targeted therapies. Rhode Isl. Med. J. 2016, 99, 23–27. [Google Scholar]
- Kalinina, O.; Louzoun, Y.; Wang, Y.; Utset, T.; Weigert, M. Origins and specificity of auto-antibodies in Sm+ SLE patients. J. Autoimmun. 2018, 90, 94–104. [Google Scholar] [CrossRef]
- Lu, R.; Munroe, M.E.; Guthridge, J.M.; Bean, K.M.; Fife, D.A.; Chen, H.; Slight-Webb, S.; Keith, M.P.; Harley, J.B.; James, J.A. Dysregulation of innate and adaptive serum mediators precedes systemic lupus erythematosus classification and improves prognostic accuracy of Autoantibodies. J. Autoimmun. 2016, 74, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Kim, S.T.; Craft, J. The pathogenesis of systemic lupus erythematosus—An update. Curr. Opin. Immunol. 2012, 24, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Munroe, M.E.; Lu, R.; Zhao, Y.D.; Fife, D.A.; Robertson, J.M.; Guthridge, J.M.; Niewold, T.B.; Tsokos, G.C.; Keith, M.P.; Harley, J.B.; et al. Altered type II interferon precedes autoantibody accrual and elevated type I interferon activity prior to systemic lupus erythematosus classification. Ann. Rheum. Dis. 2016, 75, 2014–2021. [Google Scholar] [CrossRef]
- Lewis, M.J.; McAndrew, M.B.; Wheeler, C.; Workman, N.; Agashe, P.; Koopmann, J.; Uddin, E.; Morris, D.L.; Zou, L.; Stark, R.; et al. Autoantibodies targeting TLR and SMAD pathways define new subgroups in systemic lupus erythematosus. J. Autoimmun. 2018, 91, 1–12. [Google Scholar] [CrossRef]
- Anaya, J.M.; Leon, M.; Rojas, M.; Rodriguez, Y.; Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Ramirez-Santana, C. Progress towards precision medicine for lupus: The role of genetic biomarkers. Expert Rev. Precis. Med. Drug Dev. 2018, 3, 119–135. [Google Scholar] [CrossRef]
- Gupta, V.; Kumar, S.; Pratap, A.; Singh, R.; Kumari, R.; Kumar, S.; Aggarwal, A.; Misra, R. Association of ITGAM, TNFSF4, TNFAIP3 and STAT4 gene polymorphisms with risk of systemic lupus erythematosus in a North Indian population. Lupus 2018, 27, 1973–1979. [Google Scholar] [CrossRef]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.D.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Jarvinen, T.M.; Hellquist, A.; Zucchelli, M.; Koskenmies, S.; Panelius, J.; Hasan, T.; Julkunen, H.; D’Amato, M.; Kere, J. Replication of GWAS-identified systemic lupus erythematosus susceptibility genes affirms B-cell receptor pathway signalling and strengthens the role of IRF5 in disease susceptibility in a Northern European population. Rheumatology 2012, 51, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Shipman, L. Systemic lupus erythematosus: New GWAS loci and insights into ancestry. Nat. Rev. Rheumatol. 2016, 12, 499. [Google Scholar]
- Vyse, T.J. Genome-wide association metaanalysis in Chinese and European individuals identifies ten new loci associated with systemic lupus erythematosus. Nat. Genet. 2016, 48, 940–946. [Google Scholar]
- Grennan, D.M.; Parfitt, A.; Manolios, N.; Huang, Q.; Hyland, V.; Dunckley, H.; Doran, T.; Gatenby, P.; Badcock, C. Family and twin studies in systemic lupus erythematosus. Dis. Markers 1997, 13, 93–98. [Google Scholar]
- Ulff-Moller, C.J.; Svendsen, A.J.; Viemose, L.N.; Jacobsen, S. Concordance of autoimmune disease in a nationwide Danish systemic lupus erythematosus twin cohort. Semin. Arthritis Rheum. 2018, 47, 538–544. [Google Scholar] [CrossRef] [PubMed]
- Tsokos, G.C. Systemic lupus erythematosus. N Engl J Med. 2011, 365, 2110–2121. [Google Scholar] [CrossRef] [PubMed]
- Sundar, K.; Jacques, S.; Gottlieb, P.; Villars, R.; Benito, M.E.; Taylor, D.K.; Spatz, L.A. Expression of the Epstein-Barr virus nuclear antigen-1 (EBNA-1) in the mouse can elicit the production of anti-dsDNA and anti-Sm antibodies. J. Autoimmun. 2004, 23, 127–140. [Google Scholar] [CrossRef]
- Barzilai, O.; Ram, M.; Shoenfeld, Y. Viral infection can induce the production of autoantibodies. Curr. Opin. Rheumatol. 2007, 19, 636–643. [Google Scholar] [CrossRef]
- James, J.A.; Kaufman, K.M.; Farris, A.D.; Taylor-Albert, E.; Lehman, T.J.; Harley, J.B. An increased prevalence of Epstein-Barr virus infection in young patients suggests a possible etiology for systemic lupus erythematosus. J. Clin. Investig. 1997, 100, 3019–3026. [Google Scholar] [CrossRef] [PubMed]
- Poole, B.D.; Gross, T.; Maier, S.; Harley, J.B.; James, J.A. Lupus-like autoantibody development in rabbits and mice after immunization with EBNA-1 fragments. J. Autoimmun. 2008, 31, 362–371. [Google Scholar] [CrossRef]
- Sabbatini, A.; Bombardieri, S.; Migliorini, P. Autoantibodies from patients with systemic lupus erythematosus bind a shared sequence of SmD and Epstein-Barr virus-encoded nuclear antigen EBNA I. Eur. J. Immunol. 1993, 23, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- James, J.A.; Gross, T.; Scofield, R.H.; Harley, J.B. Immunoglobulin epitope spreading and autoimmune disease after peptide immunization: Sm B/B’-derived PPPGMRPP and PPPGIRGP induce spliceosome autoimmunity. J. Exp. Med. 1995, 181, 453–461. [Google Scholar] [CrossRef]
- Emiliani, Y.; Muzi, G.; Sánchez, A.; Sánchez, J.; Munera, M. Prediction of molecular mimicry between proteins from Trypanosoma sp. and human antigens associated with systemic lupus erythematosus. Microb. Pathog. 2022, 172, 105760. [Google Scholar] [CrossRef] [PubMed]
- Múnera, M.; Farak, J.; Pérez, M.; Rojas, J.; Villero, J.; Sánchez, A.; Sánchez, J.; Emiliani, Y. Prediction of molecular mimicry between antigens from Leishmania sp. and human: Implications for autoimmune response in systemic lupus erythematosus. Microb. Pathog. 2020, 148, 104444. [Google Scholar] [CrossRef]
- Fox, N.C.; Jenkins, R.; Leary, S.M.; Stevenson, V.L.; Losseff, N.A.; Crum, W.R.; Harvey, R.J.; Rossor, M.N.; Miller, D.H.; Thompson, A.J. Progressive cerebral atrophy in MS: A serial study using registered, volumetric MRI. Neurology 2000, 54, 807–812. [Google Scholar] [CrossRef]
- Ramagopalan, S.V.; Dobson, R.; Meier, U.C.; Giovannoni, G. Multiple sclerosis: Risk factors, prodromes, and potential causal pathways. Lancet Neurol. 2010, 9, 727–739. [Google Scholar] [CrossRef]
- Libbey, J.E.; Cusick, M.F.; Fujinami, R.S. Role of pathogens in multiple sclerosis. Int. Rev. Immunol. 2014, 33, 266–283. [Google Scholar] [CrossRef]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schroder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of CD8(+) T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef]
- Saxena, A.; Martin-Blondel, G.; Mars, L.T.; Liblau, R.S. Role of CD8 T cell subsets in the pathogenesis of multiple sclerosis. FEBS Lett. 2011, 585, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, B.M.; Noyce, A.J.; Bestwick, J.; Belete, D.; Giovannoni, G.; Dobson, R. Gene-Environment Interactions in Multiple Sclerosis: A UK Biobank Study. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e1007. [Google Scholar] [CrossRef]
- Alfredsson, L.; Olsson, T. Lifestyle and Environmental Factors in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a028944. [Google Scholar] [CrossRef] [PubMed]
- Otto, C.; Hofmann, J.; Rubrecht, K. Antibody producing B lineage cells invade the central nervous system predominantly at the time of and triggered by acute Epstein-Barr virus infection: A hypothesis on the origin of intrathecal immunoglobulin synthesis in multiple sclerosis. Med. Hypotheses 2016, 91, 109–113. [Google Scholar] [CrossRef]
- Bjornevik, K.; Munz, C.; Cohen, J.I.; Ascherio, A. Epstein-Barr virus as a leading cause of multiple sclerosis: Mechanisms and implications. Nat. Rev. Neurol. 2023, 19, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Hottenrott, T.; Dersch, R.; Berger, B.; Rauer, S.; Huzly, D.; Stich, O. The MRZ reaction in primary progressive multiple sclerosis. Fluids Barriers CNS 2017, 14, 2. [Google Scholar] [CrossRef] [PubMed]
- Houen, G.; Heiden, J.; Trier, N.H.; Draborg, A.H.; Benros, M.E.; Zinkevičiūtė, R.; Petraitytė-Burneikienė, R.; Ciplys, E.; Slibinskas, R.; Frederiksen, J.L. Antibodies to Epstein-Barr virus and neurotropic viruses in multiple sclerosis and optic neuritis. J. Neuro Immunol. 2020, 346, 577314. [Google Scholar] [CrossRef]
- Gåsland, H.; Trier, N.H.; Kyllesbech, C.; Draborg, A.H.; Slibinskas, R.; Ciplys, E.; Frederiksen, J.L.; Houen, G. Antibodies to expanded virus antigen panels show elevated diagnostic sensitivities in multiple sclerosis and optic neuritis. Immunol. Lett. 2023, 254, 54–64. [Google Scholar] [CrossRef]
- Kyllesbech, C.; Trier, N.H.; Slibinskas, R.; Ciplys, E.; Tsakiri, A.; Frederiksen, J.L.; Houen, G. Virus-specific antibody indices may supplement the total IgG index in diagnostics of multiple sclerosis. J. Neuroimmunol. 2022, 367, 577868. [Google Scholar] [CrossRef]
- Hedström, A.K.; Huang, J.; Michel, S.; Butt, J.; Brenner, N.; Hillert, J.; Waterboer, T.; Kochum, I.; Olsson, T.; Alfredsson, L. High levels of Epstein-Barr virus nuclear antigen-1-specific antibodies and infectious mononucleosis act both independently and synergistically to increase multiple sclerosis risk. Front. Neurol. 2019, 10, 1368. [Google Scholar] [CrossRef]
- Tengvall, K.; Huang, J.; Hellström, C.; Krammer, P.; Biström, M.; Ayoglu, B.; Bomfim, I.L.; Stridh, P.; Bitt, J.; Brenner, N.; et al. Molecular mimicry between Anoctamin 2 and Epstein-Barr virus nuclear antigen 1 associates with multiple sclerosis risk. Proc. Natl. Acad. Sci. USA 2019, 116, 16955–16960. [Google Scholar] [CrossRef]
- Lanz, T.V.; Brewer, C.R.; Ho, P.P.; Moon, J.-S.; Jude, K.M.; Fernandez, D.; Gomez, M.A.; Nadj, G.-S.; Bartley, C.M.; Schubert, R.D.; et al. Clonally expanded B cells in multiple sclerosis bind EBV EBNA1 and GlialCAM. Nature 2022, 603, 321–327. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The enigmatic role of viruses in multiple sclerosis: Molecular mimicry or disturbed immune surveillance? Trends Immunol. 2017, 38, 451–498. [Google Scholar] [CrossRef]
- Holmoy, T.; Kvale, E.O.; Vartdal, F. Cerebrospinal fluid CD4+ T cells from a multiple sclerosis patient cross-recognize Epstein-Barr virus and myelin basic protein. J Neurovirol 2004, 10, 278–283. [Google Scholar] [CrossRef]
- Lunemann, J.D.; Jelcic, I.; Roberts, S.; Lutterotti, A.; Tackenberg, B.; Martin, R.; Munz, C. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J. Exp. Med. 2008, 205, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Wucherpfennig, K.W.; Strominger, J.L. Molecular mimicry in T cell-mediated autoimmunity: Viral peptides activate human T cell clones specific for myelin basic protein. Cell 1995, 80, 695–705. [Google Scholar] [CrossRef] [PubMed]
- van Sechel, A.C.; Bajramovic, J.J.; van Stipdonk, M.J.; Persoon-Deen, C.; Geutskens, S.B.; van Noort, J.M. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J. Immunol. 1999, 162, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Capone, G.; Calabró, M.; Lucchese, G.; Fasano, C.; Girardi, B.; Polimeno, L.; Kanduc, D. Peptide matching between EpsteinBarr virus and human proteins. Pathog. Dis. 2013, 69, 205–212. [Google Scholar] [CrossRef]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol Disord. Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef]
- Sola, P.; Merelli, E.; Marasca, R.; Poggi, M.; Luppi, M.; Montorsi, M.; Torelli, G. Human herpesvirus 6 and multiple sclerosis: Survey of anti-HHV-6 antibodies by immunofluorescence analysis and of viral sequences by polymerase chain reaction. J. Neurol. Neurosurg. Psychiatry 1993, 56, 917–919. [Google Scholar] [CrossRef]
- Wilborn, F.; Schmidt, C.A.; Brinkmann, V.; Jendroska, K.; Oettle, H.; Siegert, W. A potential role for human herpesvirus type 6 in nervous system disease. J. Neuroimmunol. 1994, 49, 213–214. [Google Scholar] [CrossRef]
- Cirone, M.; Cuomo, L.; Zompetta, C.; Ruggieri, S.; Frati, L.; Faggioni, A.; Ragona, G. Human herpesvirus 6 and multiple sclerosis: A study of T cell cross-reactivity to viral and myelin basic protein antigens. J. Med. Virol. 2002, 68, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Harkiolaki, M.; Holmes, S.L.; Svendsen, P.; Gregersen, J.W.; Jensen, L.T.; McMahon, R.; Friese, M.A.; van Boxel, G.; Etzensperger, R.; Tzartos, J.S.; et al. T cell-mediated autoimmune disease due to low-affinity cross-reactivity to common microbial peptides. Immunity 2009, 30, 348–357. [Google Scholar] [CrossRef]
- Kahaly, G.J.; Hansen, M.P. Type 1 diabetes associated autoimmunity. Autoimmun. Rev. 2016, 15, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Gianani, R.; Eisenbarth, G.S. The stages of type 1A diabetes: 2005. Immunol. Rev. 2005, 204, 232–249. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, N.S.; Rui, J.; Hebrok, M.; Herold, K.C. Life and death of beta cells in Type 1 diabetes: A comprehensive review. J. Autoimmun. 2016, 71, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Karvonen, M.; Viik-Kajander, M.; Moltchanova, E.; Libman, I.; LaPorte, R.; Tuomilehto, J. Incidence of childhood type 1 diabetes worldwide. Diabetes mondiale (DiaMond) project group. Diabetes Care 2000, 23, 1516–1526. [Google Scholar] [CrossRef]
- Gepts, W. Islet changes suggesting a possible immune aetiology of human diabetes mellitus. Acta Endocrinol. Suppl. 1976, 205, 95–106. [Google Scholar]
- Bottazzo, G.F.; Dean, B.M.; McNally, J.M.; MacKay, E.H.; Swift, P.G.; Gamble, D.R. In situ characterization of autoimmune phenomena and expression of HLA molecules in the pancreas in diabetic insulitis. N. Engl. J. Med. 1985, 313, 353–360. [Google Scholar] [CrossRef]
- Roep, B.O. The role of T-cells in the pathogenesis of type 1 diabetes: From cause to cure. Diabetologia 2003, 46, 305–321. [Google Scholar] [CrossRef]
- Roep, B.O.; Thomaidou, S.; van Tienhoven, R.; Zaldumbide, A. Type 1 diabetes mellitus as a disease of the beta-cell (do not blame the immune system?). Nat. Rev. Endocrinol. 2021, 17, 150–161. [Google Scholar] [CrossRef]
- Lampeter, E.F.; McCann, S.R.; Kolb, H. Transfer of diabetes type 1 by bone-marrow transplantation. Lancet 1998, 351, 568–569. [Google Scholar] [CrossRef]
- Rojas-Villarraga, A.; Botello-Corzo, D.; Anaya, J.M. HLA-Class II in Latin American patients with type 1 diabetes. Autoimmun. Rev. 2010, 9, 666–673. [Google Scholar] [CrossRef]
- Thomson, G.; Valdes, A.M.; Noble, J.A.; Kockum, I.; Grote, M.N.; Najman, J.; Erlich, H.A.; Cucca, F.; Pugliese, A.; Steenkiste, A.; et al. Relative predispositional effects of HLA class II DRB1-DQB1 haplotypes and genotypes on type 1 diabetes: A meta-analysis. Tissue Antigens 2007, 70, 110–127. [Google Scholar] [CrossRef]
- Ye, J.; Richardson, T.G.; McArdle, W.L.; Relton, C.L.; Gillespie, K.M.; Suderman, M.; Hemani, G. Identification of loci where DNA methylation potentially mediates genetic risk of type 1 diabetes. J. Autoimmun. 2018, 93, 66–75. [Google Scholar] [CrossRef]
- Coppieters, K.T.; Wiberg, A.; von Herrath, M.G. Viral infections and molecular mimicry in type 1 diabetes. APMIS 2012, 120, 941–949. [Google Scholar] [CrossRef]
- Karvonen, M.; Jantti, V.; Muntoni, S.; Stabilini, M.; Stabilini, L.; Muntoni, S.; Tuomilehto, J. Comparison of the seasonal pattern in the clinical onset of IDDM in Finland and Sardinia. Diabetes Care 1998, 21, 1101–1109. [Google Scholar] [CrossRef]
- Rasmussen, T.; Witso, E.; Tapia, G.; Stene, L.C.; Ronningen, K.S. Self-reported lower respiratory tract infections and development of islet autoimmunity in children with the type 1 diabetes high-risk HLA genotype: The MIDIA study. Diabetes Metab. Res. Rev. 2011, 27, 834–837. [Google Scholar] [CrossRef]
- Wagenknecht, L.E.; Roseman, J.M.; Herman, W.H. Increased incidence of insulin-dependent diabetes mellitus following an epidemic of Coxsackievirus B5. Am. J. Epidemiol. 1991, 133, 1024–1031. [Google Scholar] [CrossRef]
- Yeung, W.C.; Rawlinson, W.D.; Craig, M.E. Enterovirus infection and type 1 diabetes mellitus: Systematic review and meta-analysis of observational molecular studies. BMJ 2011, 342, d35. [Google Scholar] [CrossRef]
- Dotta, F.; Censini, S.; van Halteren, A.G.; Marselli, L.; Masini, M.; Dionisi, S.; Mosca, F.; Boggi, U.; Muda, A.O.; Del Prato, S.; et al. Coxsackie B4 virus infection of beta cells and natural killer cell insulitis in recent-onset type 1 diabetic patients. Proc. Natl. Acad. Sci. USA 2007, 104, 5115–5120. [Google Scholar] [CrossRef]
- Al-Hello, H.; Paananen, A.; Eskelinen, M.; Ylipaasto, P.; Hovi, T.; Salmela, K.; Lukashev, A.N.; Bobegamage, S.; Roivainen, M. An enterovirus strain isolated from diabetic child belongs to a genetic subcluster of echovirus 11, but is also neutralised with monotypic antisera to coxsackievirus A9. J. Gen. Virol. 2008, 89, 1949–1959. [Google Scholar] [CrossRef]
- Nilsson, A.L.; Vaziri-Sani, F.; Broberg, P.; Elfaitouri, A.; Pipkorn, R.; Blomberg, J.; Ivarsson, S.A.; Elding Larsson, H.; Lernmark, A. Serological evaluation of possible exposure to Ljungan virus and related parechovirus in autoimmune (type 1) diabetes in children. J. Med. Virol. 2015, 87, 1130–1140. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Laine, D.; Zhan, Y.; Londrigan, S.; Kirkwood, C.; Harrison, L.C. Rotavirus infection induces transient pancreatic involution and hyperglycemia in weanling mice. PLoS ONE 2014, 9, e106560. [Google Scholar] [CrossRef]
- Valdes, C.; Unanue, N.; Hernandez, M.; Garcia, R.; Castro, M.; Vasquez, L.; Torres, J.P.; Mericq, V. Is there a link between influenza and type I diabetes? Increased incidence of TID during the pandemic H1N1 influenza of 2009 in Chile. Pediatr. Endocrinol. Rev. 2013, 11, 161–166. [Google Scholar]
- Kondrashova, A.; Nurminen, N.; Patrikainen, M.; Huhtala, H.; Lehtonen, J.; Toppari, J.; Ilonen, J.; Simell, O.G.; Veijola, R.; Knip, M.; et al. Influenza A virus antibodies show no association with pancreatic islet autoantibodies in children genetically predisposed to type 1 diabetes. Diabetologia 2015, 58, 2592–2595. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Isaacs, S.R.; Foskett, D.B.; Maxwell, A.J.; Ward, E.J.; Faulkner, C.L.; Luo, J.Y.X.; Rawlinson, W.D.; Craig, M.E.; Kim, K.W. Viruses and Type 1 Diabetes: From Enteroviruses to the Virome. Microorganisms 2021, 9, 1519. [Google Scholar] [CrossRef]
- Filippi, C.M.; von Herrath, M.G. Viral trigger for type 1 diabetes: Pros and cons. Diabetes 2008, 57, 2863–2871. [Google Scholar] [CrossRef]
- Andreoletti, L.; Hober, D.; Hober-Vandenberghe, C.; Belaich, S.; Vantyghem, M.C.; Lefebvre, J.; Wattre, P. Detection of coxsackie B virus RNA sequences in whole blood samples from adult patients at the onset of type I diabetes mellitus. J. Med. Virol. 1997, 52, 121–127. [Google Scholar] [CrossRef]
- Hyoty, H.; Hiltunen, M.; Knip, M.; Laakkonen, M.; Vahasalo, P.; Karjalainen, J.; Koskela, P.; Roivainen, M.; Leinikki, P.; Hovi, T.; et al. A prospective study of the role of coxsackie B and other enterovirus infections in the pathogenesis of IDDM. Childhood Diabetes in Finland (DiMe) Study Group. Diabetes 1995, 44, 652–657. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Bowman, M.A.; Campbell, L.; Darrow, B.L.; Kaufman, D.L.; Maclaren, N.K. Cellular immunity to a determinant common to glutamate decarboxylase and coxsackie virus in insulin-dependent diabetes. J. Clin. Investig. 1994, 94, 2125–2129. [Google Scholar] [CrossRef]
- Tian, J.; Lehmann, P.V.; Kaufman, D.L. T cell cross-reactivity between coxsackievirus and glutamate decarboxylase is associated with a murine diabetes susceptibility allele. J. Exp. Med. 1994, 180, 1979–1984. [Google Scholar] [CrossRef] [PubMed]
- Tracy, S.; Drescher, K.M.; Chapman, N.M.; Kim, K.S.; Carson, S.D.; Pirruccello, S.; Lane, P.H.; Romero, J.R.; Leser, J.S. Toward testing the hypothesis that group B coxsackieviruses (CVB) trigger insulin-dependent diabetes: Inoculating nonobese diabetic mice with CVB markedly lowers diabetes incidence. J. Virol. 2002, 76, 12097–12111. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, M.S.; Bradley, L.M.; Harbertson, J.; Krahl, T.; Lee, J.; Sarvetnick, N. Diabetes induced by Coxsackie virus: Initiation by bystander damage and not molecular mimicry. Nat. Med. 1998, 4, 781–785. [Google Scholar] [CrossRef] [PubMed]
- Richter, W.; Mertens, T.; Schoel, B.; Muir, P.; Ritzkowsky, A.; Scherbaum, W.A.; Boehm, B.O. Sequence homology of the diabetes-associated autoantigen glutamate decarboxylase with coxsackie B4-2C protein and heat shock protein 60 mediates no molecular mimicry of autoantibodies. J. Exp. Med. 1994, 180, 721–726. [Google Scholar] [CrossRef]
- Schloot, N.C.; Willemen, S.J.; Duinkerken, G.; Drijfhout, J.W.; de Vries, R.R.; Roep, B.O. Molecular mimicry in type 1 diabetes mellitus revisited: T-cell clones to GAD65 peptides with sequence homology to Coxsackie or proinsulin peptides do not crossreact with homologous counterpart. Hum. Immunol. 2001, 62, 299–309. [Google Scholar] [CrossRef]
- Burbelo, P.D.; Groot, S.; Dalakas, M.C.; Iadarola, M.J. High-definition profiling of autoantibodies to glutamic acid decarboxylases GAD65/GAD67 in stiff-person syndrome. Biochem. Biophys. Res. Commun. 2008, 366, 1–7. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Stone, N.L.; Harrison, L.C. T-cell epitopes in type 1 diabetes autoantigen tyrosine phosphatase IA-2: Potential for mimicry with rotavirus and other environmental agents. Mol. Med. 1998, 4, 231–239. [Google Scholar] [CrossRef]
- Honeyman, M.C.; Coulson, B.S.; Stone, N.L.; Gellert, S.A.; Goldwater, P.N.; Steele, C.E.; Couper, J.J.; Tait, B.D.; Colman, P.G.; Harrison, L.C. Association between rotavirus infection and pancreatic islet autoimmunity in children at risk of developing type 1 diabetes. Diabetes 2000, 49, 1319–1324. [Google Scholar] [CrossRef]
- Blomqvist, M.; Juhela, S.; Erkkila, S.; Korhonen, S.; Simell, T.; Kupila, A.; Vaarala, O.; Simell, O.; Knip, M.; Ilonen, J. Rotavirus infections and development of diabetes-associated autoantibodies during the first 2 years of life. Clin. Exp. Immunol. 2002, 128, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Makela, M.; Oling, V.; Marttila, J.; Waris, M.; Knip, M.; Simell, O.; Ilonen, J. Rotavirus-specific T cell responses and cytokine mRNA expression in children with diabetes-associated autoantibodies and type 1 diabetes. Clin. Exp. Immunol. 2006, 145, 261–270. [Google Scholar] [CrossRef]
- Starovasnik, M.A.; Braisted, A.C.; Wells, J.A. Structural mimicry of a native protein by a minimized binding domain. Proc. Natl. Acad. Sci. USA 1997, 94, 10080–10085. [Google Scholar] [CrossRef]
- Bergmann, A.C.; Houen, G.; Trier, N.H. Determination of crucial epitopes in the sperm protein calsperin employing synthetic peptides and monoclonal antibodies. J. Pept. Sci. 2023, 29, e3450. [Google Scholar] [CrossRef]
- Flaherty, D.K. Immunogenicity and antigenicity. Immunol. Pharm. 2012, 23–30. [Google Scholar]
- Rubinstein, N.D.; Mayrose, I.; Halperin, D.; Yekutieli, D.; Gershoni, J.M.; Pupko, T. Computational characterization of B-cell epitopes. Mol. Immunol. 2008, 45, 3477–3489. [Google Scholar] [CrossRef]
- Trost, B.; Lucchese, G.; Stufano, A.; Bickis, M.; Kusalik, A.; Kanduc, D. No human protein is exempt from bacterial motifs, not even one. Self Nonself 2010, 1, 328–334. [Google Scholar] [CrossRef]
- Dam, C.E.; Houen, G.; Trier, N.H. The dependency on neighboring amino acids for reactivity of anti-citrullinated protein antibodies to citrullinated proteins. Scand. J. Clin. Lab. Investig. 2016, 76, 417. [Google Scholar] [CrossRef]
- Mahler, M.; Fritzler, M.J. Epitope specificity and significance in systemic autoimmune diseases. Ann. N. Y. Acad. Sci. 2010, 1183, 267–287. [Google Scholar] [CrossRef]
- Ge, C.; Tong, D.; Liang, B.; Lönnblom, E.; Scheneider, N.; Hafert, C.; Viljanen, J.; Ayoglu, B.; Stawikowska, R.; Nilsson, P.; et al. Anticitrullinated protein antibodies cause arthritis by cross-reactivity to joint cartilage. JCI Insight 2017, 2, e93688. [Google Scholar] [CrossRef]
- Ge, C.; Xu, B.; Liang, B.; Lönnblom, E.; Lundström, S.L.; Zubarev, R.A.; Ayoglu, B.; Nilsson, P.; Skogh, T.; Kastbom, A.; et al. Structural Basis of Cross-Reactivity of Anti-Citrullinated Protein Antibodies. Arthritis Rheumatol. 2019, 71, 210–221. [Google Scholar] [CrossRef] [PubMed]
- Amagai, M. Non-pathogenic anti-desmoglein 3 IgG autoantibodies in Fogo Selvagem. J. Investig. Dermatol. 2006, 126, 1931–1932. [Google Scholar] [CrossRef]
- Land, J.; Rutgers, A.; Kallenberg, C.G. Anti-neutrophil cytoplasmic autoantibody pathogenicity revisited: Pathogenic versus non-pathogenic anti-neutrophil cytoplasmic autoantibody. Nephrol Dial Transplant. 2014, 29, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Graßhoff, H.; Fourlakis, K.; Comdühr, S.; Riemekasten, G. Autoantibodies as Biomarker and Therapeutic Target in Systemic Sclerosis. Biomedicines 2022, 10, 2150. [Google Scholar] [CrossRef]
- Agrawal, B. Heterologous immunity: Role in natural and vaccine-induced resistance to infections. Front Immunol 2019, 10, 2631. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.D.; Fraire, A.E.; Joris, I.; Brehm, M.A.; Welsch, R.M.; Selin, L.K. Memory CD8+ T cells in heterologous antiviral immunity an immunopathology in the lung. Nat. Immunol. 2021, 2, 1067–1076. [Google Scholar] [CrossRef]
- Nickerson, C.; Luthra, H.; David, C. Antigenic mimicry and autoimmune diseases. Int. Rev. Immunol. 1991, 7, 205–224. [Google Scholar] [CrossRef]
- Pacheco, Y.; Acosta-Ampudia, Y.; Monsalve, D.M.; Chang, C.; Gershwin, M.E.; Anaya, J.M. Bystander activation and autoimmunity. J. Autoimmun. 2019, 103, 102301. [Google Scholar] [CrossRef]

| % Sequence Identity | % Structural Identity | Type of Reactivity | Reference | |
|---|---|---|---|---|
| Epitope identity at the protein level between a host and an infectious agent, having “hijacked” a human protein | 100 | 100 | Specific | [8,90,91] |
| Similarity at the three-dimensional protein level between | 2550 | 75–100 | Cross-reactive | [8,41,91] |
| Similarity at the amino acid level in epitopes or critical hot spots (peptide level) | 75–100 | 25–50 | Cross-reactive | [8,84,85,88,91] |
| Structural “similarities” (mimotopes) | 0–25 | 75–100 | Cross-reactive | [8,89,91] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trier, N.H.; Houen, G. Antibody Cross-Reactivity in Auto-Immune Diseases. Int. J. Mol. Sci. 2023, 24, 13609. https://doi.org/10.3390/ijms241713609
Trier NH, Houen G. Antibody Cross-Reactivity in Auto-Immune Diseases. International Journal of Molecular Sciences. 2023; 24(17):13609. https://doi.org/10.3390/ijms241713609
Chicago/Turabian StyleTrier, Nicole Hartwig, and Gunnar Houen. 2023. "Antibody Cross-Reactivity in Auto-Immune Diseases" International Journal of Molecular Sciences 24, no. 17: 13609. https://doi.org/10.3390/ijms241713609
APA StyleTrier, N. H., & Houen, G. (2023). Antibody Cross-Reactivity in Auto-Immune Diseases. International Journal of Molecular Sciences, 24(17), 13609. https://doi.org/10.3390/ijms241713609