
Naegleria fowleri Extracellular Vesicles Induce Proinflammatory Immune Responses in BV-2 Microglial Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
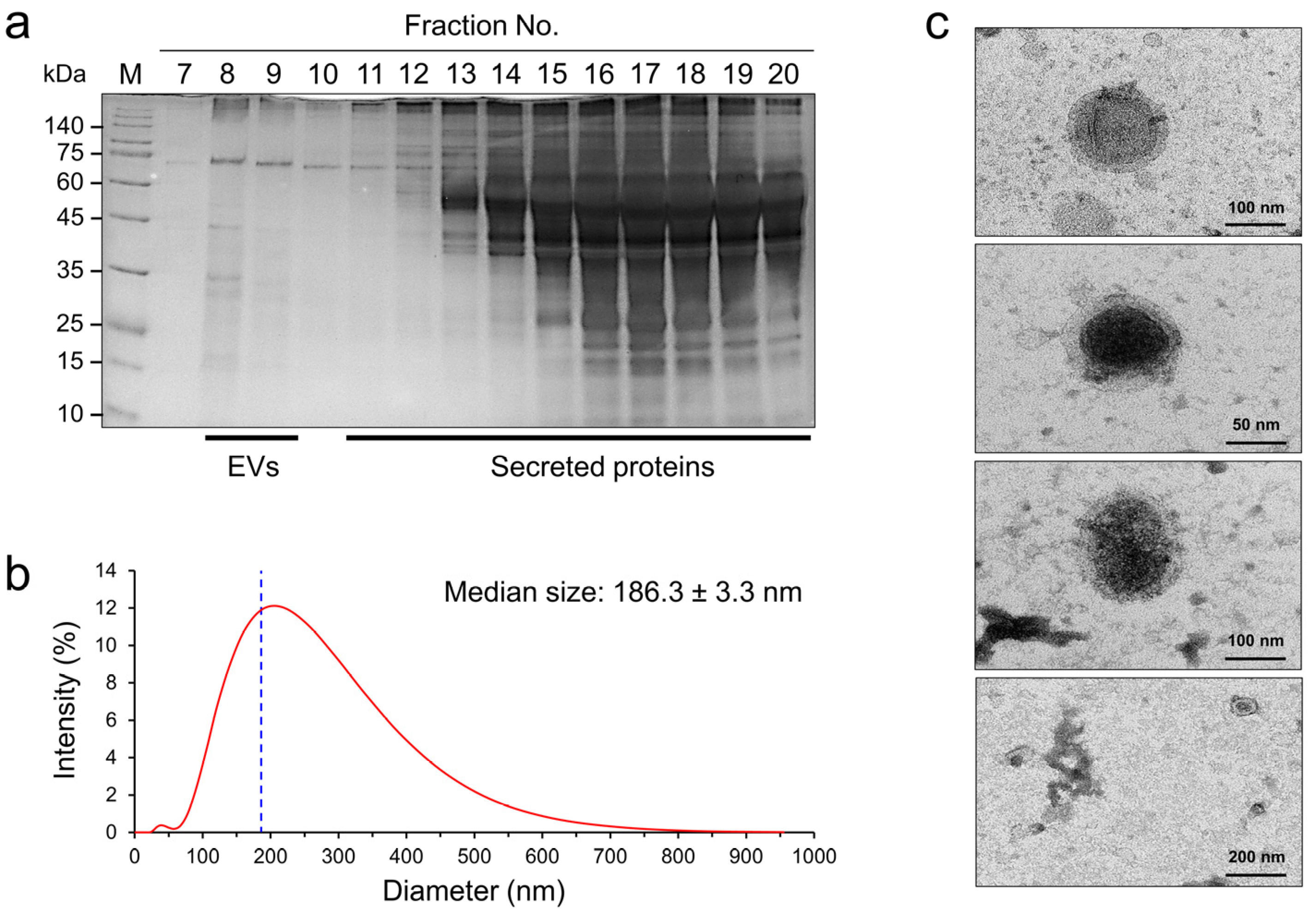
2.1. Biophysical Characteristics of NfEVs
2.2. Internalization of NfEVs into Mammalian Cells
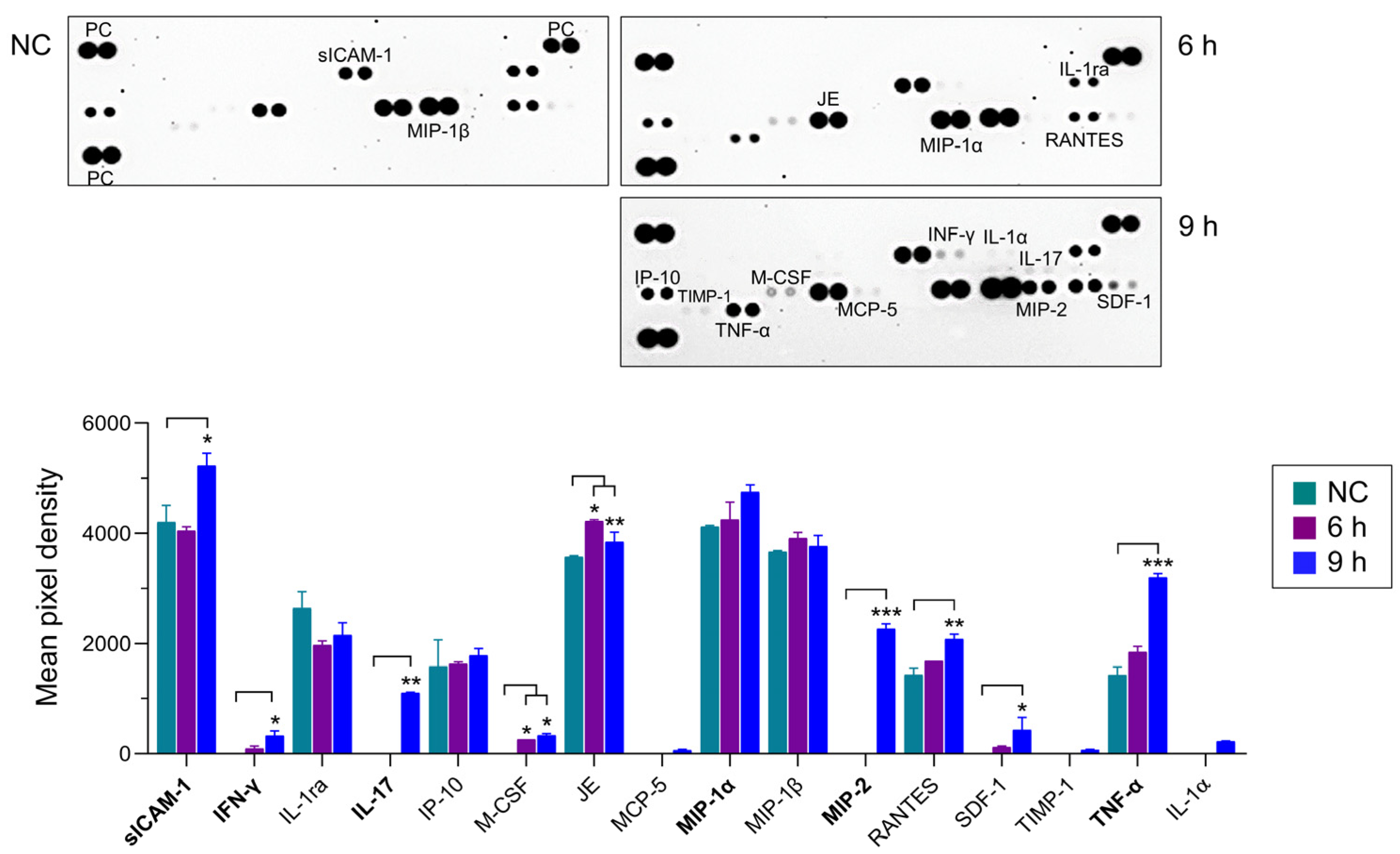
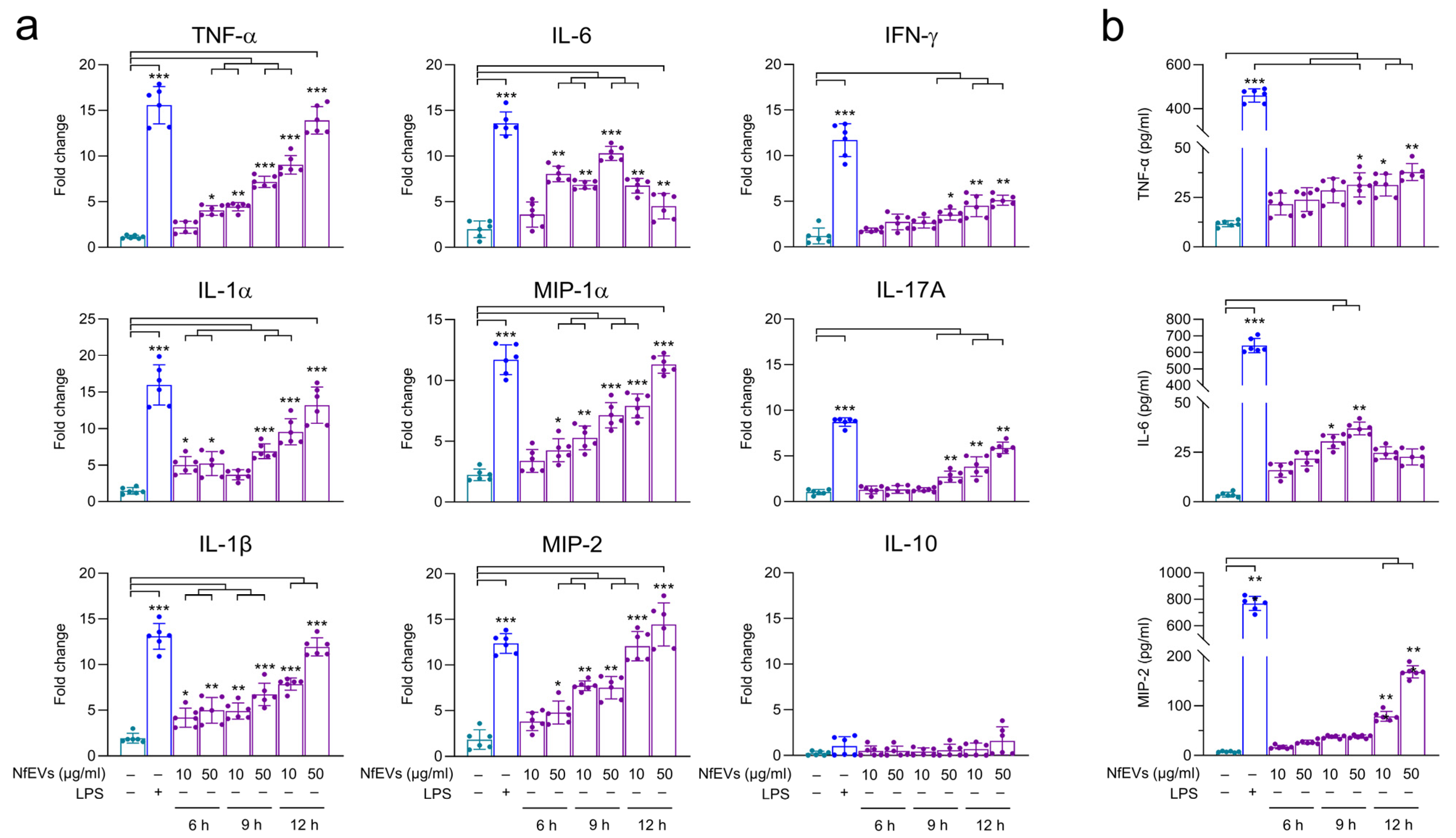
2.3. NfEVs Stimulate the Proinflammatory Immune Responses in BV-2 Microglial Cells
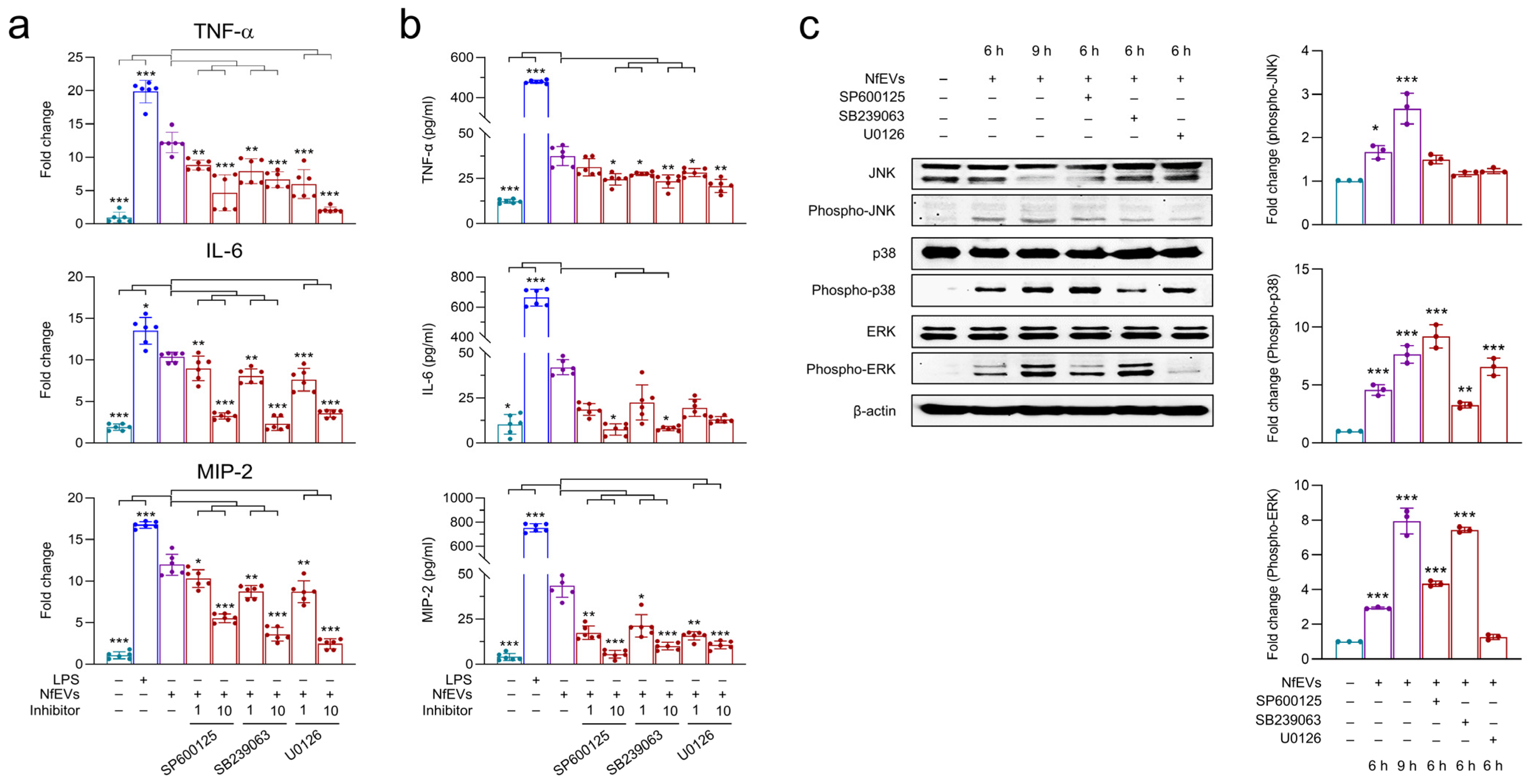
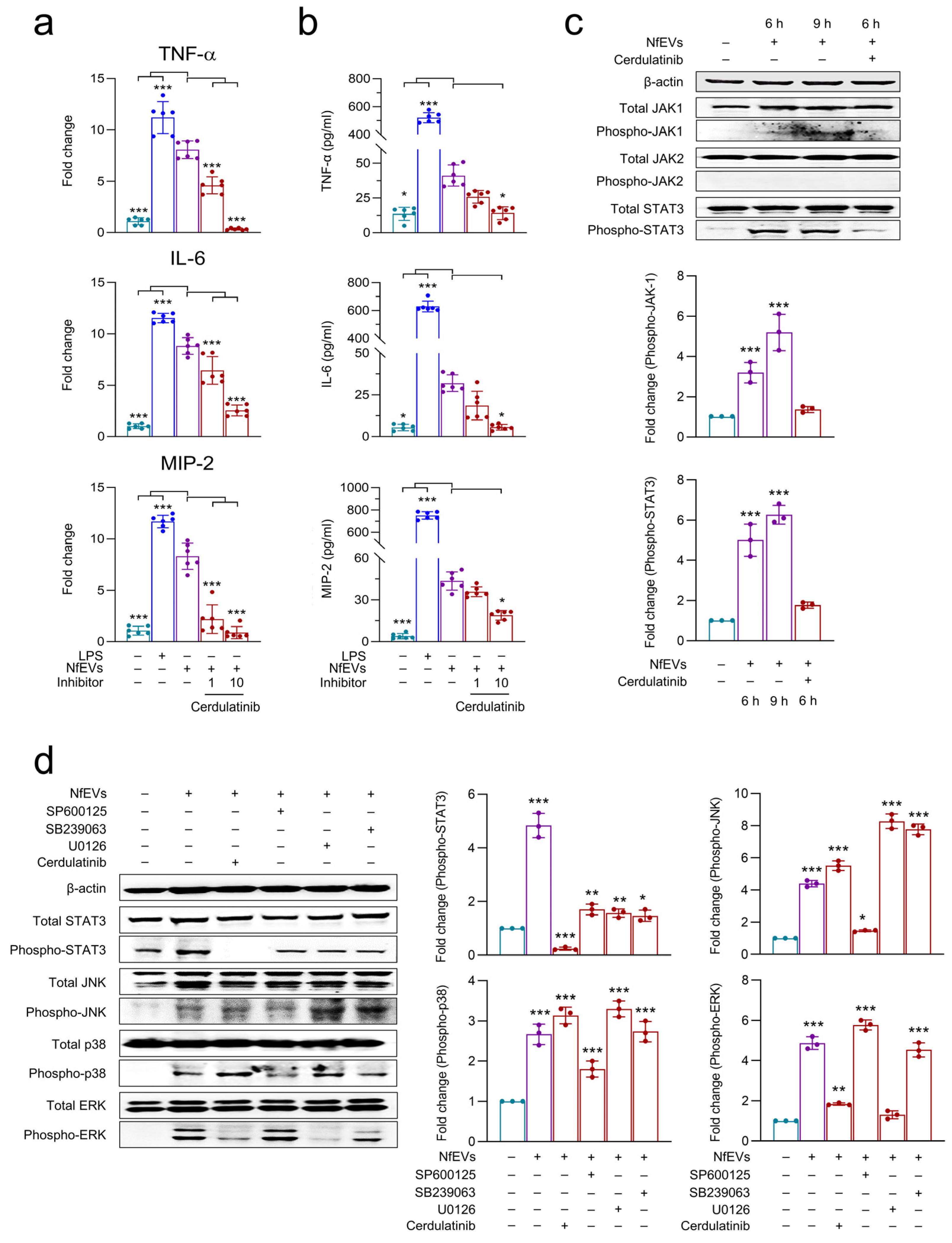
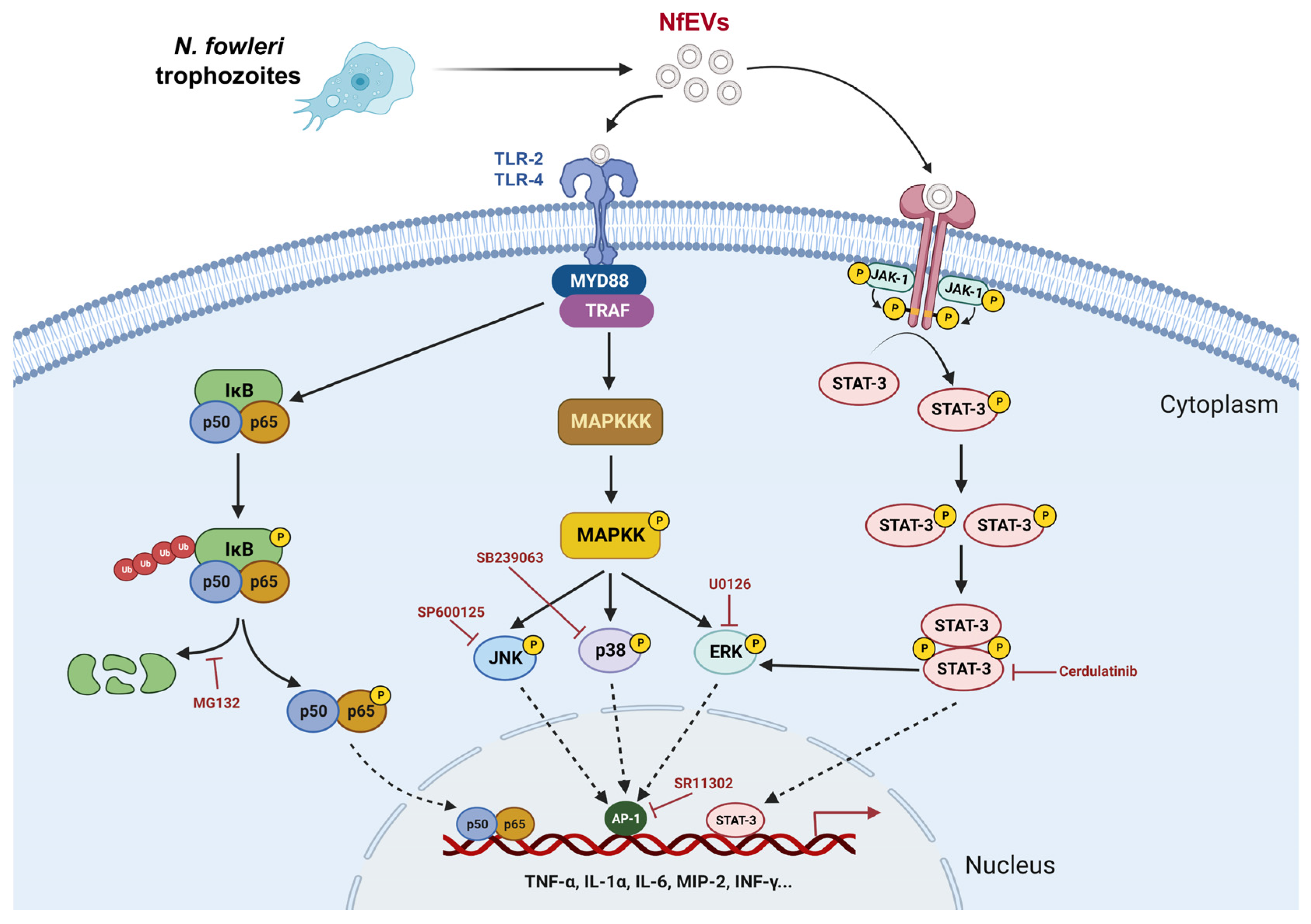
2.4. NfEVs Induce the Proinflammatory Immune Responses in BV-2 Microglial Cells via MAPK Signaling Pathway
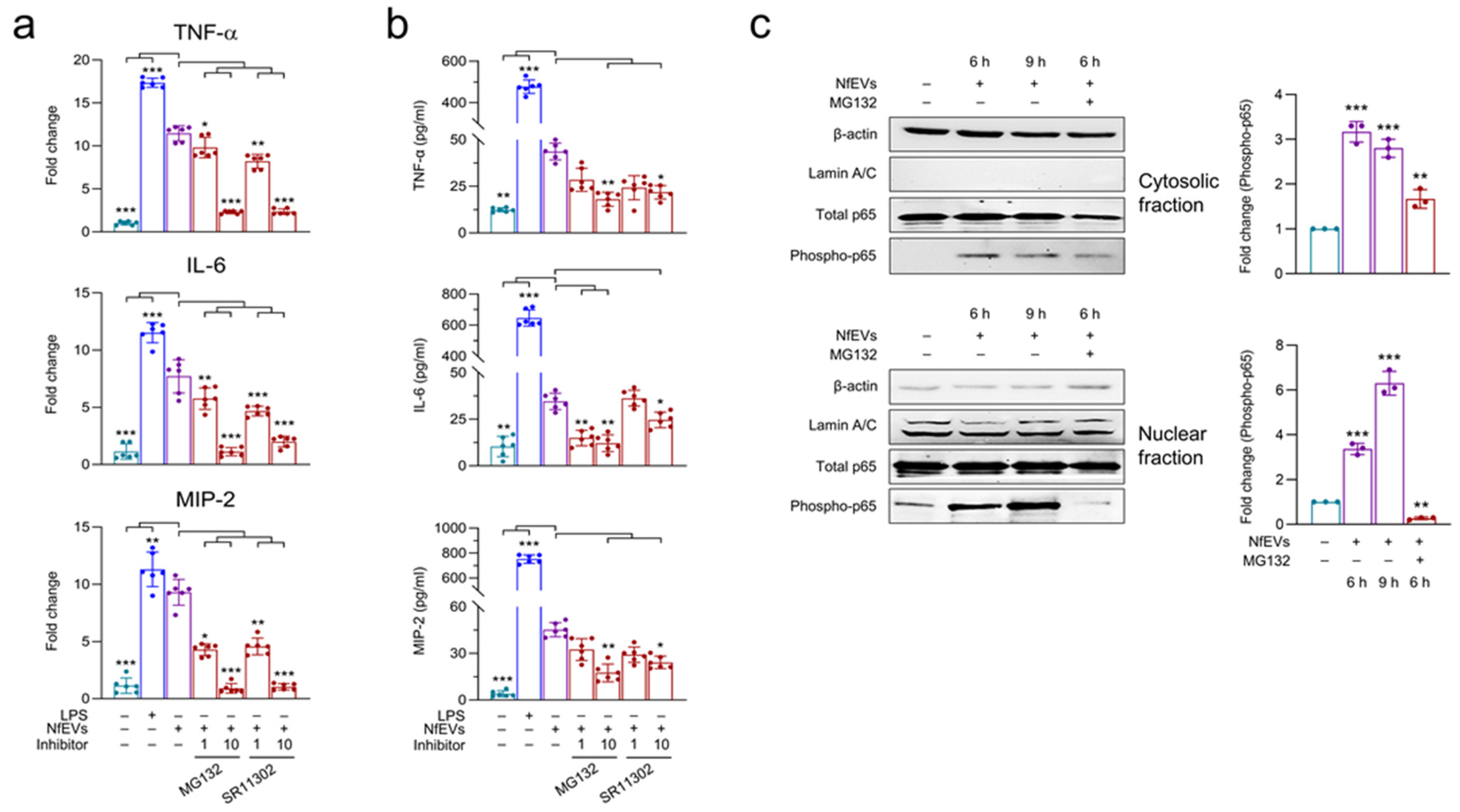
2.5. NfEVs-Induced Proinflammatory Immune Responses in BV-2 Microglial Cells Are Regulated by NF-κB Signaling Pathway
2.6. JAK-STAT Signaling Pathway Is Also Involved in NfEVs-Induced Proinflammatory Immune Responses in BV-2 Microglial Cells
3. Discussion
4. Materials and Methods
4.1. Cultivation of Naegleria fowleri
4.2. Cultivation of BV-2 Microglial Cells and C6 Glial Cells
4.3. Isolation of NfEVs
4.4. Dynamic Light Scattering (DLS) Analysis
4.5. Transmission Electron Microscopy (TEM) Analysis
4.6. Internalization Assay of NfEVs to BV-2 Microglial Cells and C6 Glial Cells
4.7. Cell Viability Assay
4.8. Analysis of Immune Responses in BV-2 Microglial Cells upon Treatment of NfEVs
4.9. Analyses of Mitogen-Activated Protein Kinases (MAPKs), Nuclear Factor κB (NF-κB), and Janus Kinase-Signal Transducer and Activator of Transcription (JAK-STAT) Signaling Pathways
4.10. Statisitcal Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tkach, M.; Théry, C. Communication by extracellular vesicles: Where we are and where we need to go. Cell 2016, 164, 1226–1232. [Google Scholar] [CrossRef]
- Yáñez-Mó, M.; Siljander, P.R.M.; Andreu, Z.; Zavec, A.B.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Zaborowski, M.P.; Balaj, L.; Breakefield, X.O.; Lai, C.P. Extracellular vesicles: Composition, biological relevance, and methods of study. Bioscience 2015, 65, 783–797. [Google Scholar] [CrossRef] [PubMed]
- Ofir-Birin, Y.; Regev-Rudzki, N. Extracellular vesicles in parasite survival. Science 2019, 363, 817–818. [Google Scholar] [CrossRef] [PubMed]
- Mugnier, M.R.; Papavasiliou, F.N.; Schulz, D. Vesicles as vehicles for virulence. Trends Parasitol. 2016, 32, 435–436. [Google Scholar] [CrossRef]
- Marti, M.; Johnson, P.J. Emerging roles for extracellular vesicles in parasitic infections. Curr. Opin. Microbiol. 2016, 32, 66–70. [Google Scholar] [CrossRef]
- Silverman, J.M.; Reiner, N.E. Leishmania exosomes deliver preemptive strikes to create an environment permissive for early infection. Front. Cell. Infect. Microbiol. 2011, 1, 26. [Google Scholar] [CrossRef]
- Silverman, J.M.; Clos, J.; De’Oliveira, C.C.; Shirvani, O.; Fang, Y.; Wang, C.; Foster, L.J.; Reiner, N.E. An exosome-based secretion pathway is responsible for protein export from Leishmania and communication with macrophages. J. Cell. Sci. 2010, 123, 842–852. [Google Scholar] [CrossRef]
- Dong, G.; Filho, A.L.; Olivier, M. Modulation of host-pathogen communication by extracellular vesicles (EVs) of the protozoan parasite Leishmania. Front. Cell. Infect. Microbiol. 2019, 9, 100. [Google Scholar] [CrossRef]
- Douanne, N.; Dong, G.; Amin, A.; Bernardo, L.; Blanchette, M.; Langlais, D.; Olivier, M.; Fernandez-Prada, C. Leishmania Parasites exchange drug-resistance genes through extracellular vesicles. Cell Rep. 2022, 40, 111121. [Google Scholar] [CrossRef]
- Twu, O.; de Miguel, N.; Lustig, G.; Stevens, G.C.; Vashisht, A.A.; Wohlschlegel, J.A.; Johnson, P.J. Trichomonas vaginalis exosomes deliver cargo to host cells and mediate host: Parasite interactions. PLoS Pathog. 2013, 9, e1003482. [Google Scholar] [CrossRef]
- Szempruch, A.J.; Sykes, S.E.; Kieft, R.; Dennison, L.; Becker, A.C.; Gartrell, A.; Martin, W.J.; Nakayasu, E.S.; Almeida, I.C.; Hajduk, S.L.; et al. Extracellular vesicles from Trypanosoma brucei mediate virulence factor transfer and cause host anemia. Cell 2016, 164, 246–257. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.C.; Tsai, C.Y.; Huang, J.M.; Wu, S.R.; Chu, L.J.; Huang, K.Y. Quantitative proteomic analysis and functional characterization of Acanthamoeba castellanii exosome-like vesicles. Parasit. Vectors 2019, 12, 467. [Google Scholar] [CrossRef] [PubMed]
- de Souza Gonçalves, D.; da Silva Ferreira, M.; Liedke, S.C.; Gomes, K.X.; de Oliveira, G.A.; Leão, P.E.L.; Cesar, G.V.; Seabra, S.H.; Cortines, J.R.; Casadevall, A.; et al. Extracellular vesicles and vesicle-free secretome of the protozoa Acanthamoeba castellanii under homeostasis and nutritional stress and their damaging potential to host cells. Virulence 2018, 9, 818–836. [Google Scholar] [CrossRef] [PubMed]
- Jahangeer, M.; Mahmood, Z.; Munir, N.; Waraich, U.E.A.; Tahir, I.M.; Akram, M.; Ali Shah, S.M.; Zulfqar, A.; Zainab, R. Naegleria fowleri: Sources of infection, pathophysiology, diagnosis, and management; a Review. Clin. Exp. Pharmacol. Physiol. 2020, 47, 199–212. [Google Scholar] [CrossRef]
- Siddiqui, R.; Ali, I.K.M.; Cope, J.R.; Khan, N.A. Biology and Pathogenesis of Naegleria fowleri. Acta Trop. 2016, 164, 375–394. [Google Scholar] [CrossRef]
- Lee, J.; Kang, J.M.; Kim, T.I.; Kim, J.H.; Sohn, H.J.; Na, B.K.; Shin, H.J. Excretory and secretory proteins of Naegleria fowleri induce inflammatory responses in BV-2 microglial cells. J. Eukaryot. Microbiol. 2017, 64, 183–192. [Google Scholar] [CrossRef]
- Marciano-Cabral, F.; Cabral, G.A. The immune response to Naegleria fowleri amebae and pathogenesis of infection. FEMS Immunol. Med. Microbiol. 2007, 51, 243–259. [Google Scholar] [CrossRef]
- Lee, Y.J.; Park, C.E.; Kim, J.H.; Sohn, H.J.; Lee, J.; Jung, S.Y.; Shin, H.J. Naegleria fowleri lysate induces strong cytopathic effects and pro-inflammatory cytokine release in rat microglial cells. Korean J. Parasitol. 2011, 49, 285–290. [Google Scholar] [CrossRef]
- Lê, H.G.; Kang, J.M.; Võ, T.C.; Na, B.K. Naegleria fowleri cathepsin B induces a pro-inflammatory immune response in BV-2 microglial cells via NF-ΚB and AP-1 dependent-MAPK signaling pathway. Int. J. Mol. Sci. 2022, 23, 8388. [Google Scholar] [CrossRef]
- Maxwell, M.J.; Chan, S.K.; Robinson, D.P.; Dwyer, D.M.; Nandan, D.; Foster, L.J.; Reiner, N.E. Proteomic analysis of the secretome of Leishmania donovani. Genome Biol. 2008, 9, R35. [Google Scholar] [CrossRef]
- Olajide, J.S.; Cai, J. Perils and promises of pathogenic protozoan extracellular vesicles. Front. Cell. Infect. Microbiol. 2020, 10, 371. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, Y.; Xiu, F.; Wang, J.; Cong, H.; He, S.; Shi, Y.; Wang, X.; Li, X.; Zhou, H. Characterization of exosomes derived from Toxoplasma gondii and their functions in modulating immune responses. Int. J. Nanomed. 2018, 13, 467–477. [Google Scholar] [CrossRef]
- Hassani, K.; Antoniak, E.; Jardim, A.; Olivier, M. Temperature-induced protein secretion by Leishmania mexicana modulates macrophage signalling and function. PLoS ONE 2011, 6, e18724. [Google Scholar] [CrossRef] [PubMed]
- Retana Moreira, L.; Steller Espinoza, M.F.; Chacón Camacho, N.; Cornet-Gomez, A.; Sáenz-Arce, G.; Osuna, A.; Lomonte, B.; Abrahams Sandí, E. Characterization of extracellular vesicles secreted by a clinical isolate of Naegleria fowleri and identification of immunogenic components within their protein cargo. Biology 2022, 11, 983. [Google Scholar] [CrossRef]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Oh, Y.H.; Jeong, S.R.; Kim, J.H.; Song, K.J.; Kim, K.; Park, S.; Sohn, S.; Shin, H.J. Cytopathic changes and pro-inflammatory cytokines induced by Naegleria fowleri trophozoites in rat microglial cells and protective effects of an anti-Nfa1 antibody. Parasite Immunol. 2005, 27, 453–459. [Google Scholar] [CrossRef]
- Thaí, T.L.; Kang, J.M.; Lê, H.G.; Lee, J.; Yoo, W.G.; Shin, H.J.; Sohn, W.M.; Na, B.K. Fowlerstefin, a cysteine protease inhibitor of Naegleria fowleri, induces inflammatory responses in BV-2 microglial cells in vitro. Parasit. Vectors 2020, 13, 41. [Google Scholar] [CrossRef]
- Beutler, B. Inferences, Questions and possibilities in toll-like receptor signalling. Nature 2004, 430, 257–263. [Google Scholar] [CrossRef]
- Ribeiro, K.S.; Vasconcellos, C.I.; Soares, R.P.; Mendes, M.T.; Ellis, C.C.; Aguilera-Flores, M.; de Almeida, I.C.; Schenkman, S.; Iwai, L.K.; Torrecilhas, A.C. Proteomic analysis reveals different composition of extracellular vesicles released by two Trypanosoma cruzi strains associated with their distinct interaction with host cells. J. Extracell. Vesicles 2018, 7, 1463779. [Google Scholar] [CrossRef]
- Nogueira, P.M.; Ribeiro, K.; Silveira, A.C.O.; Campos, J.H.; Martins-Filho, O.A.; Bela, S.R.; Campos, M.A.; Pessoa, N.L.; Colli, W.; Alves, M.J.M.; et al. Vesicles from different Trypanosoma cruzi strains trigger differential innate and chronic immune responses. J. Extracell. Vesicles 2015, 4, 28734. [Google Scholar] [CrossRef]
- Li, S.; Gong, P.; Tai, L.; Li, X.; Wang, X.; Zhao, C.; Zhang, X.; Yang, Z.; Yang, J.; Li, J.; et al. Extracellular vesicles secreted by Neospora caninum are recognized by Toll-like receptor 2 and modulate host cell innate immunity through the MAPK signaling pathway. Front. Immunol. 2018, 9, 1633. [Google Scholar] [CrossRef]
- Cronemberger-Andrade, A.; Xander, P.; Soares, R.P.; Pessoa, N.L.; Campos, M.A.; Ellis, C.C.; Grajeda, B.; Ofir-Birin, Y.; Almeida, I.C.; Regev-Rudzki, N.; et al. Trypanosoma cruzi-infected human macrophages shed proinflammatory extracellular vesicles that enhance host-cell invasion via Toll-like receptor 2. Front. Cell. Infect. Microbiol. 2020, 10, 99. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castillo, M.; Santos-Argumedo, L.; Galván-Moroyoqui, J.M.; Serrano-Luna, J.; Shibayama, M. Toll-like receptors participate in Naegleria fowleri recognition. Parasitol. Res. 2018, 117, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.Y.; Shum, A.Y.C.; Chao, C.C.; Kuo, J.S.; Wang, J.Y. Production of macrophage inflammatory protein-2 following hypoxia/reoxygenation in glial cells. Glia 2000, 32, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Abdalla, H.; Li, H.L.; Shi, F.D.; Zhu, J.; Höjberg, B.; Lindquist, L.; Wretlind, B.; Bakhiet, M.; Link, H. Neutralization of macrophage inflammatory protein 2 (MIP-2) and MIP-1α attenuates neutrophil recruitment in the central nervous system during experimental bacterial meningitis. Infect. Immun. 1999, 67, 2590–2601. [Google Scholar] [CrossRef]
- Lertjuthaporn, S.; Somkird, J.; Lekmanee, K.; Atipimonpat, A.; Sukapirom, K.; Sawasdipokin, H.; Tiewcharoen, S.; Pattanapanyasat, K.; Khowawisetsut, L. Extracellular vesicles from Naegleria fowleri induce IL-8 response in THP-1 macrophage. Pathogens 2022, 11, 632. [Google Scholar] [CrossRef]
- Kim, O.S.; Park, E.J.; Joe, E.H.; Jou, I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J. Biol. Chem. 2002, 277, 40594–40601. [Google Scholar] [CrossRef]
- Nelson, E.C.; Jones, M.M. Culture isolation of agents of primary amebic meningcencephalitis. J. Parasitol. 1970, 56, 248–249. [Google Scholar]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH image to ImageJ: 25 years of image snalysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lê, H.G.; Kang, J.-M.; Võ, T.C.; Yoo, W.G.; Na, B.-K. Naegleria fowleri Extracellular Vesicles Induce Proinflammatory Immune Responses in BV-2 Microglial Cells. Int. J. Mol. Sci. 2023, 24, 13623. https://doi.org/10.3390/ijms241713623
Lê HG, Kang J-M, Võ TC, Yoo WG, Na B-K. Naegleria fowleri Extracellular Vesicles Induce Proinflammatory Immune Responses in BV-2 Microglial Cells. International Journal of Molecular Sciences. 2023; 24(17):13623. https://doi.org/10.3390/ijms241713623
Chicago/Turabian StyleLê, Hương Giang, Jung-Mi Kang, Tuấn Cường Võ, Won Gi Yoo, and Byoung-Kuk Na. 2023. "Naegleria fowleri Extracellular Vesicles Induce Proinflammatory Immune Responses in BV-2 Microglial Cells" International Journal of Molecular Sciences 24, no. 17: 13623. https://doi.org/10.3390/ijms241713623