
Phenomic Microglia Diversity as a Druggable Target in the Hippocampus in Neurodegenerative Diseases

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction

2. Different Microglia Phenomics in Inflammaging
3. Different Microglia Phenomics in the Hippocampus in Acute Inflammation
4. Different Microglia Phenomics in the Hippocampus in Ischemia
5. Microglia Phenomics in Alzheimer’s Disease
6. Dysbiosis and Microglia
7. Spatiotemporal Differences in Microglia States in the Hippocampal Areas
8. Microglia as a Druggable Target
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s disease |
| APJs | Astrocytes branches |
| CBD | Cannabidiol |
| CNS | Central nervous system |
| GFAP | Glial fibrillary acidic protein |
| IBA1 | Ionized calcium binding adaptor molecule |
| IFN | Interferon |
| IL | Interleukine |
| JAK | Janus kinase |
| LPS | Lipopolysaccharide |
| LRP-1 | Lipoprotein receptor-related protein |
| mCAO | Middle cerebral artery occlusion |
| MHC | Major histocompatibility complex |
| MMP | Matrix metalloproteinases |
| MPJs | Microglia projections |
| NF-Kb | Nuclear factor-kappa B |
| PAI-1 | Plasminogen activator inhibitor type 1 |
| PLX | PLX5622, selective inhibitor of colony stimulating factor (CSF) 1 receptor |
| SR | Str. radiatum |
| TGF-β | Transforming growth factor-beta |
| TNF | Tumor necrosis factor |
| TRPV2 | Transient receptor potential vanilloid 2 |
References
- Paolicelli, R.C.; Sierra, A.; Stevens, B.; Tremblay, M.E.; Aguzzi, A.; Ajami, B.; Amit, I.; Audinat, E.; Bechmann, I.; Bennett, M.; et al. Microglia states and nomenclature: A field at its crossroads. Neuron 2022, 110, 3458–3483. [Google Scholar] [CrossRef]
- Salter, M.W.; Beggs, S. Sublime microglia: Expanding roles for the guardians of the CNS. Cell 2014, 158, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Stevens, B. Phagocytic glial cells: Sculpting synaptic circuits in the developing nervous system. Curr. Opin. Neurobiol. 2013, 23, 1034–1040. [Google Scholar] [CrossRef]
- Derecki, N.C.; Cardani, A.N.; Yang, C.H.; Quinnies, K.M.; Crihfield, A.; Lynch, K.R.; Kipnis, J. Regulation of learning and memory by meningeal immunity: A key role for IL-4. J. Exp. Med. 2010, 207, 1067–1080. [Google Scholar] [CrossRef] [PubMed]
- Ziv, Y.; Ron, N.; Butovsky, O.; Landa, G.; Sudai, E.; Greenberg, N.; Cohen, H.; Kipnis, J.; Schwartz, M. Immune cells contribute to the maintenance of neurogenesis and spatial learning abilities in adulthood. Nat. Neurosci. 2006, 9, 268–275. [Google Scholar] [CrossRef]
- Pfrieger, F.W. Roles of glial cells in synapse development. Cell Mol. Life Sci. 2009, 66, 2037–2047. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Rodríguez, J.J.; Verkhratsky, A. Neuroglia in neurodegeneration. Brain Res. Rev. 2010, 63, 189–211. [Google Scholar] [CrossRef]
- Frost, J.L.; Schafer, D.P. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 2016, 26, 587–597. [Google Scholar] [CrossRef]
- Aguzzi, A.; Barres, B.A.; Bennett, M.L. Microglia: Scapegoat, saboteur, or something else? Science 2013, 339, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Boche, D.; Perry, V.H.; Nicoll, J.A.R. Review: Activation patterns of microglia and their identification in the human brain. Neuropathol. Appl. Neurobiol. 2013, 39, 3–18. [Google Scholar] [CrossRef]
- Fetler, L.; Amigorena, S. Brain under surveillance: The microglia patrol. Science 2005, 309, 392–393. [Google Scholar] [CrossRef]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New Roles for the Synaptic Stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef]
- Wake, H.; Moorhouse, A.J.; Miyamoto, A.; Nabekura, J. Microglia: Actively surveying and shaping neuronal circuit structure and function. Trends Neurosci. 2013, 36, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R.; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia Sculpt Postnatal Neural Circuits in an Activity and Complement-Dependent Manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [PubMed]
- Fitzner, D.; Schnaars, M.; Van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.K.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Safaiyan, S.; Kannaiyan, N.; Snaidero, N.; Brioschi, S.; Biber, K.; Yona, S.; Edinger, A.L.; Jung, S.; Rossner, M.J.; Simons, M. Age-related myelin degradation burdens the clearance function of microglia during aging. Nat. Neurosci. 2016, 19, 995–998. [Google Scholar] [CrossRef]
- Lawson, L.J.; Perry, V.H.; Dri, P.; Gordon, S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 1990, 39, 151–170. [Google Scholar] [CrossRef]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Neuroscience: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef]
- Koenigsknecht-Talboo, J.; Landreth, G.E. Microglial phagocytosis induced by fibrillar β-amyloid and IgGs are differentially regulated by proinflammatory cytokines. J. Neurosci. 2005, 25, 8240–8249. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Gunzer, M.; Gutzeit, H.O.; Ullrich, O.; Reymann, K.G.; Dinkel, K. Microglia provide neuroprotection after ischemia. FASEB J. 2006, 20, 714–716. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Morsch, M.; Radford, R.; Lee, A.; Don, E.K.; Badrock, A.P.; Hall, T.E.; Cole, N.J.; Chung, R. In vivo characterization of microglial engulfment of dying neurons in the zebrafish spinal cord. Front. Cell. Neurosci. 2015, 9, 321. [Google Scholar] [CrossRef] [PubMed]
- Hinwood, M.; Morandini, J.; Day, T.A.; Walker, F.R. Evidence that microglia mediate the neurobiological effects of chronic psychological stress on the medial prefrontal cortex. Cereb. Cortex 2012, 22, 1442–1454. [Google Scholar] [CrossRef] [PubMed]
- Hinwood, M.; Tynan, R.J.; Charnley, J.L.; Beynon, S.B.; Day, T.A.; Walker, F.R. Chronic stress induced remodeling of the prefrontal cortex: Structural re-organization of microglia and the inhibitory effect of minocycline. Cereb. Cortex 2013, 23, 1784–1797. [Google Scholar] [CrossRef]
- Hanisch, U.K.; Kettenmann, H. Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nat. Neurosci. 2007, 10, 1387–1394. [Google Scholar] [CrossRef]
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflamm. 2012, 9, 155. [Google Scholar] [CrossRef]
- Sierra, A.; Encinas, J.M.; Deudero, J.J.P.; Chancey, J.H.; Enikolopov, G.; Overstreet-Wadiche, L.S.; Tsirka, S.E.; Maletic-Savatic, M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 2010, 7, 483–495. [Google Scholar] [CrossRef]
- Vinet, J.; van Weering, H.R.J.; Heinrich, A.; Kälin, R.E.; Wegner, A.; Brouwer, N.; Heppner, F.L.; van Rooijen, N.; Boddeke, H.W.G.M.; Biber, K. Neuroprotective function for ramified microglia in hippocampal excitotoxicity. J. Neuroinflamm. 2012, 9, 27. [Google Scholar] [CrossRef]
- Ransohoff, R.M.; Perry, V.H. Microglial Physiology: Unique Stimuli, Specialized Responses. Annu. Rev. Immunol. 2009, 27, 119–145. [Google Scholar] [CrossRef] [PubMed]
- Franco, R.; Fernández-Suárez, D. Alternatively activated microglia and macrophages in the central nervous system. Prog. Neurobiol. 2015, 131, 65–86. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Leak, R.K.; Shi, Y.; Suenaga, J.; Gao, Y.; Zheng, P.; Chen, J. Microglial and macrophage polarization—New prospects for brain repair. Nat. Rev. Neurol. 2015, 11, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Pu, H.; Hu, X.; Wei, Z.; Hong, D.; Zhang, W.; Gao, Y.; Chen, J.; Shi, Y. A Post-stroke Therapeutic Regimen with Omega-3 Polyunsaturated Fatty Acids that Promotes White Matter Integrity and Beneficial Microglial Responses after Cerebral Ischemia. Transl. Stroke Res. 2016, 7, 548–561. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.Y.; Liu, L.; Yang, Q.W. Functions and mechanisms of microglia/macrophages in neuroinflammation and neurogenesis after stroke. Prog. Neurobiol. 2016, 142, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Barres, B.A. Neuroscience: Glia—More than just brain glue. Nature 2009, 457, 675–677. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- De Biase, L.M.; Schuebel, K.E.; Fusfeld, Z.H.; Jair, K.; Hawes, I.A.; Cimbro, R.; Zhang, H.Y.; Liu, Q.R.; Shen, H.; Xi, Z.X.; et al. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron 2017, 95, 341–356.e6. [Google Scholar] [CrossRef]
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2017, 169, 1276–1290.e17. [Google Scholar] [CrossRef]
- Zhang, Y.; Barres, B.A. Astrocyte heterogeneity: An underappreciated topic in neurobiology. Curr. Opin. Neurobiol. 2010, 20, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Sofroniew, M.V. Diversity of astrocyte functions and phenotypes in neural circuits. Nat. Neurosci. 2015, 18, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Ben Haim, L.; Rowitch, D.H. Functional diversity of astrocytes in neural circuit regulation. Nat. Rev. Neurosci. 2016, 18, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Khakh, B.S.; Deneen, B. The Emerging Nature of Astrocyte Diversity. Annu. Rev. Neurosci. 2019, 42, 187–207. [Google Scholar] [CrossRef] [PubMed]
- Pestana, F.; Edwards-Faret, G.; Belgard, T.G.; Martirosyan, A.; Holt, M.G. No longer underappreciated: The emerging concept of astrocyte heterogeneity in neuroscience. Brain Sci. 2020, 10, 168. [Google Scholar] [CrossRef]
- Hughes, J.L.; Jones, P.S.; Beech, J.S.; Wang, D.; Menon, D.K.; Aigbirhio, F.I.; Fryer, T.D.; Baron, J.C. A microPET study of the regional distribution of [11C]-PK11195 binding following temporary focal cerebral ischemia in the rat. Correlation with post mortem mapping of microglia activation. Neuroimage 2012, 59, 2007–2016. [Google Scholar] [CrossRef]
- De Felice, E.; Gonçalves de Andrade, E.; Golia, M.T.; González Ibáñez, F.; Khakpour, M.; Di Castro, M.A.; Garofalo, S.; Di Pietro, E.; Benatti, C.; Brunello, N.; et al. Microglial diversity along the hippocampal longitudinal axis impacts synaptic plasticity in adult male mice under homeostatic conditions. J. Neuroinflamm. 2022, 19, 292. [Google Scholar] [CrossRef]
- Cerbai, F.; Lana, D.; Nosi, D.; Petkova-Kirova, P.; Zecchi, S.; Brothers, H.M.; Wenk, G.L.; Giovannini, M.G. The Neuron-Astrocyte-Microglia Triad in Normal Brain Ageing and in a Model of Neuroinflammation in the Rat Hippocampus. PLoS ONE 2012, 7, e45250. [Google Scholar] [CrossRef]
- Lana, D.; Gerace, E.; Magni, G.; Cialdai, F.; Monici, M.; Mannaioni, G.; Giovannini, M.G. Hypoxia/Ischemia-Induced Rod Microglia Phenotype in CA1 Hippocampal Slices. Int. J. Mol. Sci. 2022, 23, 1422. [Google Scholar] [CrossRef]
- Ugolini, F.; Lana, D.; Nardiello, P.; Nosi, D.; Pantano, D.; Casamenti, F.; Giovannini, M.G. Different Patterns of Neurodegeneration and Glia Activation in CA1 and CA3 Hippocampal Regions of TgCRND8 Mice. Front. Aging Neurosci. 2018, 10, 372. [Google Scholar] [CrossRef]
- Lana, D.; Melani, A.; Pugliese, A.M.; Cipriani, S.; Nosi, D.; Pedata, F.; Giovannini, M.G. The neuron-astrocyte-microglia triad in a rat model of chronic cerebral hypoperfusion: Protective effect of dipyridamole. Front. Aging Neurosci. 2014, 6, 322. [Google Scholar] [CrossRef]
- Martín-López, E.; García-Marques, J.; Núñez-Llaves, R.; López-Mascaraque, L. Clonal Astrocytic Response to Cortical Injury. PLoS ONE 2013, 8, e74039. [Google Scholar] [CrossRef] [PubMed]
- Bribian, A.; Pérez-Cerdá, F.; Matute, C.; López-Mascaraque, L. Clonal glial response in a multiple sclerosis mouse model. Front. Cell. Neurosci. 2018, 12, 375. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Iovino, L.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. The neuron-astrocyte-microglia triad involvement in neuroinflammaging mechanisms in the CA3 hippocampus of memory-impaired aged rats. Exp. Gerontol. 2016, 83, 71–88. [Google Scholar] [CrossRef]
- De Biase, L.M.; Bonci, A. Region-Specific Phenotypes of Microglia: The Role of Local Regulatory Cues. Neuroscientist 2019, 25, 314–333. [Google Scholar] [CrossRef] [PubMed]
- Ayata, P.; Badimon, A.; Strasburger, H.J.; Duff, M.K.; Montgomery, S.E.; Loh, Y.H.E.; Ebert, A.; Pimenova, A.A.; Ramirez, B.R.; Chan, A.T.; et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat. Neurosci. 2018, 21, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Hammond, T.R.; Dufort, C.; Dissing-Olesen, L.; Giera, S.; Young, A.; Wysoker, A.; Walker, A.J.; Gergits, F.; Segel, M.; Nemesh, J.; et al. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity 2019, 50, 253–271.e6. [Google Scholar] [CrossRef]
- Singh, N.; Benoit, M.R.; Zhou, J.; Das, B.; Davila-Velderrain, J.; Kellis, M.; Tsai, L.H.; Hu, X.; Yan, R. BACE-1 inhibition facilitates the transition from homeostatic microglia to DAM-1. Sci. Adv. 2022, 8, eabo1286. [Google Scholar] [CrossRef]
- Gomes-Leal, W. Microglial physiopathology: How to explain the dual role of microglia after acute neural disorders? Brain Behav. 2012, 2, 345–356. [Google Scholar] [CrossRef]
- Henneman, W.J.P.; Sluimer, J.D.; Barnes, J.; Van Der Flier, W.M.; Sluimer, I.C.; Fox, N.C.; Scheltens, P.; Vrenken, H.; Barkhof, F. Hippocampal atrophy rates in Alzheimer disease: Added value over whole brain volume measures. Neurology 2009, 72, 999–1007. [Google Scholar] [CrossRef]
- Cagnin, A.; Rossor, M.; Sampson, E.L.; MacKinnon, T.; Banati, R.B. In vivo detection of microglial activation in frontotemporal dementia. Ann. Neurol. 2004, 56, 894–897. [Google Scholar] [CrossRef]
- Rosso, S.M.; Landweer, E.J.; Houterman, M.; Donker Kaat, L.; Van Duijn, C.M.; Van Swieten, J.C. Medical and environmental risk factors for sporadic frontotemporal dementia: A retrospective case-control study. J. Neurol. Neurosurg. Psychiatry 2003, 74, 1574–1576. [Google Scholar] [CrossRef]
- Perry, V.H. Innate inflammation in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009373. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Franceschi, C.; Capri, M.; Monti, D.; Giunta, S.; Olivieri, F.; Sevini, F.; Panourgia, M.P.; Invidia, L.; Celani, L.; Scurti, M.; et al. Inflammaging and anti-inflammaging: A systemic perspective on aging and longevity emerged from studies in humans. Mech. Ageing Dev. 2007, 128, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Deleidi, M.; Jäggle, M.; Rubino, G. Immune ageing, dysmetabolism and inflammation in neurological diseases. Front. Neurosci. 2015, 9, 172. [Google Scholar] [CrossRef]
- von Bernhardi, R.; Eugenín-von Bernhardi, L.; Eugenín, J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015, 7, 124. [Google Scholar] [CrossRef] [PubMed]
- Norden, D.M.; Godbout, J.P. Review: Microglia of the aged brain: Primed to be activated and resistant to regulation. Neuropathol. Appl. Neurobiol. 2013, 39, 19–34. [Google Scholar] [CrossRef] [PubMed]
- Nair, S.; Sobotka, K.S.; Joshi, P.; Gressens, P.; Fleiss, B.; Thornton, C.; Mallard, C.; Hagberg, H. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia 2019, 67, 1047–1061. [Google Scholar] [CrossRef]
- Ma, J.; Zhang, J.; Hou, W.W.; Wu, X.H.; Liao, R.J.; Chen, Y.; Wang, Z.; Zhang, X.N.; Zhang, L.S.; Zhou, Y.D.; et al. Early treatment of minocycline alleviates white matter and cognitive impairments after chronic cerebral hypoperfusion. Sci. Rep. 2015, 5, 12079. [Google Scholar] [CrossRef]
- Manso, Y.; Holland, P.R.; Kitamura, A.; Szymkowiak, S.; Duncombe, J.; Hennessy, E.; Searcy, J.L.; Marangoni, M.; Randall, A.D.; Brown, J.T.; et al. Minocycline reduces microgliosis and improves subcortical white matter function in a model of cerebral vascular disease. Glia 2018, 66, 34–46. [Google Scholar] [CrossRef]
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31. [Google Scholar] [CrossRef]
- Bartsch, T.; Wulff, P. The hippocampus in aging and disease: From plasticity to vulnerability. Neuroscience 2015, 309, 1–16. [Google Scholar] [CrossRef]
- Mueller, S.G.; Schuff, N.; Yaffe, K.; Madison, C.; Miller, B.; Weiner, M.W. Hippocampal atrophy patterns in mild cognitive impairment and alzheimer’s disease. Hum. Brain Mapp. 2010, 31, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Small, S.A.; Schobel, S.A.; Buxton, R.B.; Witter, M.P.; Barnes, C.A. A pathophysiological framework of hippocampal dysfunction in ageing and disease. Nat. Rev. Neurosci. 2011, 12, 585–601. [Google Scholar] [CrossRef] [PubMed]
- Wong, W.T. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front. Cell. Neurosci. 2013, 7, 22. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Wenk, G.L.; Giovannini, M.G.; Zecchi-Orlandini, S.; Nosi, D. Microglial distribution, branching, and clearance activity in aged rat hippocampus are affected by astrocyte meshwork integrity: Evidence of a novel cell-cell interglial interaction. FASEB J. 2019, 33, 4007–4020. [Google Scholar] [CrossRef]
- Damani, M.R.; Zhao, L.; Fontainhas, A.M.; Amaral, J.; Fariss, R.N.; Wong, W.T. Age-related alterations in the dynamic behavior of microglia. Aging Cell 2011, 10, 263–276. [Google Scholar] [CrossRef]
- Faulkner, J.R.; Herrmann, J.E.; Woo, M.J.; Tansey, K.E.; Doan, N.B.; Sofroniew, M.V. Reactive Astrocytes Protect Tissue and Preserve Function after Spinal Cord Injury. J. Neurosci. 2004, 24, 2143–2155. [Google Scholar] [CrossRef]
- Myer, D.J.; Gurkoff, G.G.; Lee, S.M.; Hovda, D.A.; Sofroniew, M. V Essential Protective Roles of Reactive Astrocytes in Traumatic Brain Injury—PubMed. Brain 2006, 129, 2761–2772. [Google Scholar] [CrossRef]
- Li, L.; Lundkvist, A.; Andersson, D.; Wilhelmsson, U.; Nagai, N.; Pardo, A.C.; Nodin, C.; Ståhlberg, A.; Aprico, K.; Larsson, K.; et al. Protective role of reactive astrocytes in brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 468–481. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Cardona, A.E.; Pioro, E.P.; Sasse, M.E.; Kostenko, V.; Cardona, S.M.; Dijkstra, I.M.; Huang, D.R.; Kidd, G.; Dombrowski, S.; Dutta, R.; et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat. Neurosci. 2006, 9, 917–924. [Google Scholar] [CrossRef]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Varvel, N.H.; Konerth, M.E.; Xu, G.; Cardona, A.E.; Ransohoff, R.M.; Lamb, B.T. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am. J. Pathol. 2010, 177, 2549–2562. [Google Scholar] [CrossRef]
- Liu, Z.; Condello, C.; Schain, A.; Harb, R.; Grutzendler, J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J. Neurosci. 2010, 30, 17091–17101. [Google Scholar] [CrossRef]
- Harrison, J.K.; Jiang, Y.; Chen, S.; Xia, Y.; Maciejewski, D.; Mcnamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed]
- Chapman, G.A.; Moores, K.; Harrison, D.; Campbell, C.A.; Stewart, B.R.; Strijbos, P.J. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J. Neurosci. 2000, 20, RC87. [Google Scholar] [CrossRef]
- Noda, M.; Doi, Y.; Liang, J.; Kawanokuchi, J.; Sonobe, Y.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression. J. Biol. Chem. 2011, 286, 2308–2319. [Google Scholar] [CrossRef]
- Verge, G.M.; Milligan, E.D.; Maier, S.F.; Watkins, L.R.; Naeve, G.S.; Foster, A.C. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur. J. Neurosci. 2004, 20, 1150–1160. [Google Scholar] [CrossRef]
- Dorf, M.E.; Berman, M.A.; Tanabe, S.; Heesen, M.; Luo, Y. Astrocytes express functional chemokine receptors. J. Neuroimmunol. 2000, 111, 109–121. [Google Scholar] [CrossRef] [PubMed]
- Hatori, K.; Nagai, A.; Heisel, R.; Ryu, J.K.; Kim, S.U. Fractalkine and fractalkine receptors in human neurons and glial cells. J. Neurosci. Res. 2002, 69, 418–426. [Google Scholar] [CrossRef]
- Sheridan, G.K.; Murphy, K.J. Neuron-glia crosstalk in health and disease: Fractalkine and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef]
- Dénes, Á.; Ferenczi, S.; Halász, J.; Környei, Z.; Kovács, K.J. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J. Cereb. Blood Flow Metab. 2008, 28, 1707–1721. [Google Scholar] [CrossRef]
- Fuller, A.D.; Van Eldik, L.J. MFG-E8 regulates microglial phagocytosis of apoptotic neurons. J. Neuroimmune Pharmacol. 2008, 3, 246–256. [Google Scholar] [CrossRef] [PubMed]
- Limatola, C.; Lauro, C.; Catalano, M.; Ciotti, M.T.; Bertollini, C.; Di Angelantonio, S.; Ragozzino, D.; Eusebi, F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J. Neuroimmunol. 2005, 166, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.K.; Malcangio, M. Fractalkine/CX3CR1 signaling during neuropathic pain. Front. Cell. Neurosci. 2014, 8, 121. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Ugolini, F.; Nosi, D.; Wenk, G.L.; Giovannini, M.G. Alterations in the interplay between neurons, astrocytes and microglia in the rat dentate gyrus in experimental models of neurodegeneration. Front. Aging Neurosci. 2017, 9, 296. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed]
- Grabert, K.; Michoel, T.; Karavolos, M.H.; Clohisey, S.; Kenneth Baillie, J.; Stevens, M.P.; Freeman, T.C.; Summers, K.M.; McColl, B.W. Microglial brain regionâ ’dependent diversity and selective regional sensitivities to aging. Nat. Neurosci. 2016, 19, 504–516. [Google Scholar] [CrossRef]
- Hickman, S.E.; Kingery, N.D.; Ohsumi, T.K.; Borowsky, M.L.; Wang, L.C.; Means, T.K.; El Khoury, J. The microglial sensome revealed by direct RNA sequencing. Nat. Neurosci. 2013, 16, 1896–1905. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.A.; Boddeke, H.W.G.M.; Kettenmann, H. Microglia in Physiology and Disease. Annu. Rev. Physiol. 2017, 79, 619–643. [Google Scholar] [CrossRef] [PubMed]
- Krabbe, G.; Halle, A.; Matyash, V.; Rinnenthal, J.L.; Eom, G.D.; Bernhardt, U.; Miller, K.R.; Prokop, S.; Kettenmann, H.; Heppner, F.L. Functional Impairment of Microglia Coincides with Beta-Amyloid Deposition in Mice with Alzheimer-Like Pathology. PLoS ONE 2013, 8, e60921. [Google Scholar] [CrossRef]
- Gupta, S.; Agrawal, A.; Agrawal, S.; Su, H.; Gollapudi, S. A paradox of immunodeficiency and inflammation in human aging: Lessons learned from apoptosis. Immun. Ageing 2006, 3, 5. [Google Scholar] [CrossRef] [PubMed]
- Blume, T.; Focke, C.; Peters, F.; Deussing, M.; Albert, N.L.; Lindner, S.; Gildehaus, F.J.; Von Ungern-Sternberg, B.; Ozmen, L.; Baumann, K.; et al. Microglial response to increasing amyloid load saturates with aging: A longitudinal dual tracer in vivo µpET-study 11 Medical and Health Sciences 1109 Neurosciences. J. Neuroinflamm. 2018, 15, 307. [Google Scholar] [CrossRef]
- Gehrmann, J.; Matsumoto, Y.; Kreutzberg, G.W. Microglia: Intrinsic immuneffector cell of the brain. Brain Res. Rev. 1995, 20, 269–287. [Google Scholar] [CrossRef]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef]
- Weinhard, L.; Di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia induction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef]
- Milner, R.; Crocker, S.J.; Hung, S.; Wang, X.; Frausto, R.F.; del Zoppo, G.J. Fibronectin- and Vitronectin-Induced Microglial Activation and Matrix Metalloproteinase-9 Expression Is Mediated by Integrins α5β1 and αvβ5. J. Immunol. 2007, 178, 8158–8167. [Google Scholar] [CrossRef]
- Chen, Y.; Ju, L.; Rushdi, M.; Ge, C.; Zhu, C. Receptor-mediated cell mechanosensing. Mol. Biol. Cell 2017, 28, 3134–3155. [Google Scholar] [CrossRef]
- Ohsawa, K.; Irino, Y.; Sanagi, T.; Nakamura, Y.; Suzuki, E.; Inoue, K.; Kohsaka, S. P2Y12 receptor-mediated integrin-β1 activation regulates microglial process extension induced by ATP. Glia 2010, 58, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Jeon, H.; Kim, J.H.; Kim, J.H.; Lee, W.H.; Lee, M.S.; Suk, K. Plasminogen activator inhibitor type 1 regulates microglial motility and phagocytic activity. J. Neuroinflamm. 2012, 9, 149. [Google Scholar] [CrossRef] [PubMed]
- Jha, M.K.; Jo, M.; Kim, J.H.; Suk, K. Microglia-Astrocyte Crosstalk: An Intimate Molecular Conversation. Neuroscientist 2019, 25, 227–240. [Google Scholar] [CrossRef]
- da Fonseca, A.C.C.; Matias, D.; Garcia, C.; Amaral, R.; Geraldo, L.H.; Freitas, C.; Lima, F.R.S. The impact of microglial activation on blood-brain barrier in brain diseases. Front. Cell. Neurosci. 2014, 8, 362. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.K.; Milner, R. A critical role for microglia in maintaining vascular integrity in the hypoxic spinal cord. Proc. Natl. Acad. Sci. USA 2019, 116, 26029–26037. [Google Scholar] [CrossRef]
- Jolivel, V.; Bicker, F.; Binamé, F.; Ploen, R.; Keller, S.; Gollan, R.; Jurek, B.; Birkenstock, J.; Poisa-Beiro, L.; Bruttger, J.; et al. Perivascular microglia promote blood vessel disintegration in the ischemic penumbra. Acta Neuropathol. 2015, 129, 279–295. [Google Scholar] [CrossRef]
- Krasemann, S.; Madore, C.; Cialic, R.; Baufeld, C.; Calcagno, N.; El Fatimy, R.; Beckers, L.; O’Loughlin, E.; Xu, Y.; Fanek, Z.; et al. The TREM2-APOE Pathway Drives the Transcriptional Phenotype of Dysfunctional Microglia in Neurodegenerative Diseases. Immunity 2017, 47, 566–581.e9. [Google Scholar] [CrossRef]
- Gelderblom, M.; Sobey, C.G.; Kleinschnitz, C.; Magnus, T. Danger signals in stroke. Ageing Res. Rev. 2015, 24, 77–82. [Google Scholar] [CrossRef]
- Eltzschig, H.K.; Sitkovsky, M.V.; Robson, S.C. Purinergic signaling during inflammation. N. Engl. J. Med. 2012, 367, 2322–2333. [Google Scholar] [CrossRef]
- Koizumi, S.; Shigemoto-Mogami, Y.; Nasu-Tada, K.; Shinozaki, Y.; Ohsawa, K.; Tsuda, M.; Joshi, B.V.; Jacobson, K.A.; Kohsaka, S.; Inoue, K. UDP acting at P2Y6 receptors is a mediator of microglial phagocytosis. Nature 2007, 446, 1091–1095. [Google Scholar] [CrossRef]
- Webster, C.M.; Hokari, M.; McManus, A.; Tang, X.N.; Ma, H.; Kacimi, R.; Yenari, M.A. Microglial P2Y12 Deficiency/Inhibition Protects against Brain Ischemia. PLoS ONE 2013, 8, e70927. [Google Scholar] [CrossRef] [PubMed]
- Domercq, M.; Perez-Samartin, A.; Aparicio, D.; Alberdi, E.; Pampliega, O.; Matute, C. P2X7 receptors mediate ischemic damage to oligodendrocytes. Glia 2010, 58, 730–740. [Google Scholar] [CrossRef]
- Reimer, M.M.; McQueen, J.; Searcy, L.; Scullion, G.; Zonta, B.; Desmazieres, A.; Holland, P.R.; Smith, J.; Gliddon, C.; Wood, E.R.; et al. Rapid disruption of axon-glial integrity in response to mild cerebral hypoperfusion. J. Neurosci. 2011, 31, 18185–18194. [Google Scholar] [CrossRef] [PubMed]
- Simpson, R.A.; Hemingway, D.; Crowther, M.; Goodall, S.; Thompson, M.M. The gelatinases, their activators and inhibitors in the progression of colorectal cancer. Color. Dis. 1999, 1, 248–255. [Google Scholar] [CrossRef]
- Seo, J.H.; Miyamoto, N.; Hayakawa, K.; Pham, L.D.D.; Maki, T.; Ayata, C.; Kim, K.W.; Lo, E.H.; Arai, K. Oligodendrocyte precursors induce early blood-brain barrier opening after white matter injury. J. Clin. Investig. 2013, 123, 782–786. [Google Scholar] [CrossRef]
- Freeman, L.R.; Keller, J.N. Oxidative stress and cerebral endothelial cells: Regulation of the blood-brain-barrier and antioxidant based interventions. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 822–829. [Google Scholar] [CrossRef]
- Chandler, S.; Coates, R.; Gearing, A.; Lury, J.; Wells, G.; Bone, E. Matrix metalloproteinases degrade myelin basic protein. Neurosci. Lett. 1995, 201, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Becker, E.B.E.; Bonni, A. Cell cycle regulation of neuronal apoptosis in development and disease. Prog. Neurobiol. 2004, 72, 1–25. [Google Scholar] [CrossRef]
- Márquez-Ropero, M.; Benito, E.; Plaza-Zabala, A.; Sierra, A. Microglial Corpse Clearance: Lessons From Macrophages. Front. Immunol. 2020, 11, 506. [Google Scholar] [CrossRef]
- Michell-Robinson, M.A.; Touil, H.; Healy, L.M.; Owen, D.R.; Durafourt, B.A.; Bar-Or, A.; Antel, J.P.; Moore, C.S. Roles of microglia in brain development, tissue maintenance and repair. Brain 2015, 138, 1138–1159. [Google Scholar] [CrossRef]
- Takahashi, K.; Rochford, C.D.P.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [PubMed]
- Peri, F.; Nüsslein-Volhard, C. Live Imaging of Neuronal Degradation by Microglia Reveals a Role for v0-ATPase a1 in Phagosomal Fusion In Vivo. Cell 2008, 133, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Wakselman, S.; Béchade, C.; Roumier, A.; Bernard, D.; Triller, A.; Bessis, A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J. Neurosci. 2008, 28, 8138–8143. [Google Scholar] [CrossRef]
- Mazaheri, F.; Breus, O.; Durdu, S.; Haas, P.; Wittbrodt, J.; Gilmour, D.; Peri, F. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat. Commun. 2014, 5, 4046. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.A.; Chang, C.F.; Goods, B.A.; Hammond, M.D.; Mac Grory, B.; Ai, Y.; Steinschneider, A.F.; Renfroe, S.C.; Askenase, M.H.; Mccullough, L.D.; et al. TGF-β1 modulates microglial phenotype and promotes recovery after intracerebral hemorrhage. J. Clin. Investig. 2017, 127, 280–292. [Google Scholar] [CrossRef]
- Leclerc, J.L.; Lampert, A.S.; Loyola Amador, C.; Schlakman, B.; Vasilopoulos, T.; Svendsen, P.; Moestrup, S.K.; Doré, S. The absence of the CD163 receptor has distinct temporal influences on intracerebral hemorrhage outcomes. J. Cereb. Blood Flow Metab. 2018, 38, 262–273. [Google Scholar] [CrossRef] [PubMed]
- Bi, R.; Fang, Z.; You, M.; He, Q.; Hu, B. Microglia Phenotype and Intracerebral Hemorrhage: A Balance of Yin and Yang. Front. Cell. Neurosci. 2021, 15, 765205. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, M.; Ninomiya, I.; Hatakeyama, M.; Takahashi, T.; Shimohata, T. Microglia and Monocytes/Macrophages Polarization Reveal Novel Therapeutic Mechanism against Stroke. Int. J. Mol. Sci. 2017, 18, 2135. [Google Scholar] [CrossRef] [PubMed]
- Nissl, F. Über einige Beziehungen zwischen Nervenzellerkrankungen und gliösen Erscheinungen bei verschiedenen Psychosen. Arch. Psychiatry 1899, 32, 656–676. [Google Scholar]
- Ziebell, J.M.; Taylor, S.E.; Cao, T.; Harrison, J.L.; Lifshitz, J. Rod microglia: Elongation, alignment, and coupling to form trains across the somatosensory cortex after experimental diffuse brain injury. J. Neuroinflamm. 2012, 9, 247. [Google Scholar] [CrossRef]
- Taylor, S.E.; Morganti-Kossmann, C.; Lifshitz, J.; Ziebell, J.M. Rod microglia: A morphological definition. PLoS ONE 2014, 9, e97096. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Thomas, T.C.; Ziebell, J.M.; Pauly, J.R.; Lifshitz, J. Morphological and genetic activation of microglia after diffuse traumatic brain injury in the rat. Neuroscience 2012, 225, 65–75. [Google Scholar] [CrossRef] [PubMed]
- Yuan, T.F.; Liang, Y.X.; Peng, B.; Lin, B.; So, K.F. Local proliferation is the main source of rod microglia after optic nerve transection. Sci. Rep. 2015, 5, 10788. [Google Scholar] [CrossRef]
- Rao, Y.; Liang, Y.X.; Peng, B. A revisit of rod microglia in preclinical studies. Neural Regen. Res. 2017, 12, 56–57. [Google Scholar] [CrossRef]
- Holloway, O.G.; Canty, A.J.; King, A.E.; Ziebell, J.M. Rod microglia and their role in neurological diseases. Semin. Cell Dev. Biol. 2019, 94, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Giordano, K.R.; Denman, C.R.; Dubisch, P.S.; Akhter, M.; Lifshitz, J. An update on the rod microglia variant in experimental and clinical brain injury and disease. Brain Commun. 2021, 3, fcaa227. [Google Scholar] [CrossRef]
- Ziebell, J.M.; Ray-Jones, H.; Lifshitz, J. Nogo presence is inversely associated with shifts in cortical microglial morphology following experimental diffuse brain injury. Neuroscience 2017, 359, 209–223. [Google Scholar] [CrossRef]
- Witcher, K.G.; Bray, C.E.; Dziabis, J.E.; McKim, D.B.; Benner, B.N.; Rowe, R.K.; Kokiko-Cochran, O.N.; Popovich, P.G.; Lifshitz, J.; Eiferman, D.S.; et al. Traumatic brain injury-induced neuronal damage in the somatosensory cortex causes formation of rod-shaped microglia that promote astrogliosis and persistent neuroinflammation. Glia 2018, 66, 2719–2736. [Google Scholar] [CrossRef]
- Bachstetter, A.D.; Van Eldik, L.J.; Schmitt, F.A.; Neltner, J.H.; Ighodaro, E.T.; Webster, S.J.; Patel, E.; Abner, E.L.; Kryscio, R.J.; Nelson, P.T. Disease-related microglia heterogeneity in the hippocampus of Alzheimer’s disease, dementia with Lewy bodies, and hippocampal sclerosis of aging. Acta Neuropathol. Commun. 2015, 3, 32. [Google Scholar] [CrossRef]
- Anan’ina, T.; Kisel, A.; Kudabaeva, M.; Chernysheva, G.; Smolyakova, V.; Usov, K.; Krutenkova, E.; Plotnikov, M.; Khodanovich, M. Neurodegeneration, Myelin Loss and Glial Response in the Three-Vessel Global Ischemia Model in Rat. Int. J. Mol. Sci. 2020, 21, 6246. [Google Scholar] [CrossRef]
- Kirino, T. Delayed neuronal death. Neuropathology 2000, 20, 95–97. [Google Scholar] [CrossRef] [PubMed]
- Graeber, M.B.; Mehraein, P. Microglial rod cells. Neuropathol. Appl. Neurobiol. 1994, 20, 178–17880. [Google Scholar]
- Au, N.P.B.; Ma, C.H.E. Recent Advances in the Study of Bipolar/Rod-Shaped Microglia and their Roles in Neurodegeneration. Front. Aging Neurosci. 2017, 9, 128. [Google Scholar] [CrossRef]
- Cho, B.P.; Song, D.Y.; Sugama, S.; Shin, D.H.; Shimizu, Y.; Kim, S.S.; Kim, Y.S.; Joh, T.H. Pathological dynamics of activated microglia following medial forebrain bundle transection. Glia 2006, 53, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Spielmeyer, W. Histopathologie des Nervensystems, 1st ed.; Springer: Berlin/Heidelberg, Germany, 1922. [Google Scholar]
- Van Wageningen, T.A.; Vlaar, E.; Kooij, G.; Jongenelen, C.A.M.; Geurts, J.J.G.; Van Dam, A.M. Regulation of microglial TMEM119 and P2RY12 immunoreactivity in multiple sclerosis white and grey matter lesions is dependent on their inflammatory environment. Acta Neuropathol. Commun. 2019, 7, 206. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Ellert-Miklaszewska, A.; Lipko, M.; Sielska, M.; Frankowska, M.; Kaminska, B. Characteristics of the alternative phenotype of microglia/macrophages and its modulation in experimental gliomas. PLoS ONE 2011, 6, e23902. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Streit, W.J. Microglial senescence: Does the brain’s immune system have an expiration date? Trends Neurosci. 2006, 29, 506–510. [Google Scholar] [CrossRef]
- Bauer, J.; Sminia, T.; Wouterlood, F.G.; Dijkstra, C.D. Phagocytic activity of macrophages and microglial cells during the course of acute and chronic relapsing experimental autoimmune encephalomyelitis. J. Neurosci. Res. 1994, 38, 365–375. [Google Scholar] [CrossRef]
- Papageorgiou, I.E.; Lewen, A.; Galow, L.V.; Cesetti, T.; Scheffel, J.; Regen, T.; Hanisch, U.K.; Kann, O. TLR4-activated microglia require IFN-γ to induce severe neuronal dysfunction and death in situ. Proc. Natl. Acad. Sci. USA 2016, 113, 212–217. [Google Scholar] [CrossRef]
- Roth, T.L.; Nayak, D.; Atanasijevic, T.; Koretsky, A.P.; Latour, L.L.; McGavern, D.B. Transcranial amelioration of inflammation and cell death after brain injury. Nature 2014, 505, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Kim, C.; Sharp, F.R. Very brief focal ischemia simulating transient ischemic attacks (TIAs) can injure brain and induce Hsp70 protein. Brain Res. 2008, 1234, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Pulsinelli, W.A.; Brierley, J.B.; Plum, F. Temporal profile of neuronal damage in a model of transient forebrain ischemia. Ann. Neurol. 1982, 11, 491–498. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Freund, T.F. Selective vulnerability of the hippocampus in brain ischemia. Neuroscience 1991, 40, 599–636. [Google Scholar] [CrossRef]
- Bartsch, T.; Schönfeld, R.; Müller, F.J.; Alfke, K.; Leplow, B.; Aldenhoff, J.; Deuschl, G.; Koch, J.M. Focal lesions of human hippocampal CA1 neurons in transient global amnesia impair place memory. Science 2010, 328, 1412–1415. [Google Scholar] [CrossRef]
- Bartsch, T.; Döhring, J.; Reuter, S.; Finke, C.; Rohr, A.; Brauer, H.; Deuschl, G.; Jansen, O. Selective neuronal vulnerability of human hippocampal CA1 neurons: Lesion evolution, temporal course, and pattern of hippocampal damage in diffusion-weighted MR imaging. J. Cereb. Blood Flow Metab. 2015, 35, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Petito, C.K.; Feldmann, E.; Pulsinelli, W.A.; Plum, F. Delayed hippocampal damage in humans following cardiorespiratory arrest. Neurology 1987, 37, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Zola-Morgan, S.; Squire, L.R.; Amaral, D.G. Human amnesia and the medial temporal region: Enduring memory impairment following a bilateral lesion limited to field CA1 of the hippocampus. J. Neurosci. 1986, 6, 2950–2967. [Google Scholar] [CrossRef]
- Fu, R.; Shen, Q.; Xu, P.; Luo, J.J.; Tang, Y. Phagocytosis of microglia in the central nervous system diseases. Mol. Neurobiol. 2014, 49, 1422–1434. [Google Scholar] [CrossRef]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef]
- ElAli, A.; Rivest, S. Microglia in Alzheimer’s disease: A multifaceted relationship. Brain. Behav. Immun. 2016, 55, 138–150. [Google Scholar] [CrossRef]
- Daria, A.; Colombo, A.; Llovera, G.; Hampel, H.; Willem, M.; Liesz, A.; Haass, C.; Tahirovic, S. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017, 36, 583–603. [Google Scholar] [CrossRef]
- Bolmont, T.; Haiss, F.; Eicke, D.; Radde, R.; Mathis, C.A.; Klunk, W.E.; Kohsaka, S.; Jucker, M.; Calhoun, M.E. Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J. Neurosci. 2008, 28, 4283–4292. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ulland, T.K.; Colonna, M. TREM2-dependent effects on microglia in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 202. [Google Scholar] [CrossRef] [PubMed]
- Ulland, T.K.; Song, W.M.; Huang, S.C.C.; Ulrich, J.D.; Sergushichev, A.; Beatty, W.L.; Loboda, A.A.; Zhou, Y.; Cairns, N.J.; Kambal, A.; et al. TREM2 Maintains Microglial Metabolic Fitness in Alzheimer’s Disease. Cell 2017, 170, 649–663.e13. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Neher, J.J.; Neniskyte, U.; Zhao, J.-W.; Bal-Price, A.; Tolkovsky, A.M.; Brown, G.C. Inhibition of Microglial Phagocytosis Is Sufficient To Prevent Inflammatory Neuronal Death. J. Immunol. 2011, 186, 4973–4983. [Google Scholar] [CrossRef] [PubMed]
- Neher, J.J.; Neniskyte, U.; Brown, G.C. Primary phagocytosis of neurons by inflamed microglia: Potential roles in neurodegeneration. Front. Pharmacol. 2012, 3, 27. [Google Scholar] [CrossRef]
- Giunta, B.; Fernandez, F.; Nikolic, W.V.; Obregon, D.; Rrapo, E.; Town, T.; Tan, J. Inflammaging as a prodrome to Alzheimer’s disease. J. Neuroinflamm. 2008, 5, 51. [Google Scholar] [CrossRef]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef]
- Spangenberg, E.E.; Lee, R.J.; Najafi, A.R.; Rice, R.A.; Elmore, M.R.P.; Blurton-Jones, M.; West, B.L.; Green, K.N. Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-β pathology. Brain 2016, 139, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Raj, D.; Saiepour, N.; Van Dam, D.; Brouwer, N.; Holtman, I.R.; Eggen, B.J.L.; Möller, T.; Tamm, J.A.; Abdourahman, A.; et al. Immune hyperreactivity of Aβ plaque-associated microglia in Alzheimer’s disease. Neurobiol. Aging 2017, 55, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, X.F.; Wang, T.; Wang, Z.; Liao, C.; Wang, Z.; Huang, R.; Wang, D.; Li, X.; Wu, L.; et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J. Exp. Med. 2017, 214, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, H.; Zhang, S.; Lu, S.; Sun, J.; Qian, Y. Enhancement of LPS-induced microglial inflammation response via TLR4 under high glucose conditions. Cell. Physiol. Biochem. 2015, 35, 1571–1581. [Google Scholar] [CrossRef]
- Jay, T.R.; Hirsch, A.M.; Broihier, M.L.; Miller, C.M.; Neilson, L.E.; Ransohoff, R.M.; Lamb, B.T.; Landreth, G.E. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 2017, 37, 637–647. [Google Scholar] [CrossRef]
- Dansokho, C.; Heneka, M.T. Neuroinflammatory responses in Alzheimer’s disease. J. Neural Transm. 2018, 125, 771–779. [Google Scholar] [CrossRef]
- Michaud, J.P.; Hallé, M.; Lampron, A.; Thériault, P.; Préfontaine, P.; Filali, M.; Tribout-Jover, P.; Lanteigne, A.M.; Jodoin, R.; Cluff, C.; et al. Toll-like receptor 4 stimulation with the detoxified ligand monophosphoryl lipid A improves Alzheimer’s disease-related pathology. Proc. Natl. Acad. Sci. USA 2013, 110, 1941–1946. [Google Scholar] [CrossRef]
- Parhizkar, S.; Arzberger, T.; Brendel, M.; Kleinberger, G.; Deussing, M.; Focke, C.; Nuscher, B.; Xiong, M.; Ghasemigharagoz, A.; Katzmarski, N.; et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat. Neurosci. 2019, 22, 191–204. [Google Scholar] [CrossRef]
- Bemiller, S.M.; McCray, T.J.; Allan, K.; Formica, S.V.; Xu, G.; Wilson, G.; Kokiko-Cochran, O.N.; Crish, S.D.; Lasagna-Reeves, C.A.; Ransohoff, R.M.; et al. TREM2 deficiency exacerbates tau pathology through dysregulated kinase signaling in a mouse model of tauopathy. Mol. Neurodegener. 2017, 12, 74. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef]
- Wang, Y.; Cella, M.; Mallinson, K.; Ulrich, J.D.; Young, K.L.; Robinette, M.L.; Gilfillan, S.; Krishnan, G.M.; Sudhakar, S.; Zinselmeyer, B.H.; et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 2015, 160, 1061–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ulland, T.K.; Ulrich, J.D.; Song, W.; Tzaferis, J.A.; Hole, J.T.; Yuan, P.; Mahan, T.E.; Shi, Y.; Gilfillan, S.; et al. TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J. Exp. Med. 2016, 213, 667–675. [Google Scholar] [CrossRef]
- Clarke, L.E.; Liddelow, S.A.; Chakraborty, C.; Münch, A.E.; Heiman, M.; Barres, B.A. Normal aging induces A1-like astrocyte reactivity. Proc. Natl. Acad. Sci. USA 2018, 115, E1896–E1905. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, M.K.; Carrier, M.; González Ibáñez, F.; Šimončičová, E.; Wallman, M.J.; Vallières, L.; Parent, M.; Tremblay, M.È. Ultrastructural characterization of dark microglia during aging in a mouse model of Alzheimer’s disease pathology and in human post-mortem brain samples. J. Neuroinflamm. 2022, 19, 235. [Google Scholar] [CrossRef] [PubMed]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Gabriela Sánchez, M.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gómez-Nicola, D.; Luheshi, G.; Vallières, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef] [PubMed]
- El Hajj, H.; Savage, J.C.; Bisht, K.; Parent, M.; Vallières, L.; Rivest, S.; Tremblay, M.È. Ultrastructural evidence of microglial heterogeneity in Alzheimer’s disease amyloid pathology. J. Neuroinflamm. 2019, 16, 87. [Google Scholar] [CrossRef]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef]
- Wang, E.J.; Sun, J.; Pettoello-Mantovani, M.; Anderson, C.M.; Osiecki, K.; Zhao, M.L.; Lopez, L.; Lee, S.C.; Berman, J.W.; Goldstein, H. Microglia from mice transgenic for a provirus encoding a monocyte-tropic HIV type 1 isolate produce infectious virus and display in vitro and in vivo upregulation of lipopolysaccharide-induced chemokine gene expression. AIDS Res. Hum. Retroviruses 2003, 19, 755–765. [Google Scholar] [CrossRef]
- Hart, A.D.; Wyttenbach, A.; Hugh Perry, V.; Teeling, J.L. Age related changes in microglial phenotype vary between CNS regions: Grey versus white matter differences. Brain. Behav. Immun. 2012, 26, 754–765. [Google Scholar] [CrossRef]
- Minoretti, P.; Gazzaruso, C.; Di Vito, C.; Emanuele, E.; Bianchi, M.; Coen, E.; Reino, M.; Geroldi, D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci. Lett. 2006, 391, 147–149. [Google Scholar] [CrossRef]
- Giovannini, M.G.; Lana, D.; Pepeu, G. More than the cholinergic system: The evolving role of glia in memory, aging, and neurodegeneration. In Proceedings of the 17th International Symposium on Cholinergic Mechanisms, Dubrovnik, Croatia, 8–12 May 2022. Abstract Book p. 48. [Google Scholar]
- Fixemer, S.; Ameli, C.; Hammer, G.; Salamanca, L.; Uriarte Huarte, O.; Schwartz, C.; Gérardy, J.J.; Mechawar, N.; Skupin, A.; Mittelbronn, M.; et al. Microglia phenotypes are associated with subregional patterns of concomitant tau, amyloid-β and α-synuclein pathologies in the hippocampus of patients with Alzheimer’s disease and dementia with Lewy bodies. Acta Neuropathol. Commun. 2022, 10, 36. [Google Scholar] [CrossRef]
- Adler, D.H.; Wisse, L.E.M.; Ittyerah, R.; Pluta, J.B.; Ding, S.L.; Xie, L.; Wang, J.; Kadivar, S.; Robinson, J.L.; Schuck, T.; et al. Characterizing the human hippocampus in aging and Alzheimer’s disease using a computational atlas derived from ex vivo MRI and histology. Proc. Natl. Acad. Sci. USA 2018, 115, 4252–4257. [Google Scholar] [CrossRef] [PubMed]
- McQuade, A.; Blurton-Jones, M. Microglia in Alzheimer’s Disease: Exploring How Genetics and Phenotype Influence Risk. J. Mol. Biol. 2019, 431, 1805–1817. [Google Scholar] [CrossRef] [PubMed]
- Erny, D.; De Angelis, A.L.H.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef] [PubMed]
- Thion, M.S.; Low, D.; Silvin, A.; Chen, J.; Grisel, P.; Schulte-Schrepping, J.; Blecher, R.; Ulas, T.; Squarzoni, P.; Hoeffel, G.; et al. Microbiome Influences Prenatal and Adult Microglia in a Sex-Specific Manner. Cell 2018, 172, 500–516.e16. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.; Murdock, M.H.; Jing, D.; Won, T.H.; Chung, H.; Kressel, A.M.; Tsaava, T.; Addorisio, M.E.; Putzel, G.G.; Zhou, L.; et al. The microbiota regulate neuronal function and fear extinction learning. Nature 2019, 574, 543–548. [Google Scholar] [CrossRef]
- Pandey, S.; Kawai, T.; Akira, S. Microbial sensing by toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol. 2015, 7, a016246. [Google Scholar] [CrossRef]
- Vargas-Caraveo, A.; Sayd, A.; Maus, S.R.; Caso, J.R.; Madrigal, J.L.M.; García-Bueno, B.; Leza, J.C. Lipopolysaccharide enters the rat brain by a lipoprotein-mediated transport mechanism in physiological conditions. Sci. Rep. 2017, 7, 13113. [Google Scholar] [CrossRef]
- Liu, S.; Gao, J.; Zhu, M.; Liu, K.; Zhang, H.L. Gut Microbiota and Dysbiosis in Alzheimer’s Disease: Implications for Pathogenesis and Treatment. Mol. Neurobiol. 2020, 57, 5026–5043. [Google Scholar] [CrossRef]
- Harach, T.; Marungruang, N.; Duthilleul, N.; Cheatham, V.; Mc Coy, K.D.; Frisoni, G.; Neher, J.J.; Fåk, F.; Jucker, M.; Lasser, T.; et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci. Rep. 2017, 7, 41802. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Haq, R.; Schlachetzki, J.C.M.; Glass, C.K.; Mazmanian, S.K. Microbiome–microglia connections via the gut–brain axis. J. Exp. Med. 2019, 216, 41–59. [Google Scholar] [CrossRef] [PubMed]
- Gosselin, D.; Link, V.M.; Romanoski, C.E.; Fonseca, G.J.; Eichenfield, D.Z.; Spann, N.J.; Stender, J.D.; Chun, H.B.; Garner, H.; Geissmann, F.; et al. Environment drives selection and function of enhancers controlling tissue-specific macrophage identities. Cell 2014, 159, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Datta, M.; Staszewski, O.; Raschi, E.; Frosch, M.; Hagemeyer, N.; Tay, T.L.; Blank, T.; Kreutzfeldt, M.; Merkler, D.; Ziegler-Waldkirch, S.; et al. Histone Deacetylases 1 and 2 Regulate Microglia Function during Development, Homeostasis, and Neurodegeneration in a Context-Dependent Manner. Immunity 2018, 48, 514–529.e6. [Google Scholar] [CrossRef]
- Sharma, M.; Li, Y.; Stoll, M.L.; Tollefsbol, T.O. The Epigenetic Connection Between the Gut Microbiome in Obesity and Diabetes. Front. Genet. 2020, 10, 1329. [Google Scholar] [CrossRef]
- Masgrau, R.; Guaza, C.; Ransohoff, R.M.; Galea, E. Should We Stop Saying ‘Glia’ and ‘Neuroinflammation’? Trends Mol. Med. 2017, 23, 486–500. [Google Scholar] [CrossRef]
- Jinno, S.; Fleischer, F.; Eckel, S.; Schmidt, V.; Kosaka, T. Spatial arrangement of microglia in the mouse hippocampus: A stereological study in comparison with astrocytes. Glia 2007, 55, 1334–1347. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.L.; Yuan, Y.; Tian, L. Microglial regional heterogeneity and its role in the brain. Mol. Psychiatry 2020, 25, 351–367. [Google Scholar] [CrossRef]
- Damisah, E.C.; Hill, R.A.; Rai, A.; Chen, F.; Rothlin, C.V.; Ghosh, S.; Grutzendler, J. Astrocytes and microglia play orchestrated roles and respect phagocytic territories during neuronal corpse removal in vivo. Sci. Adv. 2020, 6, eaba3239. [Google Scholar] [CrossRef]
- Wenk, G.L.; Barnes, C.A. Regional changes in the hippocampal density of AMPA and NMDA receptors across the lifespan of the rat. Brain Res. 2000, 885, 1–5. [Google Scholar] [CrossRef]
- Hauss-Wegrzyniak, B.; Lukovic, L.; Bigaud, M.; Stoeckel, M.E. Brain inflammatory response induced by intracerebroventricular infusion of lipopolysaccharide: An immunohistochemical study. Brain Res. 1998, 794, 211–224. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Janeway, C.A. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef]
- Milligan, E.D.; Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.D.; Julien, J.P.; Rivest, S. Innate immunity: The missing link in neuroprotection and neurodegeneration? Nat. Rev. Neurosci. 2002, 3, 216–227. [Google Scholar] [CrossRef]
- Turrin, N.P.; Rivest, S. Molecular and cellular immune mediators of neuroprotection. Mol. Neurobiol. 2006, 34, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Dissing-Olesen, L.; LeDue, J.M.; Rungta, R.L.; Hefendehl, J.K.; Choi, H.B.; MacVicar, B.A. Activation of neuronal NMDA receptors triggers transient ATP-mediated microglial process outgrowth. J. Neurosci. 2014, 34, 10511–10527. [Google Scholar] [CrossRef]
- Bernier, L.P.; Bohlen, C.J.; York, E.M.; Choi, H.B.; Kamyabi, A.; Dissing-Olesen, L.; Hefendehl, J.K.; Collins, H.Y.; Stevens, B.; Barres, B.A.; et al. Nanoscale Surveillance of the Brain by Microglia via cAMP-Regulated Filopodia. Cell Rep. 2019, 27, 2895–2908.e4. [Google Scholar] [CrossRef]
- Jaturapatporn, D.; Isaac, M.G.E.K.N.; McCleery, J.; Tabet, N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst. Rev. 2012, 2, CD006378. [Google Scholar] [CrossRef]
- Zrzavy, T.; Hametner, S.; Wimmer, I.; Butovsky, O.; Weiner, H.L.; Lassmann, H. Loss of “homeostatic” microglia and patterns of their activation in active multiple sclerosis. Brain 2017, 140, 1900–1913. [Google Scholar] [CrossRef]
- Lewis, N.D.; Hill, J.D.; Juchem, K.W.; Stefanopoulos, D.E.; Modis, L.K. RNA sequencing of microglia and monocyte-derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. J. Neuroimmunol. 2014, 277, 26–38. [Google Scholar] [CrossRef]
- Butovsky, O.; Jedrychowski, M.P.; Cialic, R.; Krasemann, S.; Murugaiyan, G.; Fanek, Z.; Greco, D.J.; Wu, P.M.; Doykan, C.E.; Kiner, O.; et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann. Neurol. 2015, 77, 75–99. [Google Scholar] [CrossRef] [PubMed]
- Gao, T.; Jernigan, J.; Raza, S.A.; Dammer, E.B.; Xiao, H.; Seyfried, N.T.; Levey, A.I.; Rangaraju, S. Transcriptional regulation of homeostatic and disease-associated-microglial genes by IRF1, LXRβ, and CEBPα. Glia 2019, 67, 1958–1975. [Google Scholar] [CrossRef]
- Chu, K.; Yin, B.; Wang, J.; Peng, G.; Liang, H.; Xu, Z.; Du, Y.; Fang, M.; Xia, Q.; Luo, B. Inhibition of P2X7 receptor ameliorates transient global cerebral ischemia/reperfusion injury via modulating inflammatory responses in the rat hippocampus. J. Neuroinflamm. 2012, 9, 69. [Google Scholar] [CrossRef]
- Walter, S.; Letiembre, M.; Liu, Y.; Heine, H.; Penke, B.; Hao, W.; Bode, B.; Manietta, N.; Walter, J.; Schulz-Schüffer, W.; et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer’s disease. Cell. Physiol. Biochem. 2007, 20, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Sackett, S.; Zhang, Y. Endocannabinoid Modulation of Microglial Phenotypes in Neuropathology. Front. Neurol. 2020, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Lana, D.; Landucci, E.; Mazzantini, C.; Magni, G.; Pellegrini-Giampietro, D.E.; Giovannini, M.G. The Protective Effect of CBD in a Model of In Vitro Ischemia May Be Mediated by Agonism on TRPV2 Channel and Microglia Activation. Int. J. Mol. Sci. 2022, 23, 12144. [Google Scholar] [CrossRef]
- Landucci, E.; Mazzantini, C.; Lana, D.; Calvani, M.; Magni, G.; Giovannini, M.G.; Pellegrini-Giampietro, D.E. Cannabidiol inhibits microglia activation and mitigates neuronal damage induced by kainate in an in-vitro seizure model. Neurobiol. Dis. 2022, 174, 105895. [Google Scholar] [CrossRef]
- Juknat, A.; Rimmerman, N.; Levy, R.; Vogel, Z.; Kozela, E. Cannabidiol affects the expression of genes involved in zinc homeostasis in BV-2 microglial cells. Neurochem. Int. 2012, 61, 923–930. [Google Scholar] [CrossRef]
- Barata, L.; Arruza, L.; Rodríguez, M.J.; Aleo, E.; Vierge, E.; Criado, E.; Sobrino, E.; Vargas, C.; Ceprián, M.; Gutiérrez-Rodríguez, A.; et al. Neuroprotection by cannabidiol and hypothermia in a piglet model of newborn hypoxic-ischemic brain damage. Neuropharmacology 2019, 146, 1–11. [Google Scholar] [CrossRef]
- dos-Santos-Pereira, M.; Guimarães, F.S.; Del-Bel, E.; Raisman-Vozari, R.; Michel, P.P. Cannabidiol prevents LPS-induced microglial inflammation by inhibiting ROS/NF-κB-dependent signaling and glucose consumption. Glia 2020, 68, 561–573. [Google Scholar] [CrossRef]
- Martín-Moreno, A.M.; Reigada, D.; Ramírez, B.G.; Mechoulam, R.; Innamorato, N.; Cuadrado, A.; De Ceballos, M.L. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: Relevance to Alzheimer’s disease. Mol. Pharmacol. 2011, 79, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Mecha, M.; Feliú, A.; Iñigo, P.M.; Mestre, L.; Carrillo-Salinas, F.J.; Guaza, C. Cannabidiol provides long-lasting protection against the deleterious effects of inflammation in a viral model of multiple sclerosis: A role for A2A receptors. Neurobiol. Dis. 2013, 59, 141–150. [Google Scholar] [CrossRef]
- Yang, S.; Du, Y.; Zhao, X.; Tang, Q.; Su, W.; Hu, Y.; Yu, P. Cannabidiol Enhances Microglial Beta-Amyloid Peptide Phagocytosis and Clearance via Vanilloid Family Type 2 Channel Activation. Int. J. Mol. Sci. 2022, 23, 5367. [Google Scholar] [CrossRef] [PubMed]
- Hassan, S.; Eldeeb, K.; Millns, P.J.; Bennett, A.J.; Alexander, S.P.H.; Kendall, D.A. Cannabidiol enhances microglial phagocytosis via transient receptor potential (TRP) channel activation. Br. J. Pharmacol. 2014, 171, 2426–2439. [Google Scholar] [CrossRef]
- Egea, J.; Buendia, I.; Parada, E.; Navarro, E.; León, R.; Lopez, M.G. Anti-inflammatory role of microglial alpha7 nAChRs and its role in neuroprotection. Biochem. Pharmacol. 2015, 97, 463–472. [Google Scholar] [CrossRef]
- De Simone, R.; Ajmone-Cat, M.A.; Carnevale, D.; Minghetti, L. Activation of alpha7 nicotinic acetylcholine receptor by nicotine selectively up-regulates cyclooxygenase-2 and prostaglandin E2 in rat microglial cultures. J. Neuroinflamm. 2005, 2, 4. [Google Scholar] [CrossRef]
- Thomsen, M.S.; Mikkelsen, J.D. The α7 nicotinic acetylcholine receptor ligands methyllycaconitine, NS6740 and GTS-21 reduce lipopolysaccharide-induced TNF-α release from microglia. J. Neuroimmunol. 2012, 251, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Parada, E.; Egea, J.; Buendia, I.; Negredo, P.; Cunha, A.C.; Cardoso, S.; Soares, M.P.; López, M.G. The microglial α7-acetylcholine nicotinic receptor is a key element in promoting neuroprotection by inducing heme oxygenase-1 via nuclear factor erythroid-2-related factor 2. Antioxid. Redox Signal. 2013, 19, 1135–1148. [Google Scholar] [CrossRef]
- Takata, K. Molecular targeting and translational research for new therapeutic strategies on Alzheimer’s disease. Yakugaku Zasshi 2013, 133, 1389–1399. [Google Scholar] [CrossRef][Green Version]
- Sharma, G.; Vijayaraghavan, S. Nicotinic cholinergic signaling in hippocampal astrocytes involves calcium-induced calcium release from intracellular stores. Proc. Natl. Acad. Sci. USA 2001, 98, 4148–4153. [Google Scholar] [CrossRef]
- Rock, R.B.; Gekker, G.; Aravalli, R.N.; Hu, S.; Sheng, W.S.; Peterson, P.K. Potentiation of HIV-1 expression in microglial cells by nicotine: Involvement of transforming growth factor-beta 1. J. Neuroimmune Pharmacol. 2008, 3, 143–149. [Google Scholar] [CrossRef]
- Suzuki, T.; Hide, I.; Matsubara, A.; Hama, C.; Harada, K.; Miyano, K.; Andrä, M.; Matsubayashi, H.; Sakai, N.; Kohsaka, S.; et al. Microglial alpha7 nicotinic acetylcholine receptors drive a phospholipase C/IP3 pathway and modulate the cell activation toward a neuroprotective role. J. Neurosci. Res. 2006, 83, 1461–1470. [Google Scholar] [CrossRef] [PubMed]
- Loram, L.C.; Harrison, J.A.; Chao, L.; Taylor, F.R.; Reddy, A.; Travis, C.L.; Giffard, R.; Al-Abed, Y.; Tracey, K.; Maier, S.F.; et al. Intrathecal injection of an alpha seven nicotinic acetylcholine receptor agonist attenuates gp120-induced mechanical allodynia and spinal pro-inflammatory cytokine profiles in rats. Brain. Behav. Immun. 2010, 24, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Jonnala, R.R.; Buccafusco, J.J. Relationship between the increased cell surface alpha7 nicotinic receptor expression and neuroprotection induced by several nicotinic receptor agonists. J. Neurosci. Res. 2001, 66, 565–572. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [PubMed]
- Eder, C.; Klee, R.; Heinemann, U. Pharmacological properties of Ca2+-activated K+ currents of ramified murine brain macrophages. Naunyn. Schmiedebergs. Arch. Pharmacol. 1997, 356, 233–239. [Google Scholar] [CrossRef]
- Khanna, R.; Roy, L.; Zhu, X.; Schlichter, L.C. K+ channels and the microglial respiratory burst. Am. J. Physiol. Cell Physiol. 2001, 280, C796–C806. [Google Scholar] [CrossRef]
- Chen, Y.J.; Raman, G.; Bodendiek, S.; O’Donnell, M.E.; Wulff, H. The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J. Cereb. Blood Flow Metab. 2011, 31, 2363–2374. [Google Scholar] [CrossRef]
- Kaushal, V.; Koeberle, P.D.; Wang, Y.; Schlichter, L.C. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J. Neurosci. 2007, 27, 234–244. [Google Scholar] [CrossRef]
- Staal, R.G.W.; Khayrullina, T.; Zhang, H.; Fallon, S.M.; Zorn, S.; Dale, E.; Cambpell, B.; Möller, T.; Davis, S.; Hu, A.; et al. Inhibition of the potassium channel KCa3.1 by senicapoc reverses tactile allodynia in rats with peripheral nerve injury. Eur. J. Pharmacol. 2017, 795, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.W.; Di Lucente, J.; Nguyen, H.M.; Singh, V.; Singh, L.; Chavez, M.; Bushong, T.; Wulff, H.; Maezawa, I. Repurposing the KCa3.1 inhibitor senicapoc for Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2019, 6, 723–738. [Google Scholar] [CrossRef]
- Dale, E.; Staal, R.G.W.; Eder, C.; Möller, T. KCa 3.1-a microglial target ready for drug repurposing? Glia 2016, 64, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Staal, R.G.W.; Weinstein, J.R.; Nattini, M.; Cajina, M.; Chandresana, G.; Möller, T. Senicapoc: Repurposing a Drug to Target Microglia KCa3.1 in Stroke. Neurochem. Res. 2017, 42, 2639–2645. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
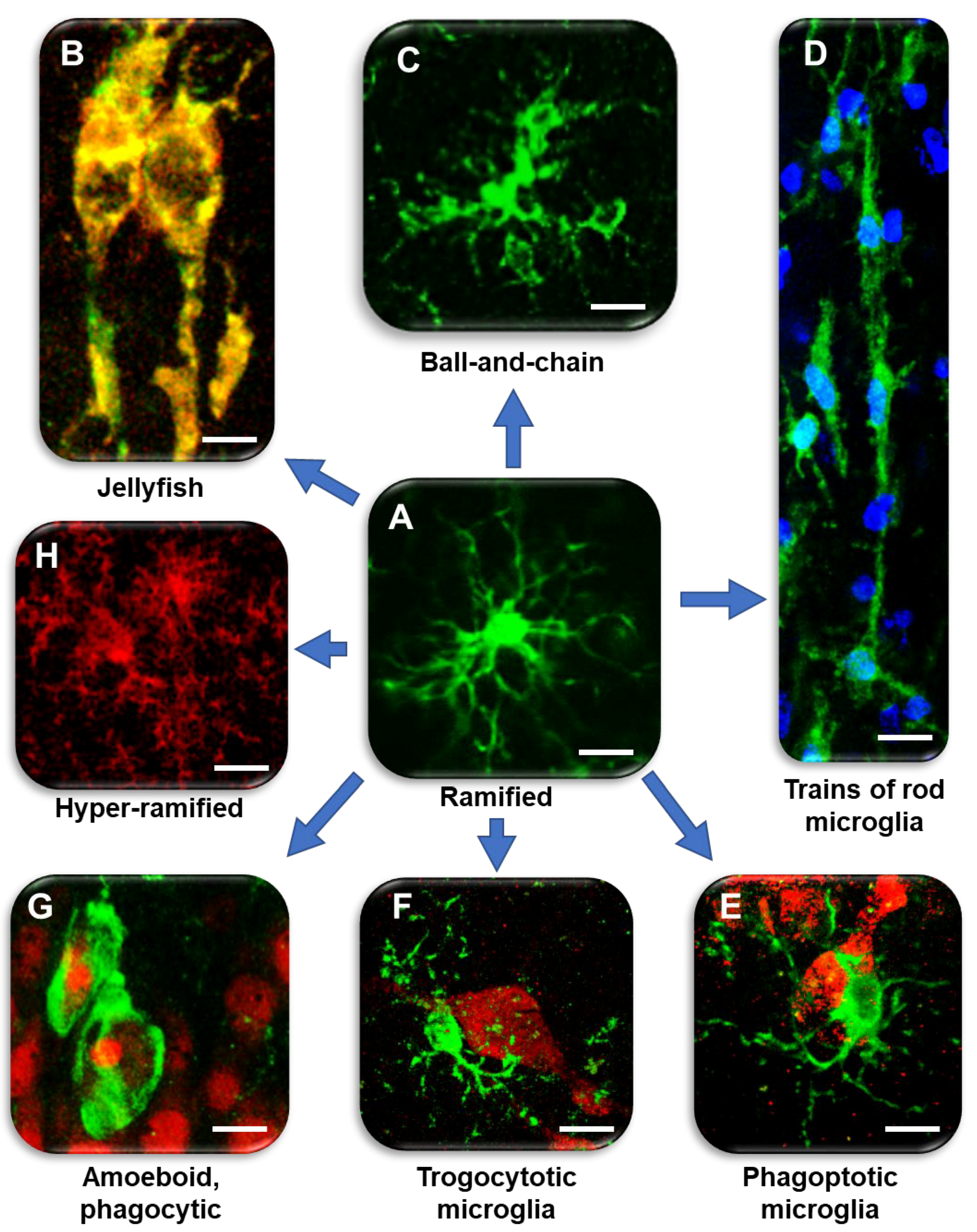
| Microglia Phenotype | CA1 SP | CA1 SR/SLM | References |
|---|---|---|---|
| Jellyfish | + | [48,162] | |
| Ball-and-chain | + | [29] | |
| Trains of rod microglia | + | + | [48,140,142,147,149,150,156] |
| Phagoptotic | + | [65,97,171,177,178,179,180] | |
| Trogocytotic | + | [108] | |
| Amoeboid phagocytic | + | + | [141,142,152,154,159,160,161] |
| Hyper-ramified | + | [206,207,208] | |
| DAM | + | [39] | |
| Plaque-associated microglia | + | [105,117,174,175,176,183,184,187,188,212] | |
| Dark microglia | + | [195,196,197] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lana, D.; Magni, G.; Landucci, E.; Wenk, G.L.; Pellegrini-Giampietro, D.E.; Giovannini, M.G. Phenomic Microglia Diversity as a Druggable Target in the Hippocampus in Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 13668. https://doi.org/10.3390/ijms241813668
Lana D, Magni G, Landucci E, Wenk GL, Pellegrini-Giampietro DE, Giovannini MG. Phenomic Microglia Diversity as a Druggable Target in the Hippocampus in Neurodegenerative Diseases. International Journal of Molecular Sciences. 2023; 24(18):13668. https://doi.org/10.3390/ijms241813668
Chicago/Turabian StyleLana, Daniele, Giada Magni, Elisa Landucci, Gary L. Wenk, Domenico Edoardo Pellegrini-Giampietro, and Maria Grazia Giovannini. 2023. "Phenomic Microglia Diversity as a Druggable Target in the Hippocampus in Neurodegenerative Diseases" International Journal of Molecular Sciences 24, no. 18: 13668. https://doi.org/10.3390/ijms241813668
APA StyleLana, D., Magni, G., Landucci, E., Wenk, G. L., Pellegrini-Giampietro, D. E., & Giovannini, M. G. (2023). Phenomic Microglia Diversity as a Druggable Target in the Hippocampus in Neurodegenerative Diseases. International Journal of Molecular Sciences, 24(18), 13668. https://doi.org/10.3390/ijms241813668