
Cross-Activation of Regulatory T Cells by Self Antigens Limits Self-Reactive and Activated CD8+ T Cell Responses
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
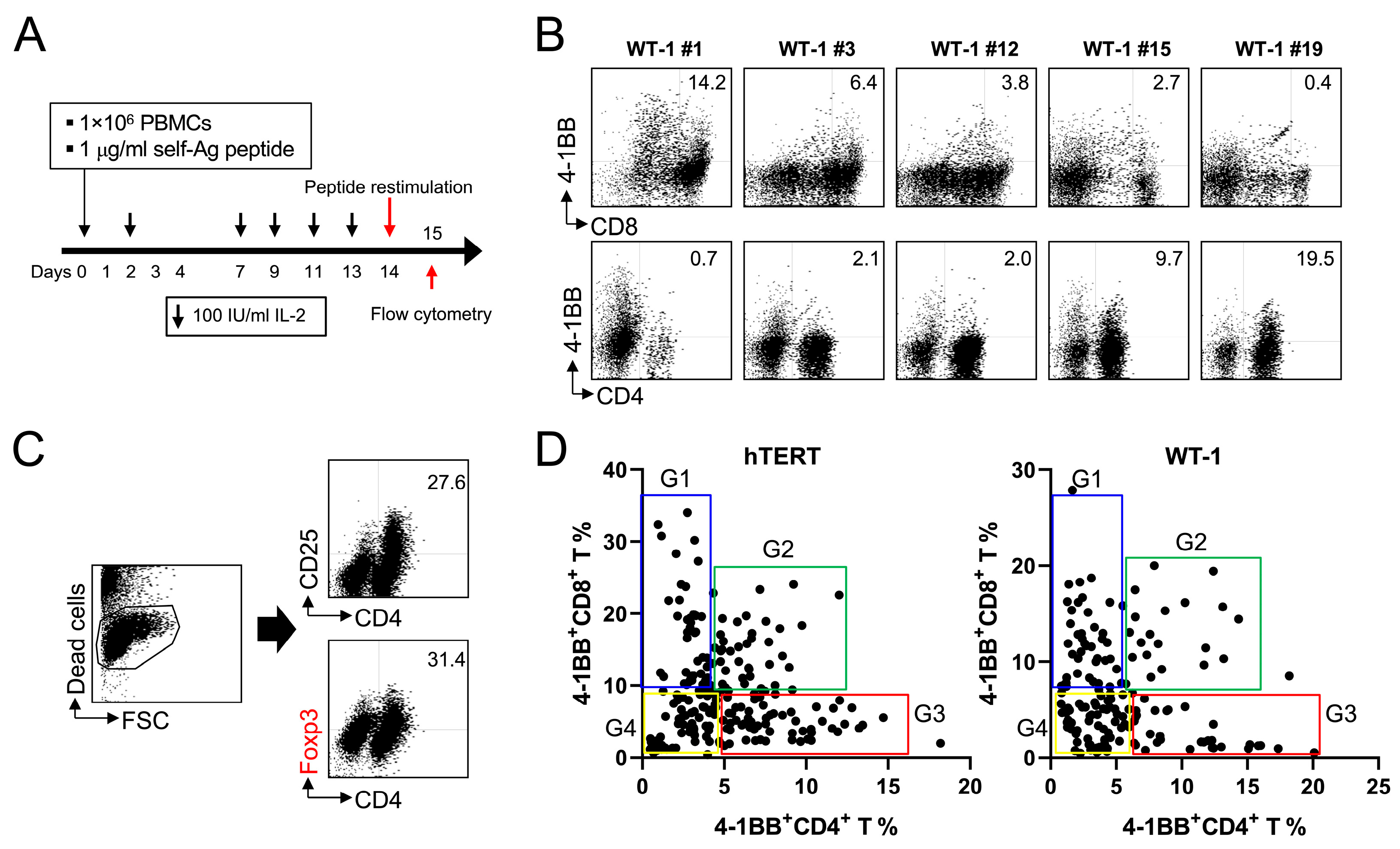
2.1. MHC Class I-Restricted Peptides Not Only Activate CD8+ T Cells but Also Cross-Activate CD4+ T Cells, including Foxp3+ Treg Cells
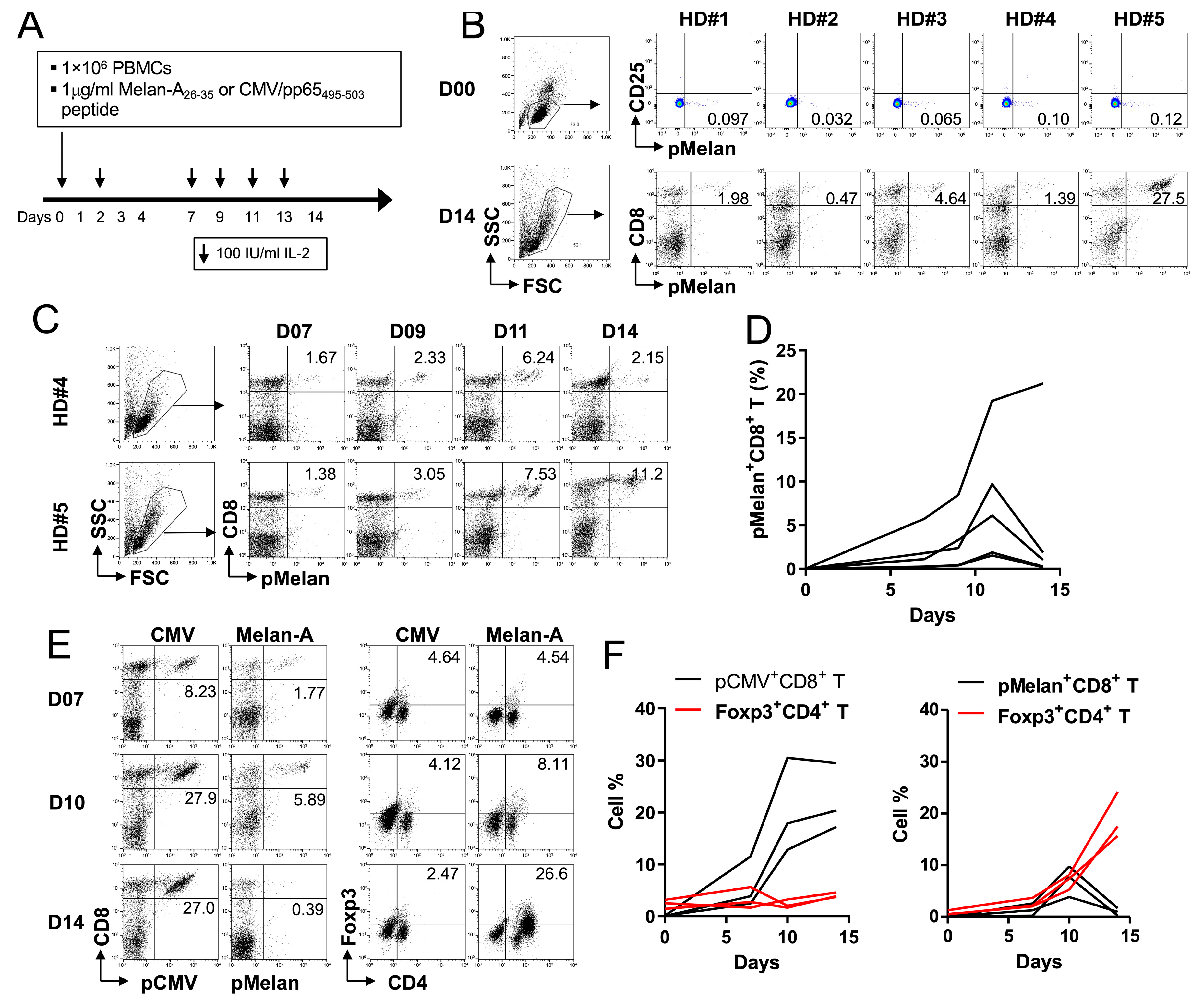
2.2. Foxp3+ Treg Cells Are Expanded by Melan-A Self-Peptide, but Not by Non-Self CMV Peptide
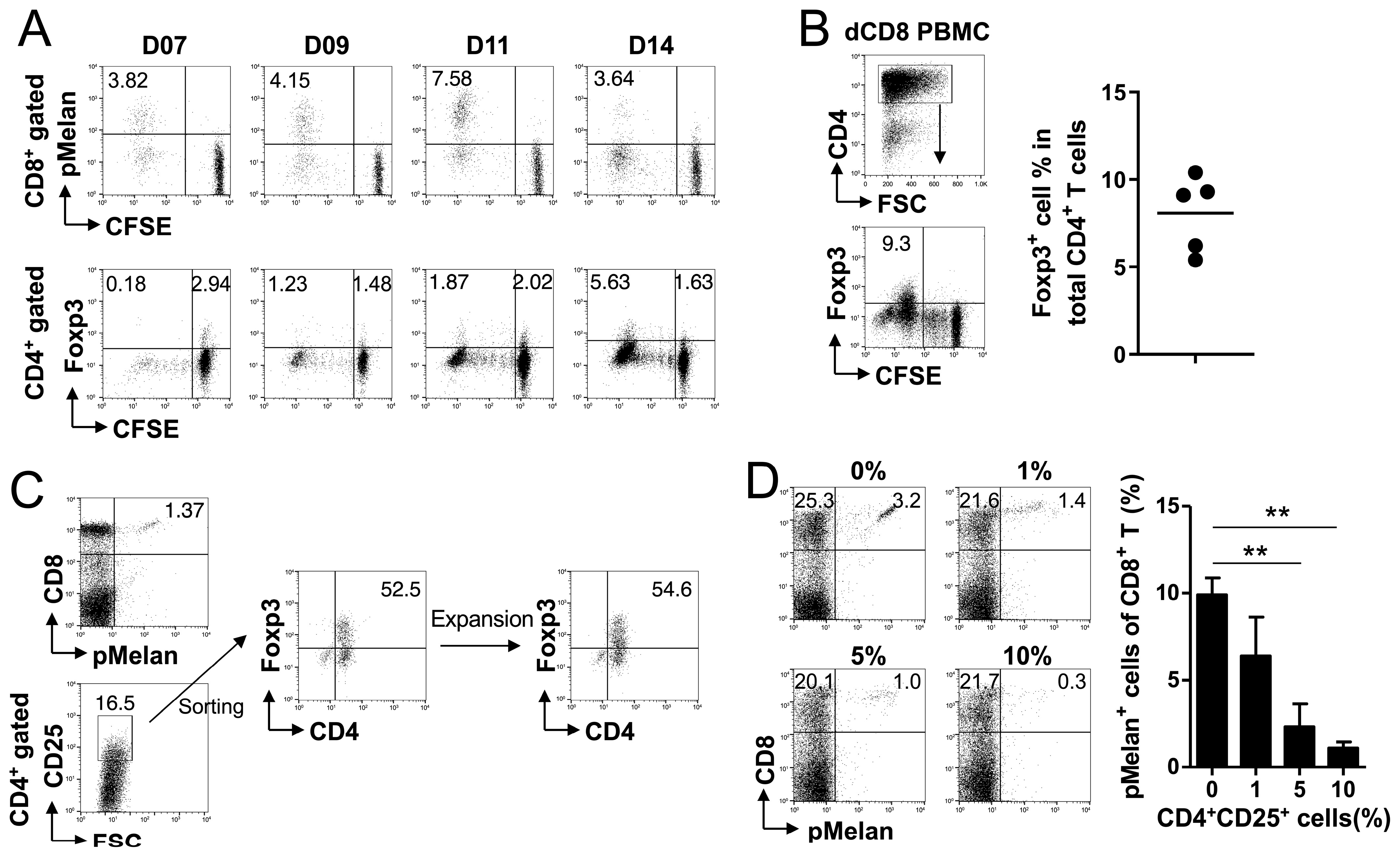
2.3. Melan-A Self-Peptide Directly Expands Foxp3+ Treg Cells
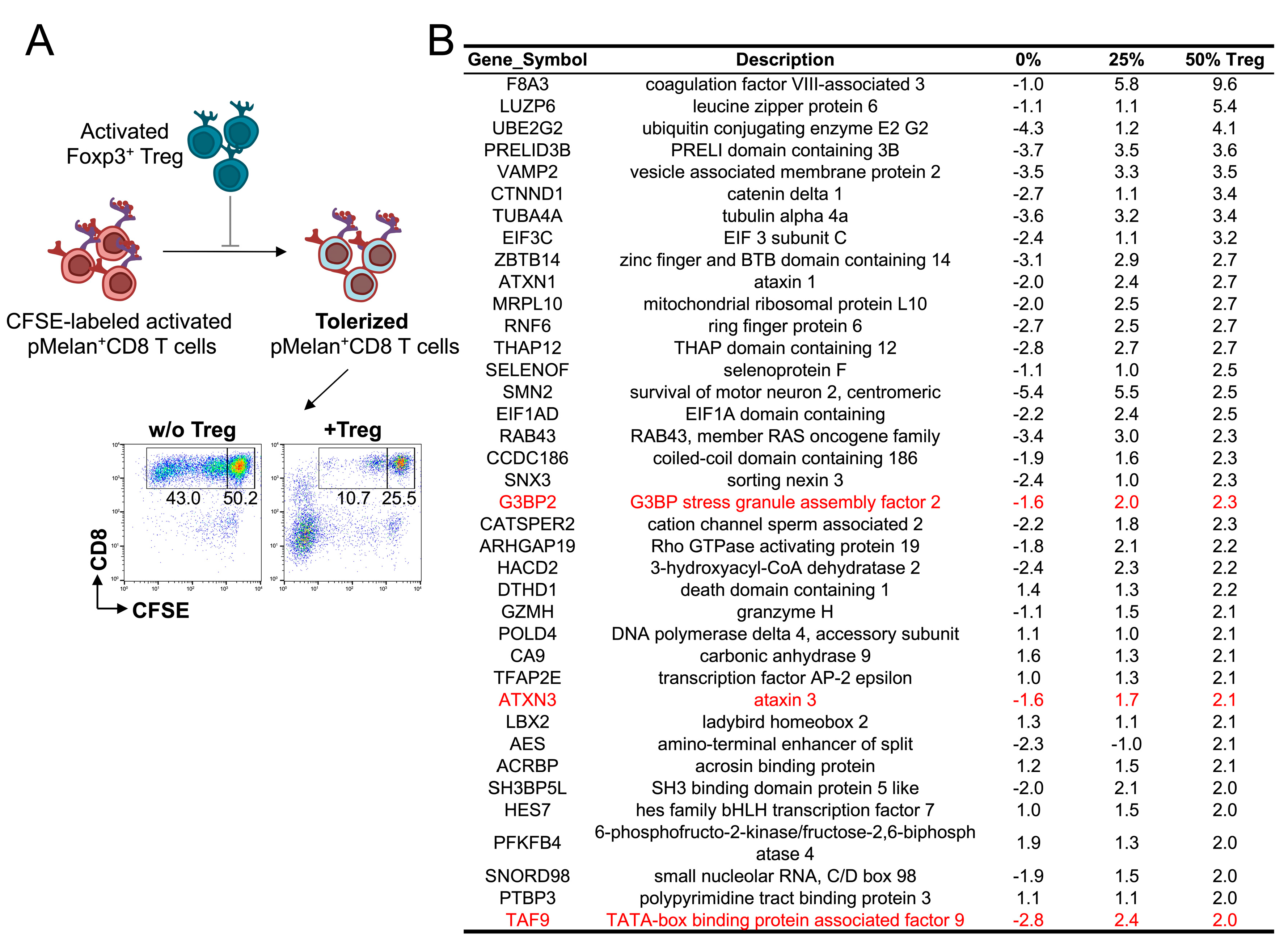
2.4. No Specific Signaling Pathway Is Induced in pMelan+CD8+ T Cells by Contact with Foxp3+ Treg Cells
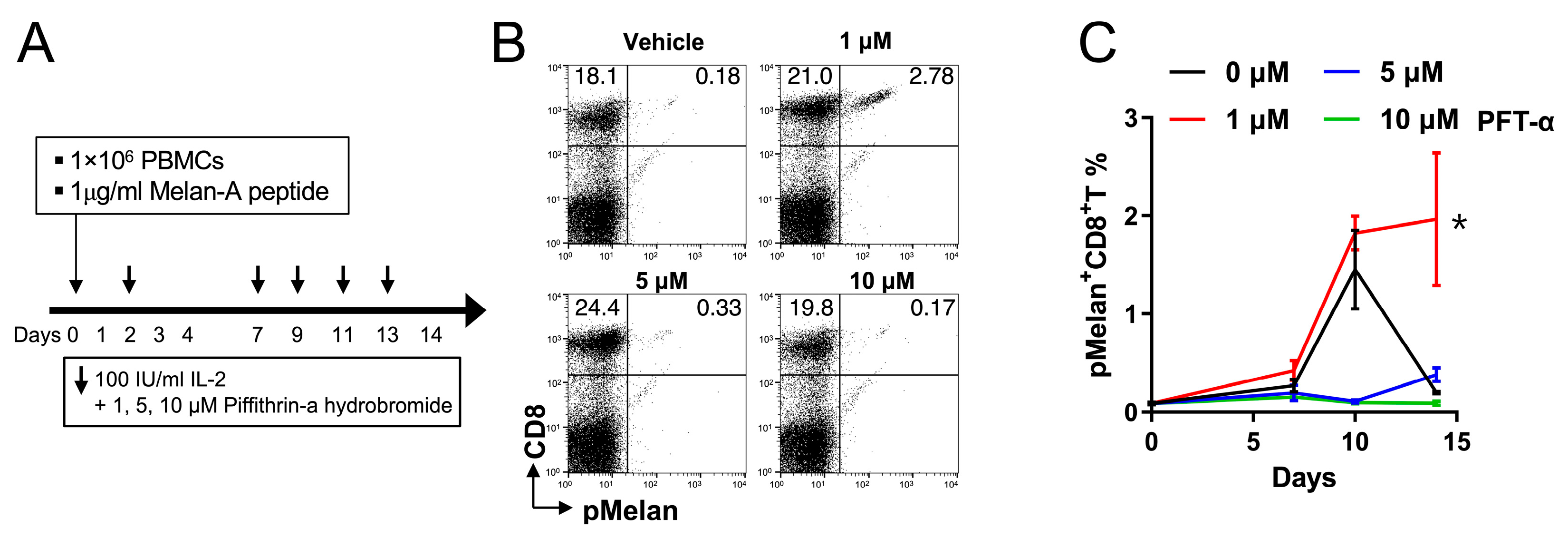
2.5. Pifithrin-α Hydrobromide Induces Continuous Growth of pMelan+CD8+ T Cells
3. Discussion
4. Materials and Methods
4.1. Reagents and Antibodies
4.2. Expansion of Peptide-Specific CD8+ T Cells from PBMCs
4.3. Induction and Detection of 4-1BB on Peptide-Specific CD8+ T Cells
4.4. Flow Cytometry
4.5. CFSE Dilution Assay
4.6. Rapid Expansion of Melan-A-Specific CD8+ T Cells and CD4+CD25+ Treg Cells
4.7. Transcriptome Analysis of Melan-A-Specific CD8+ T Cells in the Presence or Absence of Activated Treg Cells
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hogquist, K.A.; Baldwin, T.A.; Jameson, S.C. Central tolerance: Learning self-control in the thymus. Nat. Rev. Immunol. 2005, 5, 772–782. [Google Scholar] [CrossRef] [PubMed]
- Nüssing, S.; Trapani, J.A.; Parish, I.A. Revisiting T cell tolerance as a checkpoint target for cancer immunotherapy. Front. Immunol. 2020, 11, 589641. [Google Scholar] [CrossRef]
- Richards, D.M.; Kyewski, B.; Feuerer, M. Re-examining the Nature and Function of Self-Reactive T cells. Trends Immunol. 2016, 37, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Jiang, N.; Ebert, P.J.; Kidd, B.A.; Müller, S.; Lund, P.J.; Juang, J.; Adachi, K.; Tse, T.; Birnbaum, M.E.; et al. Clonal Deletion Prunes but Does Not Eliminate Self-Specific αβ CD8(+) T Lymphocytes. Immunity 2015, 42, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Su, L.F.; Kidd, B.A.; Han, A.; Kotzin, J.J.; Davis, M.M. Virus-specific CD4(+) memory-phenotype T cells are abundant in unexposed adults. Immunity 2013, 38, 373–383. [Google Scholar] [CrossRef]
- Redmond, W.L.; Sherman, L.A. Peripheral tolerance of CD8 T lymphocytes. Immunity 2005, 22, 275–284. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Matsumoto, M.; Yoshida, H.; Tsuneyama, K.; Oya, T.; Matsumoto, M. Revisiting Aire and tissue-restricted antigens at single-cell resolution. Front. Immunol. 2023, 14, 1176450. [Google Scholar] [CrossRef]
- Lyu, M.; Suzuki, H.; Kang, L.; Gaspal, F.; Zhou, W.; Goc, J.; Zhou, L.; Zhou, J.; Zhang, W.; JRI Live Cell Bank; et al. ILC3s select microbiota-specific regulatory T cells to establish tolerance in the gut. Nature 2022, 610, 744–751. [Google Scholar] [CrossRef]
- Kedmi, R.; Najar, T.A.; Mesa, K.R.; Grayson, A.; Kroehling, L.; Hao, Y.; Hao, S.; Pokrovskii, M.; Xu, M.; Talbot, J.; et al. A RORγt+ cell instructs gut microbiota-specific Treg cell differentiation. Nature 2022, 610, 737–743. [Google Scholar] [CrossRef]
- Akagbosu, B.; Tayyebi, Z.; Shibu, G.; Paucar Iza, Y.A.; Deep, D.; Parisotto, Y.F.; Fisher, L.; Pasolli, H.A.; Thevin, V.; Elmentaite, R.; et al. Novel antigen-presenting cell imparts Treg-dependent tolerance to gut microbiota. Nature 2022, 610, 752–760. [Google Scholar] [CrossRef] [PubMed]
- Josefowicz, S.Z.; Lu, L.F.; Rudensky, A.Y. Regulatory T cells: Mechanisms of differentiation and function. Annu. Rev. Immunol. 2012, 30, 531–564. [Google Scholar] [CrossRef] [PubMed]
- Schildknecht, A.; Brauer, S.; Brenner, C.; Lahl, K.; Schild, H.; Sparwasser, T.; Probst, H.C.; van den Broek, M. FoxP3+ regulatory T cells essentially contribute to peripheral CD8+ T-cell tolerance induced by steady-state dendritic cells. Proc. Natl. Acad. Sci. USA 2010, 107, 199–203. [Google Scholar] [CrossRef]
- Chen, M.L.; Pittet, M.J.; Gorelik, L.; Flavell, R.A.; Weissleder, R.; von Boehmer, H.; Khazaie, K. Regulatory T cells suppress tumor-specific CD8 T cell cytotoxicity through TGF-beta signals in vivo. Proc. Natl. Acad. Sci. USA 2005, 102, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.S.; Park, K.; Gola, A.; Baptista, A.P.; Miller, C.H.; Deep, D.; Lou, M.; Boyd, L.F.; Rudensky, A.Y.; Savage, P.A.; et al. A local regulatory T cell feedback circuit maintains immune homeostasis by pruning self-activated T cells. Cell 2021, 184, 3981–3997.e22. [Google Scholar] [CrossRef]
- Brown, C.C.; Gottschalk, R.A. Volume control: Turning the dial on regulatory T cells. Cell 2021, 184, 3847–3849. [Google Scholar] [CrossRef]
- Kastenmuller, W.; Gasteiger, G.; Subramanian, N.; Sparwasser, T.; Busch, D.H.; Belkaid, Y.; Drexler, I.; Germain, R.N. Regulatory T cells selectively control CD8+ T cell effector pool size via IL-2 restriction. J. Immunol. 2011, 187, 3186–3197. [Google Scholar] [CrossRef]
- McNally, A.; Hill, G.R.; Sparwasser, T.; Thomas, R.; Steptoe, R.J. CD4+ CD25+ regulatory T cells control CD8+ T-cell effector differentiation by modulating IL-2 homeostasis. Proc. Natl. Acad. Sci. USA 2011, 108, 7529–7534. [Google Scholar] [CrossRef]
- Pace, L.; Tempez, A.; Arnold-Schrauf, C.; Lemaitre, F.; Bousso, P.; Fetler, L.; Sparwasser, T.; Amigorena, S. Regulatory T cells increase the avidity of primary CD8+ T cell responses and promote memory. Science 2012, 338, 532–536. [Google Scholar] [CrossRef]
- Chinen, T.; Kannan, A.K.; Levine, A.G.; Fan, X.; Klein, U.; Zheng, Y.; Gasteiger, G.; Feng, Y.; Fontenot, J.D.; Rudensky, A.Y. An essential role for the IL-2 receptor in Treg cell function. Nat. Immunol. 2016, 17, 1322–1333. [Google Scholar] [CrossRef]
- Killebrew, J.R.; Perdue, N.; Kwan, A.; Thornton, A.M.; Shevach, E.M.; Campbell, D.J. A self-reactive TCR drives the development of Foxp3+ regulatory T cells that prevent autoimmune disease. J. Immunol. 2011, 187, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.K.; Lee, S.C.; Lee, M.J.; Kim, Y.H.; Kim, Y.W.; Ryu, K.W.; Lee, J.H.; Shin, S.M.; Lee, S.H.; Suzuki, S.; et al. 4-1BB-based isolation and expansion of CD8+ T cells specific for self-tumor and non-self-tumor antigens for adoptive T-cell therapy. J. Immunother. 2014, 37, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Pacholczyk, R.; Kern, J. The T-cell receptor repertoire of regulatory T cells. Immunology 2008, 125, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Pittet, M.J.; Valmori, D.; Dunbar, P.R.; Speiser, D.E.; Liénard, D.; Lejeune, F.; Fleischhauer, K.; Cerundolo, V.; Cerottini, J.C.; Romero, P. High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J. Exp. Med. 1999, 190, 705–715. [Google Scholar] [CrossRef]
- Skipper, J.C.; Gulden, P.H.; Hendrickson, R.C.; Harthun, N.; Caldwell, J.A.; Shabanowitz, J.; Engelhard, V.H.; Hunt, D.F.; Slingluff, C.L., Jr. Mass-spectrometric evaluation of HLA-A*0201-associated peptides identifies dominant naturally processed forms of CTL epitopes from MART-1 and gp100. Int. J. Cancer 1999, 82, 669–677. [Google Scholar] [CrossRef]
- Jin, J.; Sabatino, M.; Somerville, R.; Wilson, J.R.; Dudley, M.E.; Stroncek, D.F.; Rosenberg, S.A. Simplified method of the growth of human tumor infiltrating lymphocytes in gas-permeable flasks to numbers needed for patient treatment. J. Immunother. 2012, 35, 283–292. [Google Scholar] [CrossRef]
- Schmidt, A.; Oberle, N.; Krammer, P.H. Molecular mechanisms of treg-mediated T cell suppression. Front. Immunol. 2012, 3, 51. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell. Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Kim, M.M.; Wiederschain, D.; Kennedy, D.; Hansen, E.; Yuan, Z.M. Modulation of p53 and MDM2 activity by novel interaction with Ras-GAP binding proteins (G3BP). Oncogene 2007, 26, 4209–4215. [Google Scholar] [CrossRef]
- Liu, H.; Li, X.; Ning, G.; Zhu, S.; Ma, X.; Liu, X.; Liu, C.; Huang, M.; Schmitt, I.; Wüllner, U.; et al. The Machado-Joseph Disease Deubiquitinase Ataxin-3 Regulates the Stability and Apoptotic Function of p53. PLoS Biol. 2016, 14, e2000733. [Google Scholar] [CrossRef]
- Tsai, R.K.; Lin, K.L.; Huang, C.T.; Wen, Y.T. Transcriptomic Analysis Reveals That Granulocyte Colony-Stimulating Factor Trigger a Novel Signalling Pathway (TAF9-P53-TRIAP1-CASP3) to Protect Retinal Ganglion Cells after Ischemic Optic Neuropathy. Int. J. Mol. Sci. 2022, 23, 8359. [Google Scholar] [CrossRef]
- Madapura, H.S.; Salamon, D.; Wiman, K.G.; Lain, S.; Klein, G.; Klein, E.; Nagy, N. p53 contributes to T cell homeostasis through the induction of pro-apoptotic SAP. Cell Cycle 2012, 11, 4563–4569. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Moon, K.D.; Vacchio, M.S.; Hathcock, K.S.; Hodes, R.J. Downmodulation of tumor suppressor p53 by T cell receptor signalling is critical for antigen-specific CD4(+) T cell responses. Immunity 2014, 40, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Thyagarajan, K.; Chatterjee, S.; Chakraborty, P.; Kesarwani, P.; Soloshchenko, M.; Al-Hommrani, M.; Andrijauskaite, K.; Moxley, K.; Janakiraman, H.; et al. Lack of p53 Augments Antitumor Functions in Cytolytic T Cells. Cancer Res. 2016, 76, 5229–5240. [Google Scholar] [CrossRef] [PubMed]
- Barili, V.; Fisicaro, P.; Montanini, B.; Acerbi, G.; Filippi, A.; Forleo, G.; Romualdi, C.; Ferracin, M.; Guerrieri, F.; Pedrazzi, G.; et al. Targeting p53 and histone methyltransferases restores exhausted CD8+ T cells in HCV infection. Nat. Commun. 2020, 11, 604. [Google Scholar] [CrossRef]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4, a006957. [Google Scholar] [CrossRef]
- Parker, K.C.; Bednarek, M.A.; Hull, L.K.; Utz, U.; Cunningham, B.; Zweerink, H.J.; Biddison, W.E.; Coligan, J.E. Sequence motifs important for peptide binding to the human MHC class I molecule, HLA-A2. J. Immunol. 1992, 149, 3580–3587. [Google Scholar] [CrossRef]
- Sant’Angelo, D.B.; Robinson, E.; Janeway, C.A., Jr.; Denzin, L.K. Recognition of core and flanking amino acids of MHC class II-bound peptides by the T cell receptor. Eur. J. Immunol. 2002, 32, 2510–2520. [Google Scholar] [CrossRef]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef] [PubMed]
- Tynan, F.E.; Reid, H.H.; Kjer-Nielsen, L.; Miles, J.J.; Wilce, M.C.; Kostenko, L.; Borg, N.A.; Williamson, N.A.; Beddoe, T.; Purcell, A.W.; et al. A T cell receptor flattens a bulged antigenic peptide presented by a major histocompatibility complex class I molecule. Nat. Immunol. 2007, 8, 268–276. [Google Scholar] [CrossRef]
- Cole, D.K.; Yuan, F.; Rizkallah, P.J.; Miles, J.J.; Gostick, E.; Price, D.A.; Gao, G.F.; Jakobsen, B.K.; Sewell, A.K. Germ line-governed recognition of a cancer epitope by an immunodominant human T-cell receptor. J. Biol. Chem. 2009, 284, 27281–27289. [Google Scholar] [CrossRef] [PubMed]
- Tanno, H.; Gould, T.M.; McDaniel, J.R.; Cao, W.; Tanno, Y.; Durrett, R.E.; Park, D.; Cate, S.J.; Hildebrand, W.H.; Dekker, C.L.; et al. Determinants governing T cell receptor α/β-chain pairing in repertoire formation of identical twins. Proc. Natl. Acad. Sci. USA 2020, 117, 532–540. [Google Scholar] [CrossRef] [PubMed]
- Verhagen, J.; van der Meijden, E.D.; Lang, V.; Kremer, A.E.; Völkl, S.; Mackensen, A.; Aigner, M.; Kremer, A.N. Human CD4+ T cells specific for dominant epitopes of SARS-CoV-2 Spike and Nucleocapsid proteins with therapeutic potential. Clin. Exp. Immunol. 2021, 205, 363–378. [Google Scholar] [CrossRef] [PubMed]
- Foulds, K.E.; Zenewicz, L.A.; Shedlock, D.J.; Jiang, J.; Troy, A.E.; Shen, H. Cutting edge: CD4 and CD8 T cells are intrinsically different in their proliferative responses. J. Immunol. 2002, 168, 1528–1532. [Google Scholar] [CrossRef]
- Lee, W.; Lee, G.R. Transcriptional regulation and development of regulatory T cells. Exp. Mol. Med. 2018, 50, e456. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Takahashi, T.; Kuniyasu, Y.; Toda, M.; Sakaguchi, N.; Itoh, M.; Iwata, M.; Shimizu, J.; Sakaguchi, S. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int. Immunol. 1998, 10, 1969–1980. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, E.; Han, S.; Eom, H.S.; Lee, S.-J.; Han, C.; Singh, R.; Kim, S.-H.; Park, B.-M.; Kim, B.-G.; Kim, Y.H.; et al. Cross-Activation of Regulatory T Cells by Self Antigens Limits Self-Reactive and Activated CD8+ T Cell Responses. Int. J. Mol. Sci. 2023, 24, 13672. https://doi.org/10.3390/ijms241813672
Cho E, Han S, Eom HS, Lee S-J, Han C, Singh R, Kim S-H, Park B-M, Kim B-G, Kim YH, et al. Cross-Activation of Regulatory T Cells by Self Antigens Limits Self-Reactive and Activated CD8+ T Cell Responses. International Journal of Molecular Sciences. 2023; 24(18):13672. https://doi.org/10.3390/ijms241813672
Chicago/Turabian StyleCho, Eunjung, Seongeun Han, Hyeon Seok Eom, Sang-Jin Lee, Chungyong Han, Rohit Singh, Seon-Hee Kim, Bo-Mi Park, Byoung-Gie Kim, Young H. Kim, and et al. 2023. "Cross-Activation of Regulatory T Cells by Self Antigens Limits Self-Reactive and Activated CD8+ T Cell Responses" International Journal of Molecular Sciences 24, no. 18: 13672. https://doi.org/10.3390/ijms241813672