
Could Cyclosiversioside F Serve as a Dietary Supplement to Prevent Obesity and Relevant Disorders?

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
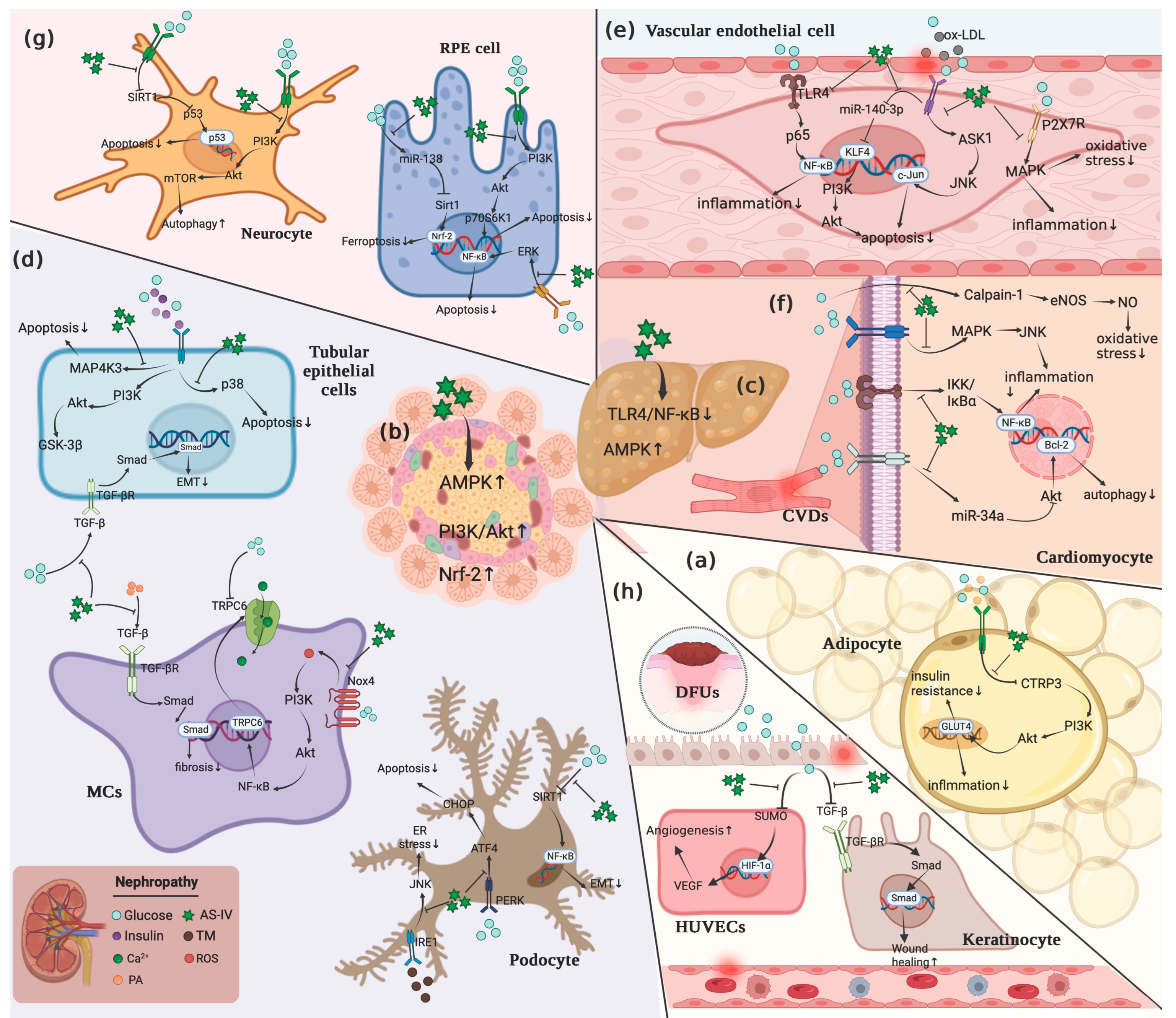
2. Effects of CSF on Signals-Associated with Obesity and Associated Disorders
2.1. PI3K/Akt Pathway
2.2. MAPK Pathway
2.3. NF-κB Pathway
2.4. Apoptotic Pathway
2.5. TGF-β Pathway
2.6. NLRP3
2.7. Nrf-2 Pathway
2.8. AMPK Pathway
2.9. Wnt Pathway
2.10. Integrin/ILK Signal
2.11. eNOS/NO Signal
2.12. Free Radicals
2.13. HIF-1α/VEGF
3. Toxicity of Cyclosiversioside F
4. Conclusions and Perspective
Funding
Conflicts of Interest
Abbreviations
| ALT | alanine aminotransferase |
| AST | Aspartate aminotransferase |
| BUN | blood urea nitrogen |
| CSF | Cyclosiversioside F |
| CTRP3 | C1q tumor necrosis factor-related protein 3 |
| DM | Diabetes Mellitus |
| DN | Diabetic Nephropathy |
| DPN | Diabetic Peripheral Neuropathy |
| DR | Diabetic Retinopathy |
| EMT | epithelial-to-mesenchymal transition |
| EPCs | endothelial progenitor cells |
| ER | endoplasmic reticulum |
| FBG | Fasting Blood Glucose |
| FFA | Free Fatty Acid |
| GA | glycated albumin |
| GBM | glomerular basement membrane |
| GSK-3β | glycogen synthase kinase-3β |
| GTA | glomerular tuft area |
| GTV | glomerular tuft volume |
| HDL | High Density Lipoprotein |
| HFD | High Fat Diet |
| HG | High glucose |
| HGF | hepatocyte growth factor |
| ILK | integrin/integrin-linked kinase |
| IR | Insulin resistance |
| LDL | Low Density Lipoprotein |
| MCV | motor nerve conduction velocity |
| NAFLD | Non-Alcoholic Fatty Liver Disease |
| NAG | N-acetyl-β-d-glucosamine-dase |
| NGAL | neutrophil gelatinase-associated lipocalin |
| NPCs | nucleus pulposus cells |
| PA | Palmitic acid |
| PAN | puromycin aminonucleoside |
| PTP1B | protein-tyrosine phosphatase 1B |
| RAS | renin-angiotensin system |
| Scr | serum creatinine |
| SCV | sensory nerve conduction velocity |
| SOCS3 | Suppressor of cytokine signaling 3 |
| STAT3 | signal transducer and activator of transcription-3 |
| STZ | streptozotocin |
| T2DM | Type 2 Diabetes Mellitus |
| TC | total cholesterol |
| TG | triglyceride |
| UA | uric acid |
References
- Oussaada, S.M.; van Galen, K.A.; Cooiman, M.I.; Kleinendorst, L.; Hazebroek, E.J.; van Haelst, M.M.; Serlie, M.J. The pathogenesis of obesity. Metabolism 2019, 92, 26–36. [Google Scholar] [CrossRef] [PubMed]
- Withrow, D.; Alter, D.A. The economic burden of obesity worldwide: A systematic review of the direct costs of obesity. Obes. Rev. 2011, 12, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Di Cesare, M.; Bentham, J.; Stevens, G.A.; Zhou, B.; Danaei, G.; Lu, Y.; Bixby, H.; Cowan, M.J.; Riley, L.M.; Hajifathalian, K.; et al. Trends in adult body-mass index in 200 countries from 1975 to 2014: A pooled analysis of 1698 population-based measurement studies with 19.2 million participants. Lancet 2016, 387, 1377–1396. [Google Scholar] [CrossRef]
- Goossens, G.H. The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function. Obes. Facts 2017, 10, 207–215. [Google Scholar] [CrossRef]
- Kusminski, C.M.; Bickel, P.E.; Scherer, P.E. Targeting adipose tissue in the treatment of obesity-associated diabetes. Nat. Rev. Drug Discov. 2016, 15, 639–660. [Google Scholar] [CrossRef]
- Li, D.; Zhang, T.; Lu, J.J.; Peng, C.; Lin, L.G. Natural constituents from food sources as therapeutic agents for obesity and metabolic diseases targeting adipose tissue inflammation. Crit. Rev. Food Sci. Nutr. 2021, 61, 1947–1965. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Chakraborty, C.; Doss, C.G.; Bandyopadhyay, S.; Agoramoorthy, G. Influence of miRNA in insulin signaling pathway and insulin resistance: Micro-molecules with a major role in type-2 diabetes. Wiley Interdiscip. Rev. RNA 2014, 5, 697–712. [Google Scholar] [CrossRef]
- He, F.; Huang, Y.; Song, Z.; Zhou, H.J.; Zhang, H.; Perry, R.J.; Shulman, G.I.; Min, W. Mitophagy-mediated adipose inflammation contributes to type 2 diabetes with hepatic insulin resistance. J. Exp. Med. 2021, 218, e20201416. [Google Scholar] [CrossRef]
- Yang, Q.; Vijayakumar, A.; Kahn, B.B. Metabolites as regulators of insulin sensitivity and metabolism. Nat. Rev. Mol. Cell Biol. 2018, 19, 654–672. [Google Scholar] [CrossRef]
- Moore, K.J.; Shah, R. Introduction to the Obesity, Metabolic Syndrome, and CVD Compendium. Circ. Res. 2020, 126, 1475–1476. [Google Scholar] [CrossRef] [PubMed]
- Schulze, M.B. Metabolic health in normal-weight and obese individuals. Diabetologia 2019, 62, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Ruze, R.; Liu, T.; Zou, X.; Song, J.; Chen, Y.; Xu, R.; Xu, Q. Obesity and type 2 diabetes mellitus: Connections in epidemiology, pathogenesis, and treatments. Front. Endocrinol. 2023, 14, 1161521. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Qin, S.; Liu, S.; Zhong, K.; Jing, Y.; Wu, X.; Peng, F.; Li, D.; Peng, C. Targeting matrix metalloproteases in diabetic wound healing. Front. Immunol. 2023, 14, 1089001. [Google Scholar] [CrossRef] [PubMed]
- Elagizi, A.; Kachur, S.; Carbone, S.; Lavie, C.J.; Blair, S.N. A Review of Obesity, Physical Activity, and Cardiovascular Disease. Curr. Obes. Rep. 2020, 9, 571–581. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.E.; do Carmo, J.M.; da Silva, A.A.; Wang, Z.; Hall, M.E. Obesity-induced hypertension: Interaction of neurohumoral and renal mechanisms. Circ. Res. 2015, 116, 991–1006. [Google Scholar] [CrossRef] [PubMed]
- Polyzos, S.A.; Kountouras, J.; Mantzoros, C.S. Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 2019, 92, 82–97. [Google Scholar] [CrossRef]
- Caballero, B. Humans against Obesity: Who Will Win? Adv. Nutr. 2019, 10 (Suppl. S1), S4–S9. [Google Scholar] [CrossRef]
- Bessesen, D.H.; Van Gaal, L.F. Progress and challenges in anti-obesity pharmacotherapy. Lancet Diabetes Endocrinol. 2018, 6, 237–248. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Dwoskin, L.P. Obesity: Current and potential pharmacotherapeutics and targets. Pharmacol. Ther. 2017, 170, 116–147. [Google Scholar] [CrossRef]
- Price, K.R.; Johnson, I.T.; Fenwick, G.R. The chemistry and biological significance of saponins in foods and feeding stuffs. Crit. Rev. Food Sci. Nutr. 1987, 26, 27–135. [Google Scholar] [CrossRef] [PubMed]
- Rios, J.L.; Waterman, P.G. A review of the pharmacology and toxicology of Astragalus. Phytother. Res. 1997, 11, 411–418. [Google Scholar] [CrossRef]
- Zhang, C.-H.; Yang, X.; Wei, J.-R.; Chen, N.-M.; Xu, J.-P.; Bi, Y.-Q.; Yang, M.; Gong, X.; Li, Z.-Y.; Ren, K.; et al. Ethnopharmacology, Phytochemistry, Pharmacology, Toxicology and Clinical Applications of Radix Astragali. Chin. J. Integr. Med. 2021, 27, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Guo, X.; Yuan, B.; Yu, W.; Suo, H.; Li, Z.; Xu, H. Disposition of Astragaloside IV via Enterohepatic Circulation Is Affected by the Activity of the Intestinal Microbiome. J. Agric. Food Chem. 2015, 63, 6084–6093. [Google Scholar] [CrossRef]
- Wei, X.; He, Y.; Wan, H.; Yin, J.; Lin, B.; Ding, Z.; Zhou, H. Integrated transcriptomics, proteomics and metabolomics to identify biomarkers of astragaloside IV against cerebral ischemic injury in rats. Food Funct. 2023, 14, 3588–3599. [Google Scholar] [CrossRef]
- Wang, Z.; Wu, Y.; Pei, C.; Wang, M.; Wang, X.; Shi, S.; Huang, D.; Wang, Y.; Li, S.; Xiao, W.; et al. Astragaloside IV pre-treatment attenuates PM2.5-induced lung injury in rats: Impact on autophagy, apoptosis and inflammation. Phytomedicine 2022, 96, 153912. [Google Scholar] [CrossRef]
- Xia, C.; He, Z.; Cai, Y. Quantitative proteomics analysis of differentially expressed proteins induced by astragaloside IV in cervical cancer cell invasion. Cell. Mol. Biol. Lett. 2020, 25, 25. [Google Scholar] [CrossRef]
- Li, X.; Wang, X.; Han, C.; Wang, X.; Xing, G.; Zhou, L.; Niu, Y. Astragaloside IV suppresses collagen production of activated hepatic stellate cells via oxidative stress-mediated p38 MAPK pathway. Free Radic. Biol. Med. 2013, 60, 168–176. [Google Scholar] [CrossRef]
- Zhou, B.; Zhou, D.L.; Wei, X.H.; Zhong, R.Y.; Xu, J.; Sun, L. Astragaloside IV attenuates free fatty acid-induced ER stress and lipid accumulation in hepatocytes via AMPK activation. Acta Pharmacol. Sin. 2017, 38, 998–1008. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, L.L.; Tang, W.J.; Tang, B. Astragaloside IV inhibits protein tyrosine phosphatase 1B and improves insulin resistance in insulin-resistant HepG2 cells and triglyceride accumulation in oleic acid (OA)-treated HepG2 cells. J. Ethnopharmacol. 2021, 268, 113556. [Google Scholar] [CrossRef]
- Wu, H.; Gao, Y.; Shi, H.-L.; Qin, L.-Y.; Huang, F.; Lan, Y.-Y.; Zhang, B.-B.; Hu, Z.-B.; Wu, X.-J. Astragaloside IV improves lipid metabolism in obese mice by alleviation of leptin resistance and regulation of thermogenic network. Sci. Rep. 2016, 6, 30190. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.F.; Su, Y.; Zhang, J.; Zhang, Y.H.; Li, Y.; Han, Y.L.; Li, W.P. Astragaloside IV alleviates liver injury in type 2 diabetes due to promotion of AMPK/mTOR-mediated autophagy. Mol. Med. Rep. 2021, 23, 437. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.Y.; Hong, F.F.; Yang, S.L. Astragaloside IV Alleviates Liver Inflammation, Oxidative Stress and Apoptosis to Protect Against Experimental Non-Alcoholic Fatty Liver Disease. Diabetes Metab. Syndr. Obes. 2021, 14, 1871–1883. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Liu, Z.; Liu, S.; Lin, N.; Deng, Y. Astragaloside IV alleviates atherosclerosis through targeting circ_0000231/miR-135a-5p/CLIC4 axis in AS cell model in vitro. Mol. Cell. Biochem. 2021, 476, 1783–1795. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhu, Y.; Zhang, Y.; Zhang, J.; Ji, T.; Li, W.; Li, W. Protective effects of AS-IV on diabetic cardiomyopathy by improving myocardial lipid metabolism in rat models of T2DM. Biomed Pharm. 2020, 127, 110081. [Google Scholar] [CrossRef]
- Zhu, Y.; Qian, X.; Li, J.; Lin, X.; Luo, J.; Huang, J.; Jin, Z. Astragaloside-IV protects H9C2(2-1) cardiomyocytes from high glucose-induced injury via miR-34a-mediated autophagy pathway. Artif. Cells Nanomed. Biotechnol. 2019, 47, 4172–4181. [Google Scholar] [CrossRef]
- Feng, H.; Zhu, X.Y.; Tang, Y.; Fu, S.Q.; Kong, B.T.; Liu, X.M. Astragaloside IV ameliorates diabetic nephropathy in db/db mice by inhibiting NLRP3 inflammasome-mediated inflammation. Int. J. Mol. Med. 2021, 48, 4996. [Google Scholar] [CrossRef]
- Lu, W.S.; Li, S.; Guo, W.W.; Chen, L.L.; Li, Y.S. Effects of Astragaloside IV on diabetic nephropathy in rats. Genet. Mol. Res. 2015, 14, 5427–5434. [Google Scholar] [CrossRef]
- Jiang, Z.H.; Wang, G.; Meng, L.L.; Tang, Y.Z.; Yang, M.; Ni, C.L. Protective Effects of Astragaloside IV on Uric Acid-Induced Pancreatic beta-Cell Injury through PI3K/AKT Pathway Activation. Evid.-Based Complement. Altern. Med. 2022, 2022, 2429162. [Google Scholar] [CrossRef]
- Li, M.C.; Yu, L.Z.; She, T.H.; Gan, Y.P.; Liu, F.X.; Hu, Z.W.; Xia, H.L. Astragaloside IV attenuates Toll-like receptor 4 expression via NF-kappa B pathway under high glucose condition in mesenchymal stem cells. Eur. J. Pharmacol. 2012, 696, 203–209. [Google Scholar] [CrossRef]
- Leng, B.; Li, C.; Sun, Y.; Zhao, K.; Zhang, L.; Lu, M.L.; Wang, H.X. Protective Effect of Astragaloside IV on High Glucose-Induced Endothelial Dysfunction via Inhibition of P2X7R Dependent P38 MAPK Signaling Pathway. Oxid Med. Cell Longev. 2020, 2020, 5070415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-N.; Zhao, S.-L.; Su, Y.-Y.; Feng, J.-X.; Wang, S.; Liao, X.-M.; Wang, L.-N.; Li, J.-C.; Meng, P.; Li, H.-Y.; et al. Astragaloside IV attenuates high glucose-induced EMT by inhibiting the TGF-beta/Smad pathway in renal proximal tubular epithelial cells. Biosci. Rep. 2020, 40, BSR20190987. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zhang, X.; Xing, B.; Zhao, J.; Zhang, P.; Shi, D.; Yang, F. Astragaloside IV attenuates gestational diabetes mellitus via targeting NLRP3 inflammasome in genetic mice. Reprod. Biol. Endocrinol. 2019, 17, 77. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Guo, H.M. Astragaloside IV ameliorates high glucose-induced HK-2 cell apoptosis and oxidative stress by regulating the Nrf2/ARE signaling pathway. Exp. Ther. Med. 2019, 17, 4409–4416. [Google Scholar] [CrossRef]
- Zeng, Y.J.; Zhang, B.; Liu, X.H.; He, L.P.; Wang, T.F.; Yu, X.W.; Li, S.M. Original Astragaloside IV alleviates puromycin aminonucleoside-induced podocyte cytoskeleton injury through the Wnt/PCP pathway. Am. J. Transl. Res. 2020, 12, 3512–3521. [Google Scholar]
- Fan, Y.; Fan, H.; Zhu, B.; Zhou, Y.; Liu, Q.; Li, P. Astragaloside IV protects against diabetic nephropathy via activating eNOS in streptozotocin diabetes-induced rats. BMC Complement. Altern. Med. 2019, 19, 355. [Google Scholar] [CrossRef]
- Jiang, P.; Ma, D.; Wang, X.; Wang, Y.; Bi, Y.; Yang, J.; Li, X. Astragaloside IV Prevents Obesity-Associated Hypertension by Improving Pro-Inflammatory Reaction and Leptin Resistance. Mol. Cells 2018, 41, 244–255. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, Y.; Zheng, X.; Zhang, J.; Liu, J.; Wu, G. Astragaloside IV Ameliorates Streptozotocin Induced Pancreatic β-Cell Apoptosis and Dysfunction Through SIRT1/P53 and Akt/GSK3β/Nrf2 Signaling Pathways. Diabetes Metab. Syndr. Obes. 2022, 15, 131–140. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, G.; Huang, B.; Chen, D.; Ye, R. Astragaloside IV Regulates Insulin Resistance and Inflammatory Response of Adipocytes via Modulating CTRP3 and PI3K/AKT Signaling. Diabetes Ther. 2022, 13, 1823–1834. [Google Scholar] [CrossRef]
- Zhu, R.F.; Zheng, J.J.; Chen, L.Z.; Gu, B.; Huang, S.L. Astragaloside IV facilitates glucose transport in C2C12 myotubes through the IRS1/AKT pathway and suppresses the palmitate-induced activation of the IKK/IB pathway. Int. J. Mol. Med. 2016, 37, 1697–1705. [Google Scholar] [CrossRef]
- Gong, P.; Xiao, X.; Wang, S.; Shi, F.; Liu, N.; Chen, X.; Yang, W.; Wang, L.; Chen, F. Hypoglycemic effect of astragaloside IV via modulating gut microbiota and regulating AMPK/SIRT1 and PI3K/AKT pathway. J. Ethnopharmacol. 2021, 281, 114558. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Li, W.P.; Li, W.Z.; Xiong, L.; Li, G.P.; Ma, R. Astragaloside IV prevents damage to human mesangial cells through the inhibition of the NADPH oxidase/ROS/Akt/NF-kappa B pathway under high glucose conditions. Int. J. Mol. Med. 2014, 34, 167–176. [Google Scholar] [CrossRef]
- Zhao, T.; Tian, J.; Xu, T.; Zhang, X.; Xiang, Q.; Xiong, J.; Xu, Y. Astragaloside IV Improves the Barrier Damage in Diabetic Glomerular Endothelial Cells Stimulated by High Glucose and High Insulin. Evid. Based Complement Altern. Med. 2022, 2022, 7647380. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Wang, W.; Han, P.; Shao, M.; Song, G.; Du, H.; Li, S. Astragaloside IV ameliorates renal injury in db/db mice. Sci. Rep. 2016, 6, 32545. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Qian, Q.; Cai, X.; Han, R.; Yang, W.; Zhang, X.; Zhu, R. Astragaloside IV inhibits oxidized low-density lipoprotein-induced endothelial damage via upregulation of miR-140-3p. Int. J. Mol. Med. 2019, 44, 847–856. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, Z.; Song, C.; Sun, L.; Sui, X.; Qu, Q.; Liu, J. Astragaloside IV protects retinal pigment epithelial cells from apoptosis by upregulating miR-128 expression in diabetic rats. Int. J. Mol. Med. 2020, 46, 340–350. [Google Scholar] [CrossRef]
- Zhao, Z.H.; Xu, M.; Fu, C.; Huang, Y.; Wang, T.H.; Zuo, Z.F.; Liu, X.Z. A Mechanistic Exploratory Study on the Therapeutic Efficacy of Astragaloside IV Against Diabetic Retinopathy Revealed by Network Pharmacology. Front. Pharmacol. 2022, 13, 903485. [Google Scholar] [CrossRef]
- Yin, Y.; Qu, H.; Yang, Q.; Fang, Z.; Gao, R. Astragaloside IV alleviates Schwann cell injury in diabetic peripheral neuropathy by regulating microRNA-155-mediated autophagy. Phytomedicine 2021, 92, 153749. [Google Scholar] [CrossRef]
- Zhang, R.X.; Xing, B.H.; Zhao, J.Y.; Zhang, X.L.; Zhou, L.; Yang, S.Y.; Yang, F.Z. Astragaloside IV relieves gestational diabetes mellitus in genetic mice through reducing hepatic gluconeogenesis. Can. J. Physiol. Pharmacol. 2020, 98, 466–472. [Google Scholar] [CrossRef]
- Wang, Q.; Shao, X.H.; Xu, W.J.; Qi, C.J.; Gu, L.; Ni, Z.H.; Mou, S. Astragalosides IV inhibits high glucose-induced cell apoptosis through HGF activation in cultured human tubular epithelial cells. Ren. Fail. 2014, 36, 400–406. [Google Scholar] [CrossRef]
- Song, G.; Han, P.; Sun, H.; Shao, M.; Yu, X.; Wang, W.; Wang, D.; Yi, W.; Ge, N.; Li, S.; et al. Astragaloside IV ameliorates early diabetic nephropathy by inhibition of MEK1/2-ERK1/2-RSK2 signaling in streptozotocin-induced diabetic mice. J. Int. Med. Res. 2018, 46, 2883–2897. [Google Scholar] [CrossRef] [PubMed]
- He, K.Q.; Li, W.Z.; Chai, X.Q.; Yin, Y.Y.; Jiang, Y.; Li, W.P. Astragaloside IV prevents kidney injury caused by iatrogenic hyperinsulinemia in a streptozotocin-induced diabetic rat model. Int. J. Mol. Med. 2018, 41, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Tang, W.; Liu, W.; Hu, Z.; Pan, C. Astragaloside IV Alleviates Renal Tubular Epithelial-Mesenchymal Transition via CX3CL1-RAF/MEK/ERK Signaling Pathway in Diabetic Kidney Disease. Drug Des. Devel. Ther. 2022, 16, 1605–1620. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.S.; Xiong, F.; Xie, X.H.; Chen, D.; Pan, J.H.; Cheng, L. Astragaloside IV attenuates proteinuria in streptozotocin-induced diabetic nephropathy via the inhibition of endoplasmic reticulum stress. BMC Nephrol. 2015, 16, 44. [Google Scholar] [CrossRef]
- Guo, H.; Wang, Y.; Zhang, X.; Zang, Y.; Zhang, Y.; Wang, L.; Wang, H.; Wang, Y.; Cao, A.; Peng, W. Astragaloside IV protects against podocyte injury via SERCA2-dependent ER stress reduction and AMPKalpha-regulated autophagy induction in streptozotocin-induced diabetic nephropathy. Sci. Rep. 2017, 7, 6852. [Google Scholar] [CrossRef]
- Guo, H.J.; Cao, A.L.; Chu, S.; Wang, Y.; Zang, Y.J.; Mao, X.D.; Peng, W. Astragaloside IV Attenuates Podocyte Apoptosis Mediated by Endoplasmic Reticulum Stress through Upregulating Sarco/Endoplasmic Reticulum Ca2+-ATPase 2 Expression in Diabetic Nephropathy. Front. Pharmacol. 2016, 7, 500. [Google Scholar] [CrossRef]
- Fan, Y.Y.; Fan, H.Y.; Li, P.; Liu, Q.S.; Huang, L.X.; Zhou, Y.L. Mitogen-activating protein kinase kinase kinase kinase-3, inhibited by Astragaloside IV through H3 lysine 4 monomethylation, promotes the progression of diabetic nephropathy by inducing apoptosis. Bioengineered 2022, 13, 11517–11529. [Google Scholar] [CrossRef]
- Deng, L.-L. Astragaloside IV as Potential Antioxidant Against Diabetic Ketoacidosis in Juvenile Mice Through Activating JNK/Nrf2 Signaling Pathway. Arch. Med. Res. 2020, 51, 654–663. [Google Scholar] [CrossRef]
- You, L.; Fang, Z.; Shen, G.; Wang, Q.; He, Y.; Ye, S.; Wang, L.; Hu, M.; Lin, Y.; Liu, M.; et al. Astragaloside IV prevents high glucose-induced cell apoptosis and inflammatory reactions through inhibition of the JNK pathway in human umbilical vein endothelial cells. Mol. Med. Rep. 2019, 19, 1603–1612. [Google Scholar] [CrossRef]
- Sun, C.; Zeng, G.; Wang, T.; Ren, H.; An, H.; Lian, C.; Liu, J.; Guo, L.; Li, W. Astragaloside IV Ameliorates Myocardial Infarction Induced Apoptosis and Restores Cardiac Function. Front. Cell. Dev. Biol. 2021, 9, 671255. [Google Scholar] [CrossRef]
- Ding, Y.; Yuan, S.; Liu, X.; Mao, P.; Zhao, C.; Huang, Q.; Zhang, R.; Fang, Y.; Song, Q.; Yuan, D.; et al. Protective effects of astragaloside IV on db/db mice with diabetic retinopathy. PLoS ONE 2014, 9, e112207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Du, M.; Wang, J.; Liu, P. Astragaloside IV Relieves Atherosclerosis and Hepatic Steatosis via MAPK/NF-kappaB Signaling Pathway in LDLR(−/−) Mice. Front. Pharmacol. 2022, 13, 828161. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.L.; Zhang, Q.Z.; Wang, Y.R.; Fu, L.N.; Han, J.S.; Zhang, J.; Wang, B.M. Astragaloside IV Improves High-Fat Diet-Induced Hepatic Steatosis in Nonalcoholic Fatty Liver Disease Rats by Regulating Inflammatory Factors Level via TLR4/NF-kappaB Signaling Pathway. Front. Pharmacol. 2020, 11, 605064. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.K.; Huang, J.H.; Guo, Y.P.; Chen, J.G.; Chen, Y.F.; Xiao, W.Z.; Wang, N.S. Astragaloside IV ameliorates renal injury in streptozotocin-induced diabetic rats through inhibiting NF-kappa B-mediated inflammatory genes expression. Cytokine 2013, 61, 970–977. [Google Scholar] [CrossRef]
- Wang, X.; Gao, Y.; Tian, N.; Zhu, Z.; Wang, T.; Xu, J.; Zhang, N. Astragaloside IV represses high glucose-induced mesangial cells activation by enhancing autophagy via SIRT1 deacetylation of NF-kappaB p65 subunit. Drug Des. Devel. Ther. 2018, 12, 2971–2980. [Google Scholar] [CrossRef]
- Wang, X.L.; Gao, Y.B.; Tian, N.X.; Wang, T.; Shi, Y.M.; Xu, J.Y.; Wu, B.J. Astragaloside IV inhibits glucose-induced epithelial-mesenchymal transition of podocytes through autophagy enhancement via the SIRT-NF-kappa B p65 axis. Sci. Rep. 2019, 9, 323. [Google Scholar] [CrossRef]
- Yin, Y.; Qi, F.; Song, Z.; Zhang, B.; Teng, J. Ferulic acid combined with astragaloside IV protects against vascular endothelial dysfunction in diabetic rats. Biosci. Trends 2014, 8, 217–226. [Google Scholar] [CrossRef]
- Leng, B.; Tang, F.; Lu, M.; Zhang, Z.; Wang, H.; Zhang, Y. Astragaloside IV improves vascular endothelial dysfunction by inhibiting the TLR4/NF-kappaB signaling pathway. Life Sci. 2018, 209, 111–121. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, R.; Yang, S.; Zhang, Y.; Shi, D. Astragaloside IV alleviates placental oxidative stress and inflammation in GDM mice. Endocr. Connect. 2020, 9, 939–945. [Google Scholar] [CrossRef]
- Chen, Y.F.; Gui, D.K.; Chen, J.G.; He, D.Y.; Luo, Y.L.; Wang, N.S. Down-Regulation of PERK-ATF4-CHOP Pathway by Astragaloside IV is Associated with the Inhibition of Endoplasmic Reticulum Stress-Induced Podocyte Apoptosis in Diabetic Rats. Cell. Physiol. Biochem. 2014, 33, 1975–1987. [Google Scholar] [CrossRef]
- Ju, Y.; Su, Y.; Chen, Q.; Ma, K.; Ji, T.; Wang, Z.; Li, W. Protective effects of Astragaloside IV on endoplasmic reticulum stress-induced renal tubular epithelial cells apoptosis in type 2 diabetic nephropathy rats. Biomed Pharm. 2019, 109, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Zang, Y.; Liu, S.; Cao, A.; Shan, X.; Deng, W.; Li, Z.; Wang, H.; Wang, Y.; Wang, L.; Peng, W. Astragaloside IV inhibits palmitic acid-induced apoptosis through regulation of calcium homeostasis in mice podocytes. Mol. Biol. Rep. 2021, 48, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.-M.; Liu, Y.-J.; Wang, Y.-M.; Wang, H.; Zhu, B.-B.; Liang, Y.-P.; Yao, W.-G.; Yu, H.; Wang, N.-S.; Zhang, X.-M.; et al. Astragaloside IV prevents high glucose-induced podocyte apoptosis via downregulation of TRPC6. Mol. Med. Rep. 2016, 13, 5149–5156. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.Q.; Su, Y.; Ju, Y.H.; Ma, K.K.; Li, W.Z.; Li, W.P. Astragalosides IV protected the renal tubular epithelial cells from free fatty acids-induced injury by reducing oxidative stress and apoptosis. BioMedicine 2018, 108, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Gui, D.; Guo, Y.; Wang, F.; Liu, W.; Chen, J.; Chen, Y.; Huang, J.; Wang, N. Astragaloside IV, a novel antioxidant, prevents glucose-induced podocyte apoptosis in vitro and in vivo. PLoS ONE 2012, 7, e39824. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Zhang, L.; Li, Z.; Ren, J. Astragaloside IV/lncRNA-TUG1/TRAF5 signaling pathway participates in podocyte apoptosis of diabetic nephropathy rats. Drug Des. Devel Ther. 2018, 12, 2785–2793. [Google Scholar] [CrossRef]
- Hong, H.; Xiao, J.; Guo, Q.; Du, J.; Jiang, Z.; Lu, S.; Zhang, H.; Zhang, X.; Wang, X. Cycloastragenol and Astragaloside IV activate telomerase and protect nucleus pulposus cells against high glucose-induced senescence and apoptosis. Exp. Ther. Med. 2021, 22, 1326. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, J.; Zhu, Y.; Zhang, H.; Wang, H. Astragaloside IV alleviates myocardial damage induced by type 2 diabetes via improving energy metabolism. Mol. Med. Rep. 2019, 20, 4612–4622. [Google Scholar] [CrossRef]
- Ben, Y.; Hao, J.; Zhang, Z.; Xiong, Y.; Zhang, C.; Chang, Y.; Yang, F.; Li, H.; Zhang, T.; Wang, X.; et al. Astragaloside IV Inhibits Mitochondrial-Dependent Apoptosis of the Dorsal Root Ganglion in Diabetic Peripheral Neuropathy Rats Through Modulation of the SIRT1/p53 Signaling Pathway. Diabetes Metab. Syndr. Obes.-Targets Ther. 2021, 14, 1647–1661. [Google Scholar] [CrossRef]
- Su, Y.; Chen, Q.; Ma, K.; Ju, Y.; Ji, T.; Wang, Z.; Li, W. Astragaloside IV inhibits palmitate-mediated oxidative stress and fibrosis in human glomerular mesangial cells via downregulation of CD36 expression. Pharmacol. Rep. 2019, 71, 319–329. [Google Scholar] [CrossRef]
- Mao, Q.; Chen, C.; Liang, H.; Zhong, S.; Cheng, X.; Li, L. Astragaloside IV inhibits excessive mesangial cell proliferation and renal fibrosis caused by diabetic nephropathy via modulation of the TGF-beta1/Smad/miR-192 signaling pathway. Exp. Ther. Med. 2019, 18, 3053–3061. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Gao, Y.; Tian, N.; Zou, D.; Shi, Y.; Zhang, N. Astragaloside IV improves renal function and fibrosis via inhibition of miR-21-induced podocyte dedifferentiation and mesangial cell activation in diabetic mice. Drug Des. Devel. Ther. 2018, 12, 2431–2442. [Google Scholar] [CrossRef]
- Gao, Q.; Pan, L.; Li, Y.; Zhang, X. Astragaloside IV attenuates high glucose-induced human keratinocytes injury via TGF-beta/Smad signaling pathway. J. Tissue Viability 2022, 31, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Qian, W.; Cai, X.; Qian, Q.; Zhuang, Q.; Yang, W.; Zhang, X.; Zhao, L. Astragaloside IV protects endothelial progenitor cells from the damage of ox-LDL via the LOX-1/NLRP3 inflammasome pathway. Drug Des. Devel. Ther. 2019, 13, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.; Li, X.; Zhang, D.; Han, W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 2022, 13, 8240–8254. [Google Scholar] [CrossRef]
- Zhang, Y.; Yuan, Y.; Zhang, J.; Zhao, Y.; Zhang, Y.; Fu, J. Astragaloside IV supplementation attenuates cognitive impairment by inhibiting neuroinflammation and oxidative stress in type 2 diabetic mice. Front. Aging Neurosci. 2022, 14, 1004557. [Google Scholar] [CrossRef]
- Wang, C.; Li, Y.; Hao, M.; Li, W. Astragaloside IV Inhibits Triglyceride Accumulation in Insulin-Resistant HepG2 Cells via AMPK-Induced SREBP-1c Phosphorylation. Front. Pharmacol. 2018, 9, 345. [Google Scholar] [CrossRef]
- Wang, E.; Wang, L.; Ding, R.; Zhai, M.; Ge, R.; Zhou, P.; Wang, T.; Fang, H.; Wang, J.; Huang, J. Astragaloside IV acts through multi-scale mechanisms to effectively reduce diabetic nephropathy. Pharmacol. Res. 2020, 157, 104831. [Google Scholar] [CrossRef]
- Chen, J.; Chen, Y.; Luo, Y.; Gui, D.; Huang, J.; He, D. Astragaloside IV ameliorates diabetic nephropathy involving protection of podocytes in streptozotocin induced diabetic rats. Eur. J. Pharmacol. 2014, 736, 86–94. [Google Scholar] [CrossRef]
- Zhang, Y.; Mao, X.-D.; Cao, A.-L.; Chu, S.; Li, Z.-J.; Wang, Y.-M.; Peng, W.; Wang, L.; Wang, H. Astragaloside IV prevents endothelial dysfunction by improving oxidative stress in streptozotocin-induced diabetic mouse aortas. Exp. Ther. Med. 2021, 22, 1197. [Google Scholar] [CrossRef]
- Nie, Q.; Zhu, L.; Zhang, L.; Leng, B.; Wang, H. Astragaloside IV protects against hyperglycemia-induced vascular endothelial dysfunction by inhibiting oxidative stress and Calpain-1 activation. Life Sci. 2019, 232, 116662. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.W.; Niu, J.Y.; Qin, Q.J.; Qiao, Z.D.; Gu, Y. Astragaloside IV attenuates glycated albumin-induced epithelial-to-mesenchymal transition by inhibiting oxidative stress in renal proximal tubular cells. Cell Stress Chaperones 2014, 19, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Fan, C.L.; Tang, M.K. Astragaloside IV protects rat retinal capillary endothelial cells against high glucose-induced oxidative injury. Drug Des. Devel Ther. 2017, 11, 3567–3577. [Google Scholar] [CrossRef] [PubMed]
- Han, D. Treatment with astragaloside IV reduced blood glucose, regulated blood lipids, and protected liver function in diabetic rats. J. Int. Med. Res. 2021, 49, 300060519841165. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Huang, P.; Yuan, B.; Liu, T.; Lan, F.; Lu, X.; Dai, L.; Liu, Y.; Yin, H. Astragaloside IV enhances diabetic wound healing involving upregulation of alternatively activated macrophages. Int. Immunopharmacol. 2016, 35, 22–28. [Google Scholar] [CrossRef]
- Wang, B.S.; Ma, X.F.; Zhang, C.Y.; Li, Y.X.; Liu, X.Z.; Hu, C.Q. Astragaloside IV Improves Angiogenesis and Promotes Wound Healing in Diabetic Rats via the Activation of the SUMOylation Pathway. Biomed Environ. Sci. 2021, 34, 124–129. [Google Scholar] [CrossRef]
- Li, L.; Hou, X.; Xu, R.; Liu, C.; Tu, M. Research review on the pharmacological effects of astragaloside IV. Fundam. Clin. Pharm. 2017, 31, 17–36. [Google Scholar] [CrossRef]
- Zhu, J.; Wan, X.; Zhu, Y.; Ma, X.; Zheng, Y.; Zhang, T. Effect of astragaloside IV on the embryo-fetal development of Sprague-Dawley rats and New Zealand White rabbits. J. Appl. Toxicol. 2009, 29, 381–385. [Google Scholar] [CrossRef]
- Wan, X.; Zhu, J.; Zhu, Y.; Ma, X.; Zheng, Y.; Zhang, T.; Zhang, W. Effect of astragaloside IV on the general and peripartum reproductive toxicity in Sprague-Dawley rats. Int. J. Toxicol. 2010, 29, 505–516. [Google Scholar] [CrossRef]
- Chen, J.; Zhong, K.; Jing, Y.; Liu, S.; Qin, S.; Peng, F.; Li, D.; Peng, C. Procyanidin B2: A promising multi-functional food-derived pigment for human diseases. Food Chem. 2023, 420, 136101. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qin, S.; Chen, J.; Zhong, K.; Li, D.; Peng, C. Could Cyclosiversioside F Serve as a Dietary Supplement to Prevent Obesity and Relevant Disorders? Int. J. Mol. Sci. 2023, 24, 13762. https://doi.org/10.3390/ijms241813762
Qin S, Chen J, Zhong K, Li D, Peng C. Could Cyclosiversioside F Serve as a Dietary Supplement to Prevent Obesity and Relevant Disorders? International Journal of Molecular Sciences. 2023; 24(18):13762. https://doi.org/10.3390/ijms241813762
Chicago/Turabian StyleQin, Siqi, Junren Chen, Kexin Zhong, Dan Li, and Cheng Peng. 2023. "Could Cyclosiversioside F Serve as a Dietary Supplement to Prevent Obesity and Relevant Disorders?" International Journal of Molecular Sciences 24, no. 18: 13762. https://doi.org/10.3390/ijms241813762
APA StyleQin, S., Chen, J., Zhong, K., Li, D., & Peng, C. (2023). Could Cyclosiversioside F Serve as a Dietary Supplement to Prevent Obesity and Relevant Disorders? International Journal of Molecular Sciences, 24(18), 13762. https://doi.org/10.3390/ijms241813762