
Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications


Abstract
1. Introduction
2. JAK/STAT Signaling Pathway
2.1. Overview of JAK/STAT Signaling
2.2. JAKs and STATs
2.3. Cytokines Activating the JAK/STAT Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | JAK | STAT | Reference |
|---|---|---|---|
| IL-2 family (IL-2, 4, 7, 9, 15, 21) | JAK1, JAK3 | STAT1, STAT3, STAT5, STAT6 | [27,36,55,56,57] |
| IL-6 family (IL-6, 11, 27, 31, LIF) | JAK1, JAK2, TYK2 | STAT1, STAT3 | [27,36,55,56,58] |
| IL-10 family (IL-10, 19, 20, 22, 24, 26) | JAK1, TYK2 | STAT1, STAT3, STAT5 | [27,36,59,60,61] |
| IL-12 family (IL-12, 23) | JAK2 | STAT3, STAT4 | [36,55,56] |
| IL-17 family (IL-17A-F) | JAK2 | STAT3 | [62,63] |
| β common cytokine family (IL-3, 5, GM-CSF) | JAK2 | STAT3, STAT5 | [27,36,55,56] |
| Hematopoietic growth factors (EPO, G-CSF, TPO) | JAK2 | STAT3, STAT5 | [36,55,56,57] |
| Type1 IFN (α, β) | JAK1, TYK2 | STAT1, STAT2 | [36,37,55,56,58] |
| Type2 IFN (γ) | JAK1, JAK2 | STAT1 | [27,36,37,55,56,57,58] |
2.4. Receptors and Negative Regulators of the JAK/STAT Signaling Pathway
3. Roles of JAK/STAT Signaling in HCC
3.1. Dysregulation of the JAK/STAT Signaling Pathway in Human HCC
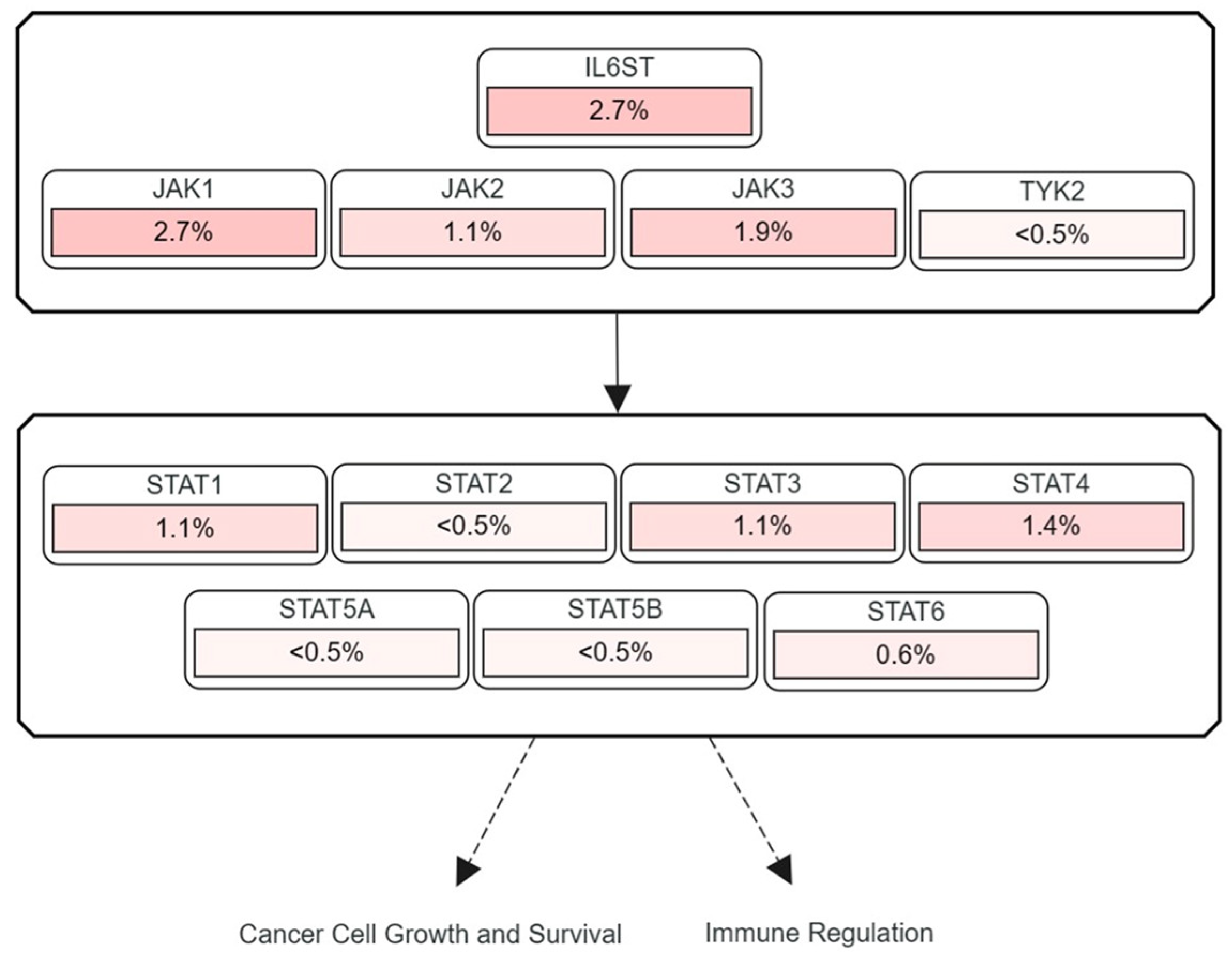
3.2. Frequencies of JAK and STAT Mutations Found in Human HCC
3.3. Activation of the JAK/STAT Signaling via Downregulation of Negative Regulators
3.4. Overproduction of Inflammatory Cytokines Activating JAK/STAT Signaling in HCC
3.4.1. IL-6 Family (IL-6, IL-11, IL-27, LIF)
3.4.2. IL-10 Family (IL-10, IL-19, IL-20, IL-22, IL-24)
3.4.3. IL-23 and IL-17
4. In Vitro and In Vivo Studies Targeting the JAK/STAT Signaling in HCC
4.1. In Vitro Studies Targeting the JAK/STAT Signaling Pathway in HCC Cells
4.2. Preclinical Animal Studies Targeting the JAK/STAT Signaling Pathway in HCC
5. Clinical Studies Targeting the JAK/STAT Signaling Pathway in HCC
6. Perspectives and Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular carcinoma. Nat. Rev. Dis. Primers 2021, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.G.; Lampertico, P.; Nahon, P. Epidemiology and surveillance for hepatocellular carcinoma: New trends. J. Hepatol. 2020, 72, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, A. Hepatocellular Carcinoma. N. Engl. J. Med. 2019, 380, 1450–1462. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Llovet, J.M.; Montal, R.; Sia, D.; Finn, R.S. Molecular therapies and precision medicine for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2018, 15, 599–616. [Google Scholar] [CrossRef]
- Moon, H.; Ro, S.W. MAPK/ERK Signaling Pathway in Hepatocellular Carcinoma. Cancers 2021, 13, 3026. [Google Scholar] [CrossRef]
- Huang, A.; Yang, X.R.; Chung, W.Y.; Dennison, A.R.; Zhou, J. Targeted therapy for hepatocellular carcinoma. Signal Transduct. Target. Ther. 2020, 5, 146. [Google Scholar] [CrossRef]
- Gallage, S.; García-Beccaria, M.; Szydlowska, M.; Rahbari, M.; Mohr, R.; Tacke, F.; Heikenwalder, M. The therapeutic landscape of hepatocellular carcinoma. Med 2021, 2, 505–552. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, H.; Zhang, L.; Zhu, A.X.; Bernards, R.; Qin, W.; Wang, C. Evolving therapeutic landscape of advanced hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 203–222. [Google Scholar] [CrossRef]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef]
- Capece, D.; Fischietti, M.; Verzella, D.; Gaggiano, A.; Cicciarelli, G.; Tessitore, A.; Zazzeroni, F.; Alesse, E. The inflammatory microenvironment in hepatocellular carcinoma: A pivotal role for tumor-associated macrophages. Biomed. Res. Int. 2013, 2013, 187204. [Google Scholar] [CrossRef]
- Moon, H.; Cho, K.; Shin, S.; Kim, D.Y.; Han, K.H.; Ro, S.W. High Risk of Hepatocellular Carcinoma Development in Fibrotic Liver: Role of the Hippo-YAP/TAZ Signaling Pathway. Int. J. Mol. Sci. 2019, 20, 581. [Google Scholar] [CrossRef] [PubMed]
- Rah, B.; Rather, R.A.; Bhat, G.R.; Baba, A.B.; Mushtaq, I.; Farooq, M.; Yousuf, T.; Dar, S.B.; Parveen, S.; Hassan, R.; et al. JAK/STAT Signaling: Molecular Targets, Therapeutic Opportunities, and Limitations of Targeted Inhibitions in Solid Malignancies. Front. Pharmacol. 2022, 13, 821344. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Signal Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef]
- Dodington, D.W.; Desai, H.R.; Woo, M. JAK/STAT - Emerging Players in Metabolism. Trends Endocrinol. Metab. 2018, 29, 55–65. [Google Scholar] [CrossRef]
- Gu, Y.J.; Sun, W.Y.; Zhang, S.; Li, X.R.; Wei, W. Targeted blockade of JAK/STAT3 signaling inhibits proliferation, migration and collagen production as well as inducing the apoptosis of hepatic stellate cells. Int. J. Mol. Med. 2016, 38, 903–911. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Plenge, R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity 2012, 36, 542–550. [Google Scholar] [CrossRef]
- O’Shea, J.J.; Holland, S.M.; Staudt, L.M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 2013, 368, 161–170. [Google Scholar] [CrossRef]
- Akada, H.; Akada, S.; Hutchison, R.E.; Sakamoto, K.; Wagner, K.U.; Mohi, G. Critical role of Jak2 in the maintenance and function of adult hematopoietic stem cells. Stem Cells 2014, 32, 1878–1889. [Google Scholar] [CrossRef]
- Fasouli, E.S.; Katsantoni, E. JAK-STAT in Early Hematopoiesis and Leukemia. Front. Cell Dev. Biol. 2021, 9, 669363. [Google Scholar] [CrossRef] [PubMed]
- Staerk, J.; Constantinescu, S.N. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. Jakstat 2012, 1, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Hin Tang, J.J.; Hao Thng, D.K.; Lim, J.J.; Toh, T.B. JAK/STAT signaling in hepatocellular carcinoma. Hepat. Oncol. 2020, 7, Hep18. [Google Scholar] [CrossRef] [PubMed]
- Svinka, J.; Mikulits, W.; Eferl, R. STAT3 in hepatocellular carcinoma: New perspectives. Hepat. Oncol. 2014, 1, 107–120. [Google Scholar] [CrossRef]
- Hu, Q.; Bian, Q.; Rong, D.; Wang, L.; Song, J.; Huang, H.S.; Zeng, J.; Mei, J.; Wang, P.Y. JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens. Front. Bioeng. Biotechnol. 2023, 11, 1110765. [Google Scholar] [CrossRef]
- Jin, W. Role of JAK/STAT3 Signaling in the Regulation of Metastasis, the Transition of Cancer Stem Cells, and Chemoresistance of Cancer by Epithelial-Mesenchymal Transition. Cells 2020, 9, 217. [Google Scholar] [CrossRef]
- Zhou, Q.; Ren, Q.; Jiao, L.; Huang, J.; Yi, J.; Chen, J.; Lai, J.; Ji, G.; Zheng, T. The potential roles of JAK/STAT signaling in the progression of osteoarthritis. Front. Endocrinol. 2022, 13, 1069057. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef]
- Seif, F.; Khoshmirsafa, M.; Aazami, H.; Mohsenzadegan, M.; Sedighi, G.; Bahar, M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun. Signal. 2017, 15, 23. [Google Scholar] [CrossRef]
- Ferrao, R.; Lupardus, P.J. The Janus Kinase (JAK) FERM and SH2 Domains: Bringing Specificity to JAK-Receptor Interactions. Front. Endocrinol. 2017, 8, 71. [Google Scholar] [CrossRef]
- Jatiani, S.S.; Baker, S.J.; Silverman, L.R.; Reddy, E.P. Jak/STAT pathways in cytokine signaling and myeloproliferative disorders: Approaches for targeted therapies. Genes Cancer 2010, 1, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Bhattacharya, S.; Braunstein, J.; Schindler, C.W. Signaling through the JAK/STAT pathway, recent advances and future challenges. Gene 2002, 285, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Xin, P.; Xu, X.; Deng, C.; Liu, S.; Wang, Y.; Zhou, X.; Ma, H.; Wei, D.; Sun, S. The role of JAK/STAT signaling pathway and its inhibitors in diseases. Int. Immunopharmacol. 2020, 80, 106210. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, N.; Liongue, C.; Ward, A.C. STAT proteins: A kaleidoscope of canonical and non-canonical functions in immunity and cancer. J. Hematol. Oncol. 2021, 14, 198. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Kiu, H.; Nicholson, S.E. Biology and significance of the JAK/STAT signalling pathways. Growth Factors 2012, 30, 88–106. [Google Scholar] [CrossRef]
- Morris, R.; Kershaw, N.J.; Babon, J.J. The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci. 2018, 27, 1984–2009. [Google Scholar] [CrossRef]
- Verhoeven, Y.; Tilborghs, S.; Jacobs, J.; De Waele, J.; Quatannens, D.; Deben, C.; Prenen, H.; Pauwels, P.; Trinh, X.B.; Wouters, A.; et al. The potential and controversy of targeting STAT family members in cancer. Semin. Cancer Biol. 2020, 60, 41–56. [Google Scholar] [CrossRef]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Rodríguez-Ruiz, M.E.; Ponz-Sarvise, M.; Castañón, E.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Kupsa, T.; Horacek, J.M.; Jebavy, L. The role of cytokines in acute myeloid leukemia: A systematic review. Biomed. Pap. Med. Fac. Univ. Palacky Olomouc Czech. Repub. 2012, 156, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Alexander, M.; Gadina, M.; O’Shea, J.J.; Meylan, F.; Schwartz, D.M. JAK-STAT signaling in human disease: From genetic syndromes to clinical inhibition. J. Allergy Clin. Immunol. 2021, 148, 911–925. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Leonard, W.J. Biology of IL-2 and its therapeutic modulation: Mechanisms and strategies. J. Leukoc. Biol. 2018, 103, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 2011, 23, 598–604. [Google Scholar] [CrossRef]
- Jones, D.M.; Read, K.A.; Oestreich, K.J. Dynamic Roles for IL-2-STAT5 Signaling in Effector and Regulatory CD4(+) T Cell Populations. J. Immunol. 2020, 205, 1721–1730. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, L.; Hao, S.; Liu, Z.; Ding, S.; Zhang, W.; Yang, X.; Li, S. Activation of the IL-4/STAT6 Signaling Pathway Promotes Lung Cancer Progression by Increasing M2 Myeloid Cells. Front. Immunol. 2019, 10, 2638. [Google Scholar] [CrossRef]
- Unver, N.; McAllister, F. IL-6 family cytokines: Key inflammatory mediators as biomarkers and potential therapeutic targets. Cytokine Growth Factor Rev. 2018, 41, 10–17. [Google Scholar] [CrossRef]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Lokau, J.; Schoeder, V.; Haybaeck, J.; Garbers, C. Jak-Stat Signaling Induced by Interleukin-6 Family Cytokines in Hepatocellular Carcinoma. Cancers 2019, 11, 1704. [Google Scholar] [CrossRef]
- Jones, S.A.; Scheller, J.; Rose-John, S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J. Clin. Investig. 2011, 121, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lin, H.; Wu, G.; Zhu, M.; Li, M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front. Oncol. 2021, 11, 760971. [Google Scholar] [CrossRef] [PubMed]
- Kourko, O.; Seaver, K.; Odoardi, N.; Basta, S.; Gee, K. IL-27, IL-30, and IL-35: A Cytokine Triumvirate in Cancer. Front. Oncol. 2019, 9, 969. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.W.; Hill, D.G.; Cardus, A.; Jones, S.A. IL-27: A double agent in the IL-6 family. Clin. Exp. Immunol. 2018, 193, 37–46. [Google Scholar] [CrossRef] [PubMed]
- O’Shea, J.J.; Schwartz, D.M.; Villarino, A.V.; Gadina, M.; McInnes, I.B.; Laurence, A. The JAK-STAT pathway: Impact on human disease and therapeutic intervention. Annu. Rev. Med. 2015, 66, 311–328. [Google Scholar] [CrossRef]
- Banerjee, S.; Biehl, A.; Gadina, M.; Hasni, S.; Schwartz, D.M. JAK-STAT Signaling as a Target for Inflammatory and Autoimmune Diseases: Current and Future Prospects. Drugs 2017, 77, 521–546. [Google Scholar] [CrossRef]
- Aittomäki, S.; Pesu, M. Therapeutic targeting of the Jak/STAT pathway. Basic Clin. Pharmacol. Toxicol. 2014, 114, 18–23. [Google Scholar] [CrossRef]
- Aaronson, D.S.; Horvath, C.M. A road map for those who don’t know JAK-STAT. Science 2002, 296, 1653–1655. [Google Scholar] [CrossRef]
- Wang, X.; Xin, W.; Zhang, H.; Zhang, F.; Gao, M.; Yuan, L.; Xu, X.; Hu, X.; Zhao, M. Aberrant expression of p-STAT3 in peripheral blood CD4+ and CD8+ T cells related to hepatocellular carcinoma development. Mol. Med. Rep. 2014, 10, 2649–2656. [Google Scholar] [CrossRef]
- Wang, C.J.; Xiao, C.W.; You, T.G.; Zheng, Y.X.; Gao, W.; Zhou, Z.Q.; Chen, J.; Xue, X.B.; Fan, J.; Zhang, H. Interferon-α enhances antitumor activities of oncolytic adenovirus-mediated IL-24 expression in hepatocellular carcinoma. Mol. Cancer 2012, 11, 31. [Google Scholar] [CrossRef]
- Andoh, A.; Shioya, M.; Nishida, A.; Bamba, S.; Tsujikawa, T.; Kim-Mitsuyama, S.; Fujiyama, Y. Expression of IL-24, an activator of the JAK1/STAT3/SOCS3 cascade, is enhanced in inflammatory bowel disease. J. Immunol. 2009, 183, 687–695. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.M.; Li, Q.L.; Gao, Q.; Jiang, J.H.; Zhu, K.; Huang, X.Y.; Pan, J.F.; Yan, J.; Hu, J.H.; Wang, Z.; et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol. Cancer 2011, 10, 150. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, M.; Yan, K.; Yang, Y.; Li, H.; Xu, X. IL-17 promotes hepatocellular carcinoma through inhibiting apoptosis induced by IFN-γ. Biochem. Biophys. Res. Commun. 2020, 522, 525–531. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, L.A.; Liongue, C.; Lewis, R.S.; Stephenson, S.E.; Ward, A.C. Cytokine receptor signaling through the Jak-Stat-Socs pathway in disease. Mol. Immunol. 2007, 44, 2497–2506. [Google Scholar] [CrossRef]
- Bedke, T.; Muscate, F.; Soukou, S.; Gagliani, N.; Huber, S. Title: IL-10-producing T cells and their dual functions. Semin. Immunol. 2019, 44, 101335. [Google Scholar] [CrossRef]
- Wilbers, R.H.P.; van Raaij, D.R.; Westerhof, L.B.; Bakker, J.; Smant, G.; Schots, A. Re-evaluation of IL-10 signaling reveals novel insights on the contribution of the intracellular domain of the IL-10R2 chain. PLoS ONE 2017, 12, e0186317. [Google Scholar] [CrossRef]
- Hutchins, A.P.; Diez, D.; Miranda-Saavedra, D. The IL-10/STAT3-mediated anti-inflammatory response: Recent developments and future challenges. Brief Funct. Genom. 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Li, L.; Zhang, J.; Chen, J.; Xu-Monette, Z.Y.; Miao, Y.; Xiao, M.; Young, K.H.; Wang, S.; Medeiros, L.J.; Wang, M.; et al. B-cell receptor-mediated NFATc1 activation induces IL-10/STAT3/PD-L1 signaling in diffuse large B-cell lymphoma. Blood 2018, 132, 1805–1817. [Google Scholar] [CrossRef]
- von Essen, M.R.; Søndergaard, H.B.; Petersen, E.R.S.; Sellebjerg, F. IL-6, IL-12, and IL-23 STAT-Pathway Genetic Risk and Responsiveness of Lymphocytes in Patients with Multiple Sclerosis. Cells 2019, 8, 285. [Google Scholar] [CrossRef]
- Floss, D.M.; Moll, J.M.; Scheller, J. IL-12 and IL-23-Close Relatives with Structural Homologies but Distinct Immunological Functions. Cells 2020, 9, 2184. [Google Scholar] [CrossRef]
- Dougan, M.; Dranoff, G.; Dougan, S.K. GM-CSF, IL-3, and IL-5 Family of Cytokines: Regulators of Inflammation. Immunity 2019, 50, 796–811. [Google Scholar] [CrossRef] [PubMed]
- Hercus, T.R.; Kan, W.L.T.; Broughton, S.E.; Tvorogov, D.; Ramshaw, H.S.; Sandow, J.J.; Nero, T.L.; Dhagat, U.; Thompson, E.J.; Shing, K.; et al. Role of the β Common (βc) Family of Cytokines in Health and Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a028514. [Google Scholar] [CrossRef] [PubMed]
- Thorn, M.; Guha, P.; Cunetta, M.; Espat, N.J.; Miller, G.; Junghans, R.P.; Katz, S.C. Tumor-associated GM-CSF overexpression induces immunoinhibitory molecules via STAT3 in myeloid-suppressor cells infiltrating liver metastases. Cancer Gene Ther. 2016, 23, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Liu, J.; Yuan, C.; Ji, Q.; Chen, D.; Zhao, H.; Jiang, W.; Ma, K.; Liu, L. JAK2/STAT3 is associated with the inflammatory process in periapical granuloma. Int. J. Clin. Exp. Pathol. 2019, 12, 190–197. [Google Scholar]
- Reusswig, F.; Fazel Modares, N.; Brechtenkamp, M.; Wienands, L.; Krüger, I.; Behnke, K.; Lee-Sundlov, M.M.; Herebian, D.; Scheller, J.; Hoffmeister, K.M.; et al. Efficiently Restored Thrombopoietin Production by Ashwell-Morell Receptor and IL-6R Induced Janus Kinase 2/Signal Transducer and Activator of Transcription Signaling Early After Partial Hepatectomy. Hepatology 2021, 74, 411–427. [Google Scholar] [CrossRef] [PubMed]
- Mu, W.; Ao, J.; Li, Y.; Zhang, J.; Duan, C. Exploring the protective mechanisms of total tannins from Geum japonicum var. chinense F.Bolle in mice with hematopoietic dysfunction via the JAK2/STAT3/5 signaling pathway. J. Ethnopharmacol. 2022, 296, 115507. [Google Scholar] [CrossRef]
- He, K.; Liu, X.; Hoffman, R.D.; Shi, R.Z.; Lv, G.Y.; Gao, J.L. G-CSF/GM-CSF-induced hematopoietic dysregulation in the progression of solid tumors. FEBS Open Bio 2022, 12, 1268–1285. [Google Scholar] [CrossRef]
- Karagiannidis, I.; Salataj, E.; Said Abu Egal, E.; Beswick, E.J. G-CSF in tumors: Aggressiveness, tumor microenvironment and immune cell regulation. Cytokine 2021, 142, 155479. [Google Scholar] [CrossRef]
- Tóthová, Z.; Tomc, J.; Debeljak, N.; Solár, P. STAT5 as a Key Protein of Erythropoietin Signalization. Int. J. Mol. Sci. 2021, 22, 7109. [Google Scholar] [CrossRef]
- Gao, B.; Wang, H.; Lafdil, F.; Feng, D. STAT proteins—Key regulators of anti-viral responses, inflammation, and tumorigenesis in the liver. J. Hepatol. 2012, 57, 430–441. [Google Scholar] [CrossRef]
- Jorgovanovic, D.; Song, M.; Wang, L.; Zhang, Y. Roles of IFN-γ in tumor progression and regression: A review. Biomark Res. 2020, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Bousoik, E.; Montazeri Aliabadi, H. "Do We Know Jack" About JAK? A Closer Look at JAK/STAT Signaling Pathway. Front. Oncol. 2018, 8, 287. [Google Scholar] [CrossRef] [PubMed]
- Broughton, S.E.; Dhagat, U.; Hercus, T.R.; Nero, T.L.; Grimbaldeston, M.A.; Bonder, C.S.; Lopez, A.F.; Parker, M.W. The GM-CSF/IL-3/IL-5 cytokine receptor family: From ligand recognition to initiation of signaling. Immunol. Rev. 2012, 250, 277–302. [Google Scholar] [CrossRef]
- Durham, G.A.; Williams, J.J.L.; Nasim, M.T.; Palmer, T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol. Sci. 2019, 40, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Tamiya, T.; Kashiwagi, I.; Takahashi, R.; Yasukawa, H.; Yoshimura, A. Suppressors of cytokine signaling (SOCS) proteins and JAK/STAT pathways: Regulation of T-cell inflammation by SOCS1 and SOCS3. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Ungureanu, D.; Vanhatupa, S.; Grönholm, J.; Palvimo, J.J.; Silvennoinen, O. SUMO-1 conjugation selectively modulates STAT1-mediated gene responses. Blood 2005, 106, 224–226. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Qu, C.K. Protein tyrosine phosphatases in the JAK/STAT pathway. Front. Biosci. 2008, 13, 4925–4932. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Conner, E.A.; Lee, J.S.; Factor, V.M.; Thorgeirsson, S.S. Ubiquitous activation of Ras and Jak/Stat pathways in human HCC. Gastroenterology 2006, 130, 1117–1128. [Google Scholar] [CrossRef]
- Dawson, M.A.; Bannister, A.J.; Göttgens, B.; Foster, S.D.; Bartke, T.; Green, A.R.; Kouzarides, T. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009, 461, 819–822. [Google Scholar] [CrossRef]
- Thomas, S.J.; Snowden, J.A.; Zeidler, M.P.; Danson, S.J. The role of JAK/STAT signalling in the pathogenesis, prognosis and treatment of solid tumours. Br. J. Cancer 2015, 113, 365–371. [Google Scholar] [CrossRef]
- Wang, X.; Liao, X.; Yu, T.; Gong, Y.; Zhang, L.; Huang, J.; Yang, C.; Han, C.; Yu, L.; Zhu, G.; et al. Analysis of clinical significance and prospective molecular mechanism of main elements of the JAK/STAT pathway in hepatocellular carcinoma. Int. J. Oncol. 2019, 55, 805–822. [Google Scholar] [CrossRef] [PubMed]
- Kusaba, M.; Nakao, K.; Goto, T.; Nishimura, D.; Kawashimo, H.; Shibata, H.; Motoyoshi, Y.; Taura, N.; Ichikawa, T.; Hamasaki, K.; et al. Abrogation of constitutive STAT3 activity sensitizes human hepatoma cells to TRAIL-mediated apoptosis. J. Hepatol. 2007, 47, 546–555. [Google Scholar] [CrossRef] [PubMed]
- Alas, S.; Bonavida, B. Rituximab inactivates signal transducer and activation of transcription 3 (STAT3) activity in B-non-Hodgkin’s lymphoma through inhibition of the interleukin 10 autocrine/paracrine loop and results in down-regulation of Bcl-2 and sensitization to cytotoxic drugs. Cancer Res. 2001, 61, 5137–5144. [Google Scholar] [PubMed]
- Carbajo-Pescador, S.; Ordoñez, R.; Benet, M.; Jover, R.; García-Palomo, A.; Mauriz, J.L.; González-Gallego, J. Inhibition of VEGF expression through blockade of Hif1α and STAT3 signalling mediates the anti-angiogenic effect of melatonin in HepG2 liver cancer cells. Br. J. Cancer 2013, 109, 83–91. [Google Scholar] [CrossRef]
- Won, C.; Kim, B.H.; Yi, E.H.; Choi, K.J.; Kim, E.K.; Jeong, J.M.; Lee, J.H.; Jang, J.J.; Yoon, J.H.; Jeong, W.I.; et al. Signal transducer and activator of transcription 3-mediated CD133 up-regulation contributes to promotion of hepatocellular carcinoma. Hepatology 2015, 62, 1160–1173. [Google Scholar] [CrossRef]
- Lee, T.K.; Castilho, A.; Cheung, V.C.; Tang, K.H.; Ma, S.; Ng, I.O. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell 2011, 9, 50–63. [Google Scholar] [CrossRef]
- Sun, X.; Sui, Q.; Zhang, C.; Tian, Z.; Zhang, J. Targeting blockage of STAT3 in hepatocellular carcinoma cells augments NK cell functions via reverse hepatocellular carcinoma-induced immune suppression. Mol. Cancer Ther. 2013, 12, 2885–2896. [Google Scholar] [CrossRef]
- Zhang, C.H.; Guo, F.L.; Xu, G.L.; Jia, W.D.; Ge, Y.S. STAT3 activation mediates epithelial-to-mesenchymal transition in human hepatocellular carcinoma cells. Hepatogastroenterology 2014, 61, 1082–1089. [Google Scholar]
- Huang, L.; Jian, Z.; Gao, Y.; Zhou, P.; Zhang, G.; Jiang, B.; Lv, Y. RPN2 promotes metastasis of hepatocellular carcinoma cell and inhibits autophagy via STAT3 and NF-κB pathways. Aging 2019, 11, 6674–6690. [Google Scholar] [CrossRef]
- Lee, T.K.; Man, K.; Poon, R.T.; Lo, C.M.; Yuen, A.P.; Ng, I.O.; Ng, K.T.; Leonard, W.; Fan, S.T. Signal transducers and activators of transcription 5b activation enhances hepatocellular carcinoma aggressiveness through induction of epithelial-mesenchymal transition. Cancer Res. 2006, 66, 9948–9956. [Google Scholar] [CrossRef]
- Kan, Z.; Zheng, H.; Liu, X.; Li, S.; Barber, T.D.; Gong, Z.; Gao, H.; Hao, K.; Willard, M.D.; Xu, J.; et al. Whole-genome sequencing identifies recurrent mutations in hepatocellular carcinoma. Genome Res. 2013, 23, 1422–1433. [Google Scholar] [CrossRef] [PubMed]
- Rebouissou, S.; Amessou, M.; Couchy, G.; Poussin, K.; Imbeaud, S.; Pilati, C.; Izard, T.; Balabaud, C.; Bioulac-Sage, P.; Zucman-Rossi, J. Frequent in-frame somatic deletions activate gp130 in inflammatory hepatocellular tumours. Nature 2009, 457, 200–204. [Google Scholar] [CrossRef]
- Xie, H.J.; Bae, H.J.; Noh, J.H.; Eun, J.W.; Kim, J.K.; Jung, K.H.; Ryu, J.C.; Ahn, Y.M.; Kim, S.Y.; Lee, S.H.; et al. Mutational analysis of JAK1 gene in human hepatocellular carcinoma. Neoplasma 2009, 56, 136–140. [Google Scholar] [CrossRef]
- Yang, S.; Luo, C.; Gu, Q.; Xu, Q.; Wang, G.; Sun, H.; Qian, Z.; Tan, Y.; Qin, Y.; Shen, Y.; et al. Activating JAK1 mutation may predict the sensitivity of JAK-STAT inhibition in hepatocellular carcinoma. Oncotarget 2016, 7, 5461–5469. [Google Scholar] [CrossRef] [PubMed]
- Pilati, C.; Zucman-Rossi, J. Mutations leading to constitutive active gp130/JAK1/STAT3 pathway. Cytokine Growth Factor Rev. 2015, 26, 499–506. [Google Scholar] [CrossRef]
- Pilati, C.; Amessou, M.; Bihl, M.P.; Balabaud, C.; Nhieu, J.T.; Paradis, V.; Nault, J.C.; Izard, T.; Bioulac-Sage, P.; Couchy, G.; et al. Somatic mutations activating STAT3 in human inflammatory hepatocellular adenomas. J. Exp. Med. 2011, 208, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-κB and STAT3 - key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef]
- Yoshikawa, H.; Matsubara, K.; Qian, G.S.; Jackson, P.; Groopman, J.D.; Manning, J.E.; Harris, C.C.; Herman, J.G. SOCS-1, a negative regulator of the JAK/STAT pathway, is silenced by methylation in human hepatocellular carcinoma and shows growth-suppression activity. Nat. Genet. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, J.; Cheng, J.; Ding, S.; Li, M.; Sun, S.; Zhang, L.; Liu, S.; Chen, X.; Zhuang, H.; et al. An integrated analysis of SOCS1 down-regulation in HBV infection-related hepatocellular carcinoma. J. Viral Hepat. 2014, 21, 264–271. [Google Scholar] [CrossRef]
- Calvisi, D.F.; Ladu, S.; Gorden, A.; Farina, M.; Lee, J.S.; Conner, E.A.; Schroeder, I.; Factor, V.M.; Thorgeirsson, S.S. Mechanistic and prognostic significance of aberrant methylation in the molecular pathogenesis of human hepatocellular carcinoma. J. Clin. Investig. 2007, 117, 2713–2722. [Google Scholar] [CrossRef]
- Calvisi, D.F. Dr. Jekyll and Mr. Hyde: A paradoxical oncogenic and tumor suppressive role of signal transducer and activator of transcription 3 in liver cancer. Hepatology 2011, 54, 9–12. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.I.; Moon, H.; Ju, H.L.; Cho, K.J.; Kim, D.Y.; Han, K.H.; Eun, J.W.; Nam, S.W.; Ribback, S.; Dombrowski, F.; et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J. Hepatol. 2016, 64, 618–627. [Google Scholar] [CrossRef]
- Refolo, M.G.; Messa, C.; Guerra, V.; Carr, B.I.; D’Alessandro, R. Inflammatory Mechanisms of HCC Development. Cancers 2020, 12, 641. [Google Scholar] [CrossRef]
- Zhang, W.; Liu, Y.; Yan, Z.; Yang, H.; Sun, W.; Yao, Y.; Chen, Y.; Jiang, R. IL-6 promotes PD-L1 expression in monocytes and macrophages by decreasing protein tyrosine phosphatase receptor type O expression in human hepatocellular carcinoma. J. Immunother. Cancer 2020, 8, e000285. [Google Scholar] [CrossRef]
- Lin, Y.; He, Z.; Ye, J.; Liu, Z.; She, X.; Gao, X.; Liang, R. Progress in Understanding the IL-6/STAT3 Pathway in Colorectal Cancer. Onco Targets Ther. 2020, 13, 13023–13032. [Google Scholar] [CrossRef] [PubMed]
- Manore, S.G.; Doheny, D.L.; Wong, G.L.; Lo, H.W. IL-6/JAK/STAT3 Signaling in Breast Cancer Metastasis: Biology and Treatment. Front. Oncol. 2022, 12, 866014. [Google Scholar] [CrossRef]
- Xiong, S.; Wang, R.; Chen, Q.; Luo, J.; Wang, J.; Zhao, Z.; Li, Y.; Wang, Y.; Wang, X.; Cheng, B. Cancer-associated fibroblasts promote stem cell-like properties of hepatocellular carcinoma cells through IL-6/STAT3/Notch signaling. Am. J. Cancer Res. 2018, 8, 302–316. [Google Scholar]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef]
- Aleksandrova, K.; Boeing, H.; Nöthlings, U.; Jenab, M.; Fedirko, V.; Kaaks, R.; Lukanova, A.; Trichopoulou, A.; Trichopoulos, D.; Boffetta, P.; et al. Inflammatory and metabolic biomarkers and risk of liver and biliary tract cancer. Hepatology 2014, 60, 858–871. [Google Scholar] [CrossRef] [PubMed]
- Soresi, M.; Giannitrapani, L.; D’Antona, F.; Florena, A.M.; La Spada, E.; Terranova, A.; Cervello, M.; D’Alessandro, N.; Montalto, G. Interleukin-6 and its soluble receptor in patients with liver cirrhosis and hepatocellular carcinoma. World J. Gastroenterol. 2006, 12, 2563–2568. [Google Scholar] [CrossRef]
- Yu, S.; Chen, J.; Quan, M.; Li, L.; Li, Y.; Gao, Y. CD63 negatively regulates hepatocellular carcinoma development through suppression of inflammatory cytokine-induced STAT3 activation. J. Cell. Mol. Med. 2021, 25, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.F.; He, M.L.; Fu, W.M.; Wang, H.; Chen, L.Z.; Zhu, X.; Chen, Y.; Xie, D.; Lai, P.; Chen, G.; et al. Primate-specific microRNA-637 inhibits tumorigenesis in hepatocellular carcinoma by disrupting signal transducer and activator of transcription 3 signaling. Hepatology 2011, 54, 2137–2148. [Google Scholar] [CrossRef] [PubMed]
- Aroucha, D.C.; do Carmo, R.F.; Moura, P.; Silva, J.L.; Vasconcelos, L.R.; Cavalcanti, M.S.; Muniz, M.T.; Aroucha, M.L.; Siqueira, E.R.; Cahú, G.G.; et al. High tumor necrosis factor-α/interleukin-10 ratio is associated with hepatocellular carcinoma in patients with chronic hepatitis C. Cytokine 2013, 62, 421–425. [Google Scholar] [CrossRef]
- Swiątek, B.J. Is interleukin-10 gene polymorphism a predictive marker in HCV infection? Cytokine Growth Factor Rev. 2012, 23, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gan, X.; Qiu, J.; Ju, Z.; Gao, J.; Zhou, J.; Shi, C.; Zhu, Y.; Li, Z. IL-10 derived from Hepatocarcinoma cells improves human induced regulatory T cells function via JAK1/STAT5 pathway in tumor microenvironment. Mol. Immunol. 2021, 133, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Buchert, M.; Burns, C.J.; Ernst, M. Targeting JAK kinase in solid tumors: Emerging opportunities and challenges. Oncogene 2016, 35, 939–951. [Google Scholar] [CrossRef]
- Cui, C.; Fu, K.; Yang, L.; Wu, S.; Cen, Z.; Meng, X.; Huang, Q.; Xie, Z. Hypoxia-inducible gene 2 promotes the immune escape of hepatocellular carcinoma from nature killer cells through the interleukin-10-STAT3 signaling pathway. J. Exp. Clin. Cancer Res. 2019, 38, 229. [Google Scholar] [CrossRef]
- Sui, Q.; Zhang, J.; Sun, X.; Zhang, C.; Han, Q.; Tian, Z. NK cells are the crucial antitumor mediators when STAT3-mediated immunosuppression is blocked in hepatocellular carcinoma. J. Immunol. 2014, 193, 2016–2023. [Google Scholar] [CrossRef]
- Langer, J.A.; Cutrone, E.C.; Kotenko, S. The Class II cytokine receptor (CRF2) family: Overview and patterns of receptor-ligand interactions. Cytokine Growth Factor Rev. 2004, 15, 33–48. [Google Scholar] [CrossRef]
- Sabat, R. IL-10 family of cytokines. Cytokine Growth Factor Rev. 2010, 21, 315–324. [Google Scholar] [CrossRef]
- Hsu, Y.H.; Li, H.H.; Sung, J.M.; Chen, W.T.; Hou, Y.C.; Chang, M.S. Interleukin-19 mediates tissue damage in murine ischemic acute kidney injury. PLoS ONE 2013, 8, e56028. [Google Scholar] [CrossRef]
- Caparrós, E.; Francés, R. The Interleukin-20 Cytokine Family in Liver Disease. Front. Immunol. 2018, 9, 1155. [Google Scholar] [CrossRef] [PubMed]
- Hsing, C.H.; Cheng, H.C.; Hsu, Y.H.; Chan, C.H.; Yeh, C.H.; Li, C.F.; Chang, M.S. Upregulated IL-19 in breast cancer promotes tumor progression and affects clinical outcome. Clin. Cancer Res. 2012, 18, 713–725. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, Y.; Kuramoto, N.; Yoneyama, M.; Azuma, Y.T. Interleukin-19 as an Immunoregulatory Cytokine. Curr. Mol. Pharmacol. 2021, 14, 191–199. [Google Scholar] [CrossRef]
- Wang, H.H.; Hsu, Y.H.; Chang, M.S. IL-20 bone diseases involvement and therapeutic target potential. J. Biomed. Sci. 2018, 25, 38. [Google Scholar] [CrossRef]
- Wegenka, U.M.; Dikopoulos, N.; Reimann, J.; Adler, G.; Wahl, C. The murine liver is a potential target organ for IL-19, IL-20 and IL-24: Type I Interferons and LPS regulate the expression of IL-20R2. J. Hepatol. 2007, 46, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Chiu, Y.S.; Wei, C.C.; Lin, Y.J.; Hsu, Y.H.; Chang, M.S. IL-20 and IL-20R1 antibodies protect against liver fibrosis. Hepatology 2014, 60, 1003–1014. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Xuefeng, Y.; Jianhua, X. Systematic review of the roles of interleukins in hepatocellular carcinoma. Clin. Chim. Acta 2020, 506, 33–43. [Google Scholar] [CrossRef]
- Ding, W.Z.; Han, G.Y.; Jin, H.H.; Zhan, C.F.; Ji, Y.; Huang, X.L. Anti-IL-20 monoclonal antibody suppresses hepatocellular carcinoma progression. Oncol. Lett. 2018, 16, 6156–6162. [Google Scholar] [CrossRef]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef]
- Silvagni, E.; Missiroli, S.; Perrone, M.; Patergnani, S.; Boncompagni, C.; Bortoluzzi, A.; Govoni, M.; Giorgi, C.; Alivernini, S.; Pinton, P.; et al. From Bed to Bench and Back: TNF-α, IL-23/IL-17A, and JAK-Dependent Inflammation in the Pathogenesis of Psoriatic Synovitis. Front. Pharmacol. 2021, 12, 672515. [Google Scholar] [CrossRef]
- Kang, Y.; Su, G.; Sun, J.; Zhang, Y. Activation of the TLR4/MyD88 signaling pathway contributes to the development of human hepatocellular carcinoma via upregulation of IL-23 and IL-17A. Oncol. Lett. 2018, 15, 9647–9654. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lu, L.; Luo, Z.; Li, W.; Lu, Y.; Tang, Q.; Pu, J. miR-383 inhibits cell growth and promotes cell apoptosis in hepatocellular carcinoma by targeting IL-17 via STAT3 signaling pathway. Biomed. Pharmacother. 2019, 120, 109551. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Guo, J.; Cao, Q.; Wang, Y.; Chen, J.; Wang, Z.; Yuan, Z. Autophagy impacts on oxaliplatin-induced hepatocarcinoma apoptosis via the IL-17/IL-17R-JAK2/STAT3 signaling pathway. Oncol. Lett. 2017, 13, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Kuang, D.M.; Zhao, Q.; Wu, Y.; Peng, C.; Wang, J.; Xu, Z.; Yin, X.Y.; Zheng, L. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J. Hepatol. 2011, 54, 948–955. [Google Scholar] [CrossRef] [PubMed]
- He, B.; You, L.; Uematsu, K.; Zang, K.; Xu, Z.; Lee, A.Y.; Costello, J.F.; McCormick, F.; Jablons, D.M. SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 14133–14138. [Google Scholar] [CrossRef]
- Khan, F.S.; Ali, I.; Afridi, U.K.; Ishtiaq, M.; Mehmood, R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol. Int. 2017, 11, 45–53. [Google Scholar] [CrossRef]
- Sanaei, M.; Kavoosi, F.; Pourahmadi, M. Effect of Decitabine (5-aza-2′-deoxycytidine, 5-aza-CdR) in Comparison with Vorinostat (Suberoylanilide Hydroxamic Acid, SAHA) on DNMT1, DNMT3a and DNMT3b, HDAC 1-3, SOCS 1, SOCS 3, JAK2, and STAT3 Gene Expression in Hepatocellular Carcinoma HLE and LCL-PI 11 Cell Lines. Asian Pac. J. Cancer Prev. 2021, 22, 2089–2098. [Google Scholar] [CrossRef]
- Mao, J.; Hu, X.; Pang, P.; Zhou, B.; Li, D.; Shan, H. miR-30e acts as a tumor suppressor in hepatocellular carcinoma partly via JAK1/STAT3 pathway. Oncol. Rep. 2017, 38, 393–401. [Google Scholar] [CrossRef]
- Norman, P. Selective JAK inhibitors in development for rheumatoid arthritis. Expert Opin. Investig. Drugs 2014, 23, 1067–1077. [Google Scholar] [CrossRef]
- Yang, L.; Xue, H.; Sun, Y.; Zhang, L.; Xue, F.; Ge, R. CircularRNA-9119 protects hepatocellular carcinoma cells from apoptosis by intercepting miR-26a/JAK1/STAT3 signaling. Cell Death Dis. 2020, 11, 605. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, L.; Li, N.; Dai, J.; Zhang, R.; Yao, F.; Zhou, S.; Wu, Z.; Zhou, H.; Zhou, L.; et al. Etomidate elicits anti-tumor capacity by disrupting the JAK2/STAT3 signaling pathway in hepatocellular carcinoma. Cancer Lett. 2023, 552, 215970. [Google Scholar] [CrossRef] [PubMed]
- Cherng, Y.G.; Chu, Y.C.; Yadav, V.K.; Huang, T.Y.; Hsieh, M.S.; Lee, K.F.; Lee, W.H.; Yeh, C.T.; Ong, J.R. Induced Mitochondrial Alteration and DNA Damage via IFNGR-JAK2-STAT1-PARP1 Pathway Facilitates Viral Hepatitis Associated Hepatocellular Carcinoma Aggressiveness and Stemness. Cancers 2021, 13, 2755. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.X.; Gao, P.J.; Sun, B.G. Pravastatin attenuates interferon-gamma action via modulation of STAT1 to prevent aortic atherosclerosis in apolipoprotein E-knockout mice. Clin. Exp. Pharmacol. Physiol. 2009, 36, 373–379. [Google Scholar] [CrossRef]
- Sutter, A.P.; Maaser, K.; Höpfner, M.; Huether, A.; Schuppan, D.; Scherübl, H. Cell cycle arrest and apoptosis induction in hepatocellular carcinoma cells by HMG-CoA reductase inhibitors. Synergistic antiproliferative action with ligands of the peripheral benzodiazepine receptor. J. Hepatol. 2005, 43, 808–816. [Google Scholar] [CrossRef]
- Han, D.; Yu, T.; Dong, N.; Wang, B.; Sun, F.; Jiang, D. Napabucasin, a novel STAT3 inhibitor suppresses proliferation, invasion and stemness of glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 289. [Google Scholar] [CrossRef]
- Li, Y.; Rogoff, H.A.; Keates, S.; Gao, Y.; Murikipudi, S.; Mikule, K.; Leggett, D.; Li, W.; Pardee, A.B.; Li, C.J. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc. Natl. Acad. Sci. USA 2015, 112, 1839–1844. [Google Scholar] [CrossRef]
- Li, Y.; Han, Q.; Zhao, H.; Guo, Q.; Zhang, J. Napabucasin Reduces Cancer Stem Cell Characteristics in Hepatocellular Carcinoma. Front. Pharmacol. 2020, 11, 597520. [Google Scholar] [CrossRef]
- Shen, Y.; Zheng, X.; Qian, Y.; Liu, M.; Nian, Z.; Cui, Q.; Zhou, Y.; Fu, B.; Sun, R.; Tian, Z.; et al. Interactions between driver genes shape the signaling pathway landscape and direct hepatocellular carcinoma therapy. Cancer Sci. 2023, 114, 2386–2399. [Google Scholar] [CrossRef]
- Ni, J.S.; Zheng, H.; Ou, Y.L.; Tao, Y.P.; Wang, Z.G.; Song, L.H.; Yan, H.L.; Zhou, W.P. miR-515-5p suppresses HCC migration and invasion via targeting IL6/JAK/STAT3 pathway. Surg. Oncol. 2020, 34, 113–120. [Google Scholar] [CrossRef]
- Li, H.; Zhang, B.; Ding, M.; Lu, S.; Zhou, H.; Sun, D.; Wu, G.; Gan, X. C1QTNF1-AS1 regulates the occurrence and development of hepatocellular carcinoma by regulating miR-221-3p/SOCS3. Hepatol. Int. 2019, 13, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Shi, X.; Gong, Z.; Su, Q.; Yu, R.; Wang, B.; Yang, T.; Dai, B.; Zhan, Y.; Zhang, D.; et al. Cantharidin treatment inhibits hepatocellular carcinoma development by regulating the JAK2/STAT3 and PI3K/Akt pathways in an EphB4-dependent manner. Pharmacol. Res. 2020, 158, 104868. [Google Scholar] [CrossRef]
- Liao, J.; Xu, T.; Zheng, J.X.; Lin, J.M.; Cai, Q.Y.; Yu, D.B.; Peng, J. Nitidine chloride inhibits hepatocellular carcinoma cell growth in vivo through the suppression of the JAK1/STAT3 signaling pathway. Int. J. Mol. Med. 2013, 32, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Wang, C.C.; Pai, H.T.; Tu, S.L.; Hou, P.Y.; Huang, C.Y.; Huang, M.T.; Chang, Y.J. The Natural Compound Dehydrocrenatidine Attenuates Nicotine-Induced Stemness and Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma by Regulating a7nAChR-Jak2 Signaling Pathways. Dis. Markers 2022, 2022, 8316335. [Google Scholar] [CrossRef] [PubMed]
- Mohan, C.D.; Bharathkumar, H.; Bulusu, K.C.; Pandey, V.; Rangappa, S.; Fuchs, J.E.; Shanmugam, M.K.; Dai, X.; Li, F.; Deivasigamani, A.; et al. Development of a novel azaspirane that targets the Janus kinase-signal transducer and activator of transcription (STAT) pathway in hepatocellular carcinoma in vitro and in vivo. J. Biol. Chem. 2014, 289, 34296–34307. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, F.; Sugimoto, K.; Harada, Y.; Hashimoto, N.; Ohi, N.; Kurahashi, S.; Naoe, T. A novel STAT inhibitor, OPB-31121, has a significant antitumor effect on leukemia with STAT-addictive oncokinases. Blood Cancer J. 2013, 3, e166. [Google Scholar] [CrossRef]
- Okusaka, T.; Ueno, H.; Ikeda, M.; Mitsunaga, S.; Ozaka, M.; Ishii, H.; Yokosuka, O.; Ooka, Y.; Yoshimoto, R.; Yanagihara, Y.; et al. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol. Res. 2015, 45, 1283–1291. [Google Scholar] [CrossRef]
- Hijona, E.; Banales, J.M.; Hijona, L.; Medina, J.F.; Arenas, J.; Herreros-Villanueva, M.; Aldazabal, P.; Bujanda, L. Pravastatin inhibits cell proliferation and increased MAT1A expression in hepatocarcinoma cells and in vivo models. Cancer Cell Int. 2012, 12, 5. [Google Scholar] [CrossRef]
- Covington, M.; He, X.; Scuron, M.; Li, J.; Collins, R.; Juvekar, A.; Shin, N.; Favata, M.; Gallagher, K.; Sarah, S.; et al. Preclinical characterization of itacitinib (INCB039110), a novel selective inhibitor of JAK1, for the treatment of inflammatory diseases. Eur. J. Pharmacol. 2020, 885, 173505. [Google Scholar] [CrossRef]
- Barbour, A.M.; Rockich, K.; Cimino, E.; Zhou, G.; Leonetti-Whalen, C.; Chen, X.; Yeleswaram, S.; Epstein, N.; Punwani, N. Effect of Hepatic Impairment on the Pharmacokinetics of Itacitinib. J. Clin. Pharmacol. 2021, 61, 954–960. [Google Scholar] [CrossRef]
- Jouve, J.L.; Lecomte, T.; Bouché, O.; Barbier, E.; Khemissa Akouz, F.; Riachi, G.; Nguyen Khac, E.; Ollivier-Hourmand, I.; Debette-Gratien, M.; Faroux, R.; et al. Pravastatin combination with sorafenib does not improve survival in advanced hepatocellular carcinoma. J. Hepatol. 2019, 71, 516–522. [Google Scholar] [CrossRef] [PubMed]
- Burel, S.A.; Han, S.R.; Lee, H.S.; Norris, D.A.; Lee, B.S.; Machemer, T.; Park, S.Y.; Zhou, T.; He, G.; Kim, Y.; et al. Preclinical evaluation of the toxicological effects of a novel constrained ethyl modified antisense compound targeting signal transducer and activator of transcription 3 in mice and cynomolgus monkeys. Nucleic Acid Ther. 2013, 23, 213–227. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.; Kurzrock, R.; Kim, Y.; Woessner, R.; Younes, A.; Nemunaitis, J.; Fowler, N.; Zhou, T.; Schmidt, J.; Jo, M.; et al. AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 2015, 7, 314ra185. [Google Scholar] [CrossRef]
- Lee, C.; Cheung, S.T. STAT3: An Emerging Therapeutic Target for Hepatocellular Carcinoma. Cancers 2019, 11, 1646. [Google Scholar] [CrossRef] [PubMed]
- Yim, S.Y.; Lee, J.S. Genomic Perspective on Mouse Liver Cancer Models. Cancers 2019, 11, 1648. [Google Scholar] [CrossRef] [PubMed]
- Brown, Z.J.; Heinrich, B.; Greten, T.F. Mouse models of hepatocellular carcinoma: An overview and highlights for immunotherapy research. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 536–554. [Google Scholar] [CrossRef] [PubMed]
- Macek Jilkova, Z.; Kurma, K.; Decaens, T. Animal Models of Hepatocellular Carcinoma: The Role of Immune System and Tumor Microenvironment. Cancers 2019, 11, 1487. [Google Scholar] [CrossRef]
- Llovet, J.M.; Bruix, J. Molecular targeted therapies in hepatocellular carcinoma. Hepatology 2008, 48, 1312–1327. [Google Scholar] [CrossRef]
- Dimri, M.; Satyanarayana, A. Molecular Signaling Pathways and Therapeutic Targets in Hepatocellular Carcinoma. Cancers 2020, 12, 491. [Google Scholar] [CrossRef]
- Llovet, J.M.; Castet, F.; Heikenwalder, M.; Maini, M.K.; Mazzaferro, V.; Pinato, D.J.; Pikarsky, E.; Zhu, A.X.; Finn, R.S. Immunotherapies for hepatocellular carcinoma. Nat. Rev. Clin. Oncol. 2022, 19, 151–172. [Google Scholar] [CrossRef]
- Sangro, B.; Sarobe, P.; Hervás-Stubbs, S.; Melero, I. Advances in immunotherapy for hepatocellular carcinoma. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 525–543. [Google Scholar] [CrossRef] [PubMed]
- de Miguel, M.; Calvo, E. Clinical Challenges of Immune Checkpoint Inhibitors. Cancer Cell 2020, 38, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Papadakos, S.P.; Arvanitakis, K.; Stergiou, I.E.; Lekakis, V.; Davakis, S.; Christodoulou, M.I.; Germanidis, G.; Theocharis, S. The Role of TLR4 in the Immunotherapy of Hepatocellular Carcinoma: Can We Teach an Old Dog New Tricks? Cancers 2023, 15, 2795. [Google Scholar] [CrossRef]
- Papadakos, S.P.; Dedes, N.; Kouroumalis, E.; Theocharis, S. The Role of the NLRP3 Inflammasome in HCC Carcinogenesis and Treatment: Harnessing Innate Immunity. Cancers 2022, 14, 3150. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Jin, B.; Liu, X.; Mao, Y.; Wan, X.; Du, S. An immune-related biomarker index for predicting the effectiveness of immunotherapy and prognosis in hepatocellular carcinoma. J. Cancer Res. Clin. Oncol. 2023, 149, 10319–10333. [Google Scholar] [CrossRef] [PubMed]
- Rimassa, L.; Finn, R.S.; Sangro, B. Combination immunotherapy for hepatocellular carcinoma. J. Hepatol. 2023, 79, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zhang, Y.; Zhang, M.; Zhao, K.; Feng, L.; Guan, J.; Dong, R.; Liu, J.; Tian, D.; Liu, M.; et al. Combined immunotherapy for hepatocellular carcinoma: How to maximize immune checkpoint blockade synergic anti-tumor effect. Crit. Rev. Oncol. Hematol. 2023, 189, 104070. [Google Scholar] [CrossRef]
- Rizzo, A.; Ricci, A.D.; Brandi, G. Atezolizumab in advanced hepatocellular carcinoma: Good things come to those who wait. Immunotherapy 2021, 13, 637–644. [Google Scholar] [CrossRef]




| Protein | Role | Reference |
|---|---|---|
| JAK1 | Promotion of inflammatory response Antiviral effect Regulation of neurological and lymphocytic function | [14,28,35] |
| JAK2 | Regulation of hematopoietic function | [14,28] |
| JAK3 | Regulation of lymphocyte production and function | [14,28] |
| TYK2 | Regulation of T cell response and microbial response | [14,28] |
| STAT1 | Regulation of cell growth, differentiation, and apoptosis Inhibition of viral and bacterial infection | [14,18,25,28,34,36,37,38] |
| STAT2 | Antiviral effect Immune regulation Promotion of tumorigenesis | [14,18,25,28,34,36,37,38] |
| STAT3 | Regulation of immune system Progression of cell cycle Inhibition of apoptosis Promotion of tumorigenesis Maintenance of cancer stem cells (CSCs) Regulation of hepatic glucogenesis | [14,16,18,25,28,34,36,37,38] |
| STAT4 | Regulation of immune system Regulation of T cell development and function | [14,18,25,28,34,36,37,38] |
| STAT5 | Regulation of cell growth, differentiation, and apoptosis Regulation of tumor immunity Tumor progression | [14,18,25,28,34,36,37,38] |
| STAT6 | Regulation of lymphocytes Expression of MHCs and immunoglobulins | [14,18,25,28,34,36,37,38] |
| Agent | Target Molecule (Inhibited Molecule) | HCC Cell Line Transplanted | Phenotype | Reference |
|---|---|---|---|---|
| miR-515-5p | IL-6 (STAT3) | HepG2 | Inhibited migration and invasion of HCC cells | [160] |
| Etomidate | JAK2 | HepG2 | Inhibition of tumor growth Increase in animal survival | [152] |
| C1QTNF1-AS1 | miR-221-3p (STAT3) | HepG2, Huh-7 | Reduced tumor volumes | [161] |
| Cantharidin | EphB4 receptor (JAK2, STAT3) | SMMC-7721 | Reduced tumor growth | [162] |
| Nitidine chloride | JAK1, STAT3 | HepG2 | Reduced tumor volumes | [163] |
| Dehydrocrenatidine | JAK2 | HepG2 | Reduced invasion of HCC Inhibition of CSC phenotypes | [164] |
| CIMO | JAK1, JAK2, STAT3 | Huh-7 | Reduced tumor growth | [165] |
| Napabucasin | STAT3 | Hepa1-6 | Reduced tumor volumes Tumor regression in 25% mice | [158] |
| OPB-31121 | STAT3 | Huh-7, HepG2 | Reduced tumor growth | [166] |
| Drug | Target | NCT Number | Phase | Status | Clinical Outcomes |
|---|---|---|---|---|---|
| Itacitinib | JAK1 | 04358185 | Ib | In progress, | Not yet determined |
| Pravastatin | STAT1 | 01075555 | III | Completed (September 2015) | MOS 10.7 m vs. 10.5 m (sorafenib vs. pravastatin + sorafenib) |
| Danvatirsen | STAT3 | 01839604 | I | Completed (February 2015) | Limited antitumor activity. |
| Napabucasin | STAT3 | 02279719 | I/II | Completed (October 2019) | No significant difference in overall response rates between sorafenib alone and napabucasin + sorafenib groups |
| OPB-31121 | STAT3 | 01406574 | I/II | Completed (March 2014) | Limited antitumor effects and low overall responses. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.; Lee, S.; Lee, J.; Moon, H.; Ro, S.W. Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications. Int. J. Mol. Sci. 2023, 24, 13764. https://doi.org/10.3390/ijms241813764
Park H, Lee S, Lee J, Moon H, Ro SW. Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications. International Journal of Molecular Sciences. 2023; 24(18):13764. https://doi.org/10.3390/ijms241813764
Chicago/Turabian StylePark, Hyunjung, Sangjik Lee, Jaehun Lee, Hyuk Moon, and Simon Weonsang Ro. 2023. "Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications" International Journal of Molecular Sciences 24, no. 18: 13764. https://doi.org/10.3390/ijms241813764
APA StylePark, H., Lee, S., Lee, J., Moon, H., & Ro, S. W. (2023). Exploring the JAK/STAT Signaling Pathway in Hepatocellular Carcinoma: Unraveling Signaling Complexity and Therapeutic Implications. International Journal of Molecular Sciences, 24(18), 13764. https://doi.org/10.3390/ijms241813764