
Plant Aquaporin Gating Is Reversed by Phosphorylation on Intracellular Loop D—Evidence from Molecular Dynamics Simulations



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
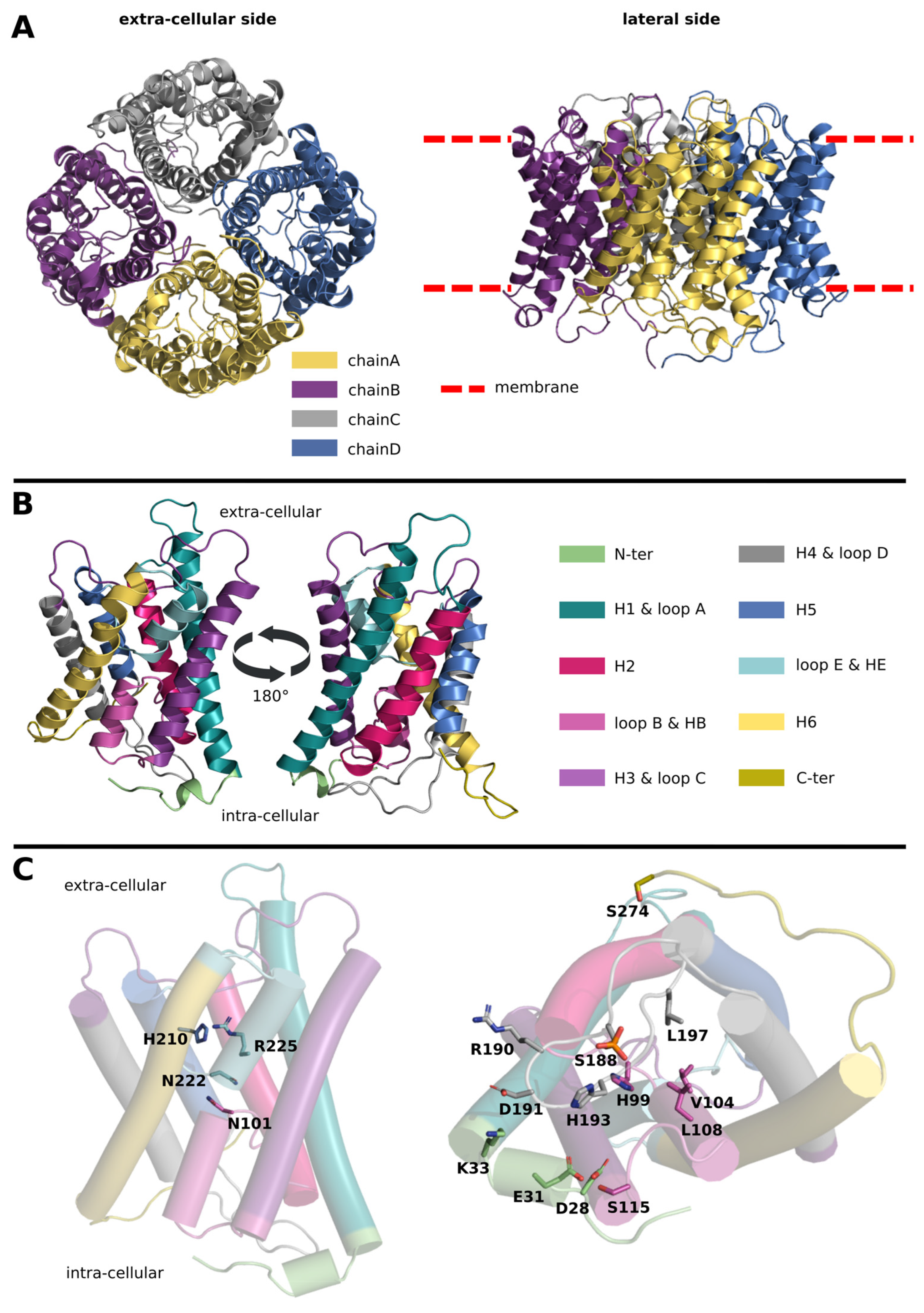
2. Results
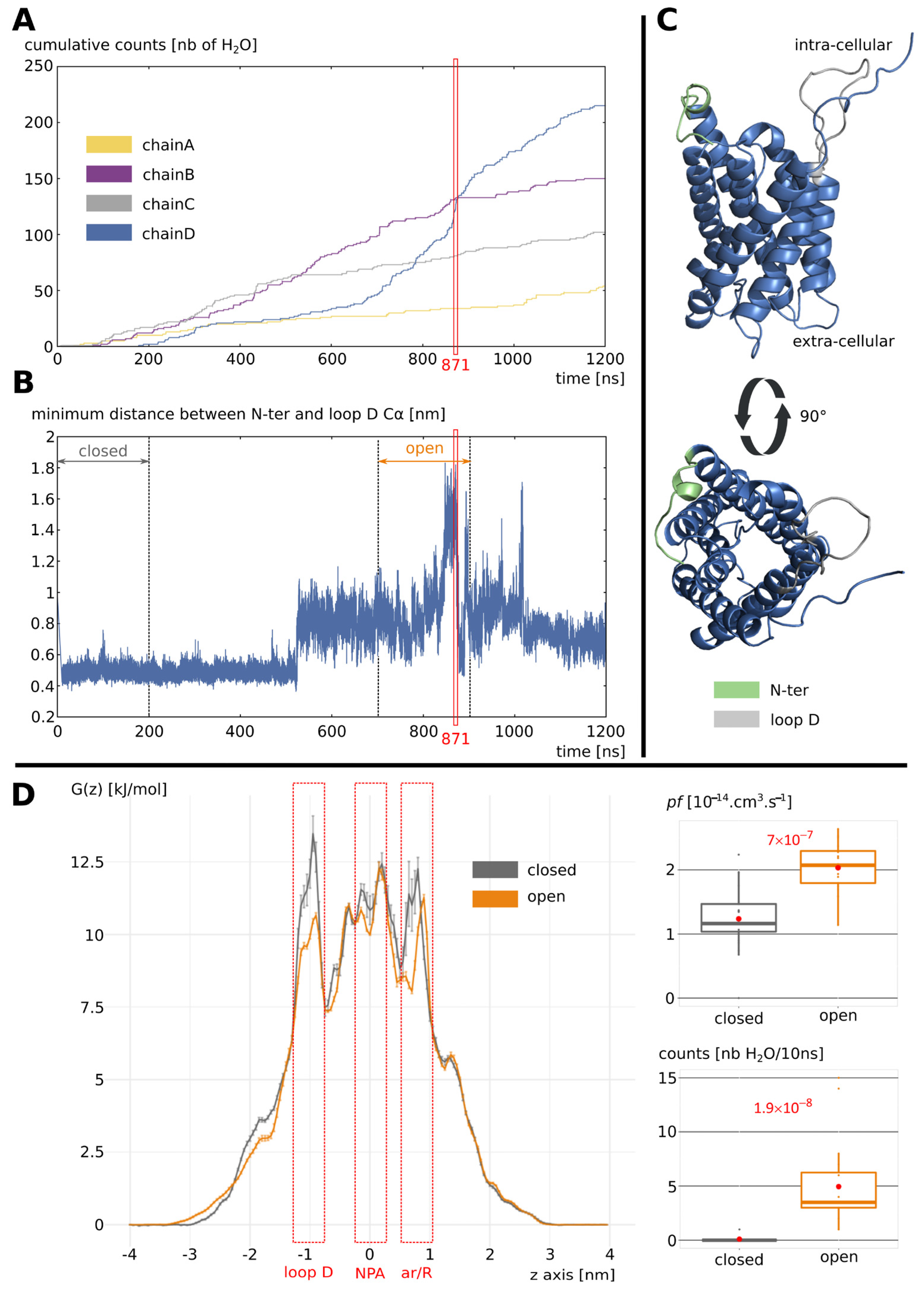
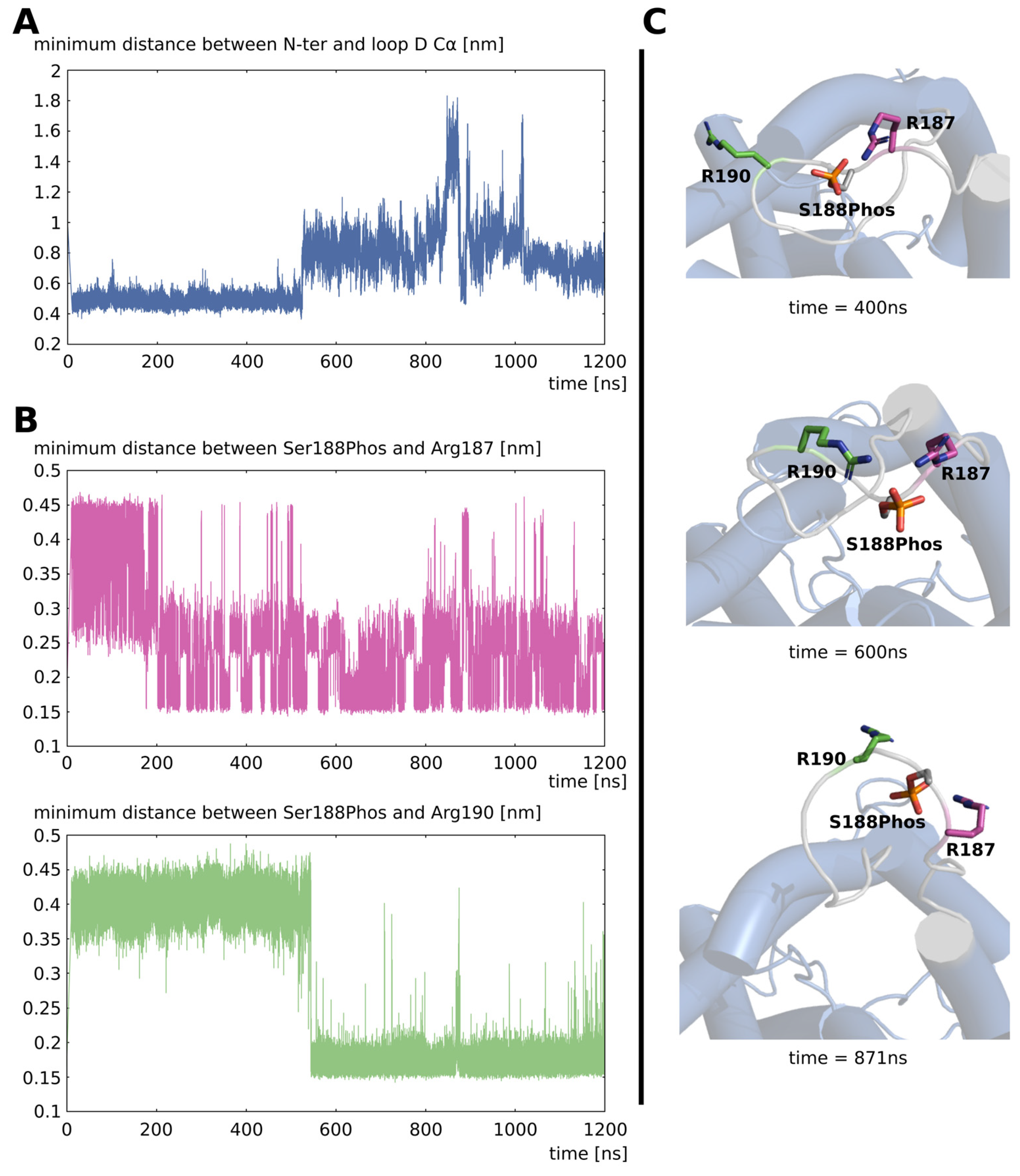
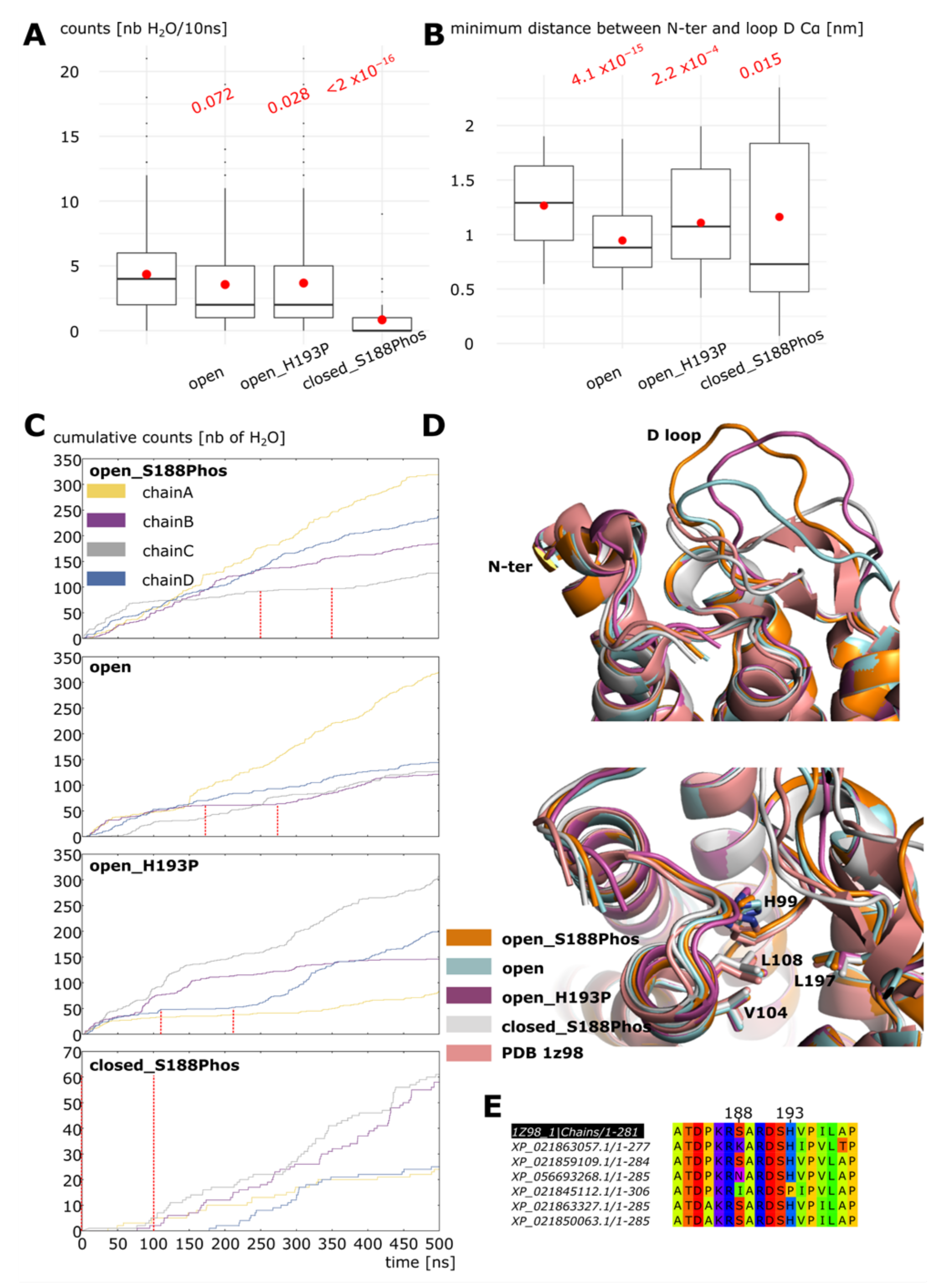
2.1. Opening of Loop D by Phosphorylation of Serine 188
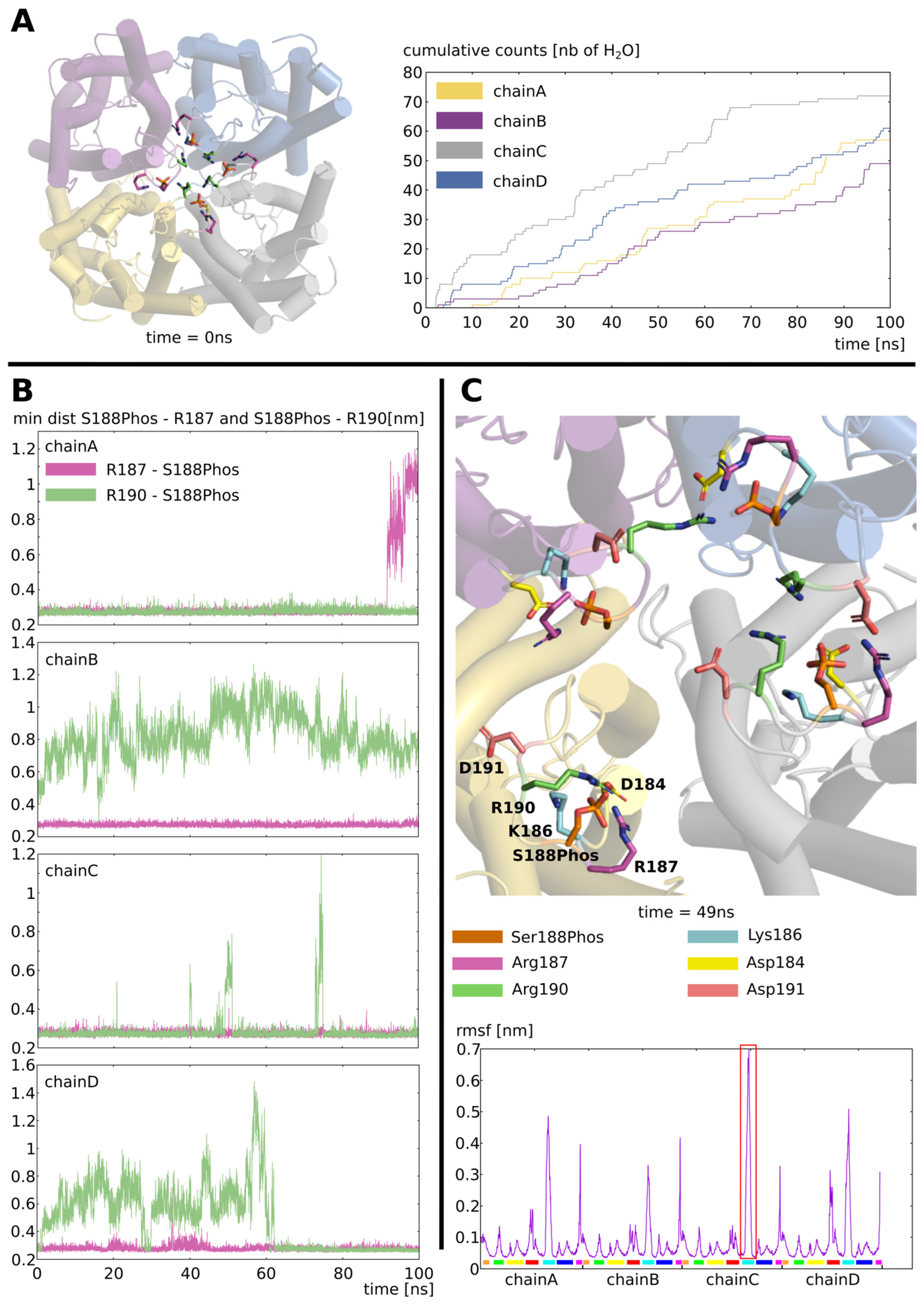
2.2. Stabilization of the Open Conformation
2.3. Dephosphorylation Triggers the Closing of Loop D
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulations
Three Experimental Setups Were Carried Out
4.2. Analysis
4.2.1. Water Permeability
4.2.2. Free Energy Profiles
4.2.3. Other Properties
4.2.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abascal, F.; Irisarri, I.; Zardoya, R. Diversity and Evolution of Membrane Intrinsic Proteins. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 1468–1481. [Google Scholar] [CrossRef] [PubMed]
- Yuan, D.; Li, W.; Hua, Y.; King, G.J.; Xu, F.; Shi, L. Genome-Wide Identification and Characterization of the Aquaporin Gene Family and Transcriptional Responses to Boron Deficiency in Brassica Napus. Front. Plant Sci. 2017, 8, 1336. [Google Scholar] [CrossRef] [PubMed]
- Johanson, U.; Karlsson, M.; Johansson, I.; Gustavsson, S.; Sjövall, S.; Fraysse, L.; Weig, A.R.; Kjellbom, P. The Complete Set of Genes Encoding Major Intrinsic Proteins in Arabidopsis Provides a Framework for a New Nomenclature for Major Intrinsic Proteins in Plants. Plant Physiol. 2001, 126, 1358–1369. [Google Scholar] [CrossRef]
- Fox, A.R.; Maistriaux, L.C.; Chaumont, F. Toward Understanding of the High Number of Plant Aquaporin Isoforms and Multiple Regulation Mechanisms. Plant Sci. 2017, 264, 179–187. [Google Scholar] [CrossRef]
- Chaumont, F.; Tyerman, S.D. (Eds.) Plant Aquaporins: From Transport to Signaling; Signaling and Communication in Plants; Springer International Publishing: Cham, Switzerland, 2017; ISBN 978-3-319-49393-0. [Google Scholar]
- Chaumont, F.; Tyerman, S.D. Aquaporins: Highly Regulated Channels Controlling Plant Water Relations. Plant Physiol. 2014, 164, 1600–1618. [Google Scholar] [CrossRef]
- Santoni, V. Plant Aquaporin Posttranslational Regulation. In Plant Aquaporins: From Transport to Signaling; Chaumont, F., Tyerman, S.D., Eds.; Signaling and Communication in Plants; Springer International Publishing: Cham, Switzerland, 2017; pp. 83–105. ISBN 978-3-319-49395-4. [Google Scholar]
- Takano, J.; Yoshinari, A.; Luu, D.-T. Plant Aquaporin Trafficking. In Plant Aquaporins: From Transport to Signaling; Chaumont, F., Tyerman, S.D., Eds.; Signaling and Communication in Plants; Springer International Publishing: Cham, Switzerland, 2017; pp. 47–81. ISBN 978-3-319-49395-4. [Google Scholar]
- Yaneff, A.; Sigaut, L.; Marquez, M.; Alleva, K.; Pietrasanta, L.I.; Amodeo, G. Heteromerization of PIP Aquaporins Affects Their Intrinsic Permeability. Proc. Natl. Acad. Sci. USA 2014, 111, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Hedfalk, K.; Törnroth-Horsefield, S.; Nyblom, M.; Johanson, U.; Kjellbom, P.; Neutze, R. Aquaporin Gating. Curr. Opin. Struct. Biol. 2006, 16, 447–456. [Google Scholar] [CrossRef]
- Li, G.; Santoni, V.; Maurel, C. Plant Aquaporins: Roles in Plant Physiology. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2014, 1840, 1574–1582. [Google Scholar] [CrossRef]
- Wspalz, T.; Fujiyoshi, Y.; Engel, A. The AQP Structure and Functional Implications. In Aquaporins; Beitz, E., Ed.; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 31–56. ISBN 978-3-540-79885-9. [Google Scholar]
- Tajkhorshid, E.; Nollert, P.; Jensen, M.Ø.; Miercke, L.J.W.; O’Connell, J.; Stroud, R.M.; Schulten, K. Control of the Selectivity of the Aquaporin Water Channel Family by Global Orientational Tuning. Science 2002, 296, 525–530. [Google Scholar] [CrossRef]
- De Groot, B.L.; Frigato, T.; Helms, V.; Grubmüller, H. The Mechanism of Proton Exclusion in the Aquaporin-1 Water Channel. J. Mol. Biol. 2003, 333, 279–293. [Google Scholar] [CrossRef]
- De Groot, B.L.; Grubmüller, H. The Dynamics and Energetics of Water Permeation and Proton Exclusion in Aquaporins. Curr. Opin. Struct. Biol. 2005, 15, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Törnroth-Horsefield, S.; Wang, Y.; Hedfalk, K.; Johanson, U.; Karlsson, M.; Tajkhorshid, E.; Neutze, R.; Kjellbom, P. Structural Mechanism of Plant Aquaporin Gating. Nature 2006, 439, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Törnroth-Horsefield, S.; Hedfalk, K.; Fischer, G.; Lindkvist-Petersson, K.; Neutze, R. Structural Insights into Eukaryotic Aquaporin Regulation. FEBS Lett. 2010, 584, 2580–2588. [Google Scholar] [CrossRef]
- Csanády, L.; Vergani, P.; Gadsby, D.C. Structure, Gating, and Regulation of the CFTR Anion Channel. Physiol. Rev. 2019, 99, 707–738. [Google Scholar] [CrossRef]
- Temmei, Y.; Uchida, S.; Hoshino, D.; Kanzawa, N.; Kuwahara, M.; Sasaki, S.; Tsuchiya, T. Water Channel Activities of Mimosa Pudica Plasma Membrane Intrinsic Proteins Are Regulated by Direct Interaction and Phosphorylation. FEBS Lett. 2005, 579, 4417–4422. [Google Scholar] [CrossRef]
- Nyblom, M.; Frick, A.; Wang, Y.; Ekvall, M.; Hallgren, K.; Hedfalk, K.; Neutze, R.; Tajkhorshid, E.; Törnroth-Horsefield, S. Structural and Functional Analysis of SoPIP2;1 Mutants Adds Insight into Plant Aquaporin Gating. J. Mol. Biol. 2009, 387, 653–668. [Google Scholar] [CrossRef] [PubMed]
- Aponte-Santamaria, C.A. Understanding the Molecular Machinery of Aquaporins through Molecular Dynamics Simulations. Ph.D. Thesis, Georg-August-Universität, Göttingen, Germany, 2012. [Google Scholar]
- Mom, R. Functional Exploration of Aquaporins through Structural Modeling. Impact on the Leaf Hydraulic Conductance in Poplar. Ph.D. Thesis, Université Clermont Auvergne, Clermont-Ferrand, France, 2020. [Google Scholar]
- Tournaire-Roux, C.; Sutka, M.; Javot, H.; Gout, E.; Gerbeau, P.; Luu, D.-T.; Bligny, R.; Maurel, C. Cytosolic PH Regulates Root Water Transport during Anoxic Stress through Gating of Aquaporins. Nature 2003, 425, 393–397. [Google Scholar] [CrossRef]
- Schrödinger, LLC. The PyMOL Molecular Graphics System, Version 1.8 2015. Available online: https://pymol.org/features.html (accessed on 23 August 2023).
- Yaneff, A.; Vitali, V.; Amodeo, G. PIP1 Aquaporins: Intrinsic Water Channels or PIP2 Aquaporin Modulators? FEBS Lett. 2015, 589, 3508–3515. [Google Scholar] [CrossRef]
- Azad, A.K.; Katsuhara, M.; Sawa, Y.; Ishikawa, T.; Shibata, H. Characterization of Four Plasma Membrane Aquaporins in Tulip Petals: A Putative Homolog Is Regulated by Phosphorylation. Plant Cell Physiol. 2008, 49, 1196–1208. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D. CHARMM36m: An Improved Force Field for Folded and Intrinsically Disordered Proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [PubMed]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Zhu, F.; Tajkhorshid, E.; Schulten, K. Collective Diffusion Model for Water Permeation through Microscopic Channels. Phys. Rev. Lett. 2004, 93, 224501. [Google Scholar] [CrossRef]
- Hadidi, H.; Kamali, R.; Binesh, A. Investigation of the Aquaporin-2 Gating Mechanism with Molecular Dynamics Simulations. Proteins: Struct. Funct. Bioinform. 2021, 89, 819–831. [Google Scholar] [CrossRef]
- Hub, J.; de Groot, B.L. Mechanism of Selectivity in Aquaporins and Aquaglyceroporins. Proc. Natl. Acad. Sci. USA 2008, 105, 1198–1203. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Ihaka, R.; Gentleman, R. R: A Language for Data Analysis and Graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mom, R.; Réty, S.; Mocquet, V.; Auguin, D. Plant Aquaporin Gating Is Reversed by Phosphorylation on Intracellular Loop D—Evidence from Molecular Dynamics Simulations. Int. J. Mol. Sci. 2023, 24, 13798. https://doi.org/10.3390/ijms241813798
Mom R, Réty S, Mocquet V, Auguin D. Plant Aquaporin Gating Is Reversed by Phosphorylation on Intracellular Loop D—Evidence from Molecular Dynamics Simulations. International Journal of Molecular Sciences. 2023; 24(18):13798. https://doi.org/10.3390/ijms241813798
Chicago/Turabian StyleMom, Robin, Stéphane Réty, Vincent Mocquet, and Daniel Auguin. 2023. "Plant Aquaporin Gating Is Reversed by Phosphorylation on Intracellular Loop D—Evidence from Molecular Dynamics Simulations" International Journal of Molecular Sciences 24, no. 18: 13798. https://doi.org/10.3390/ijms241813798