

Hardening of Respiratory Syncytial Virus Inclusion Bodies by Cyclopamine Proceeds through Perturbation of the Interactions of the M2-1 Protein with RNA and the P Protein
 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
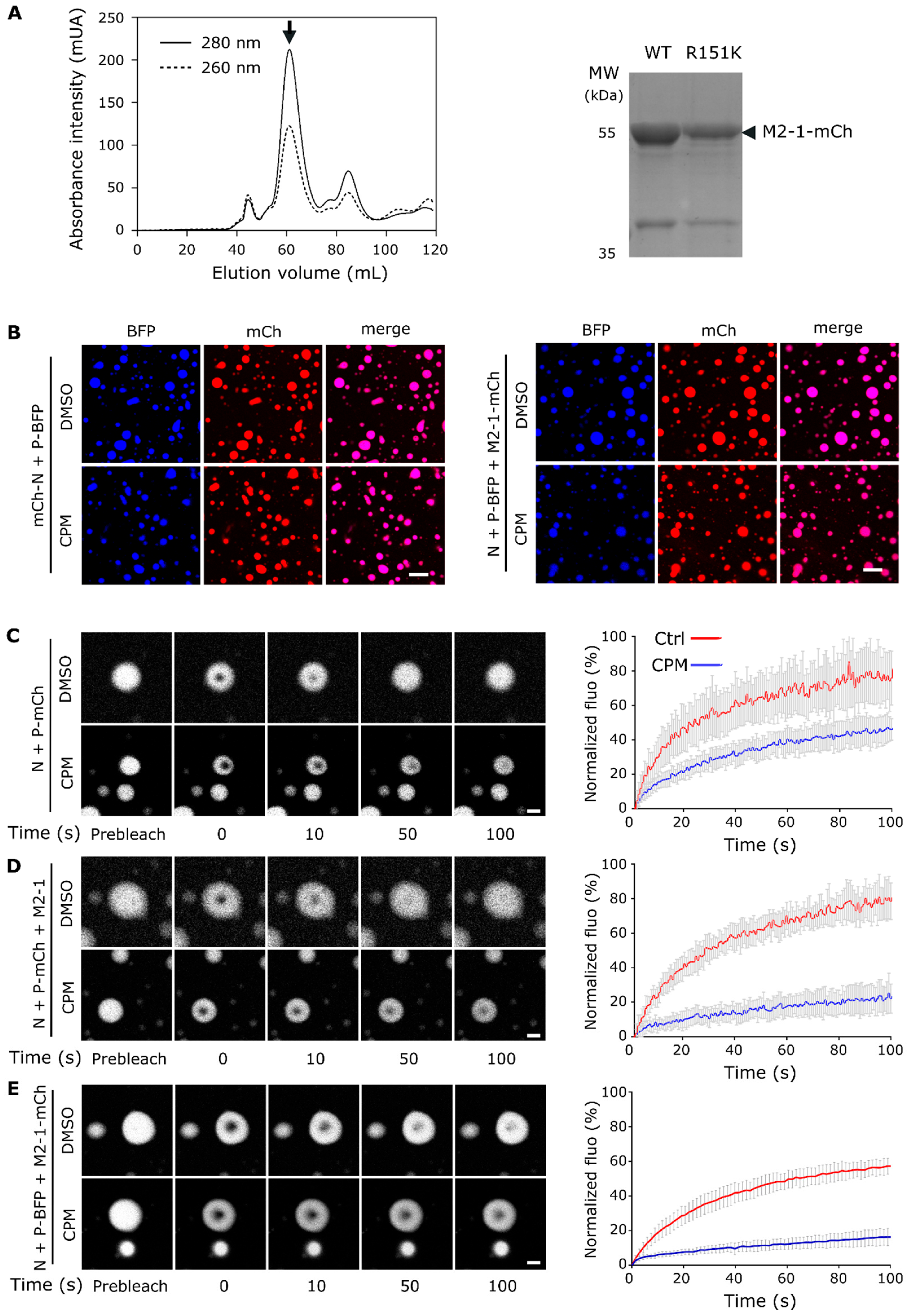
2.1. CPM Treatment Induces Hardening of Pseudo-IBs Formed in Cells
2.2. Impact of CPM on the Fluidity of Pseudo-IBs Formed In Vitro
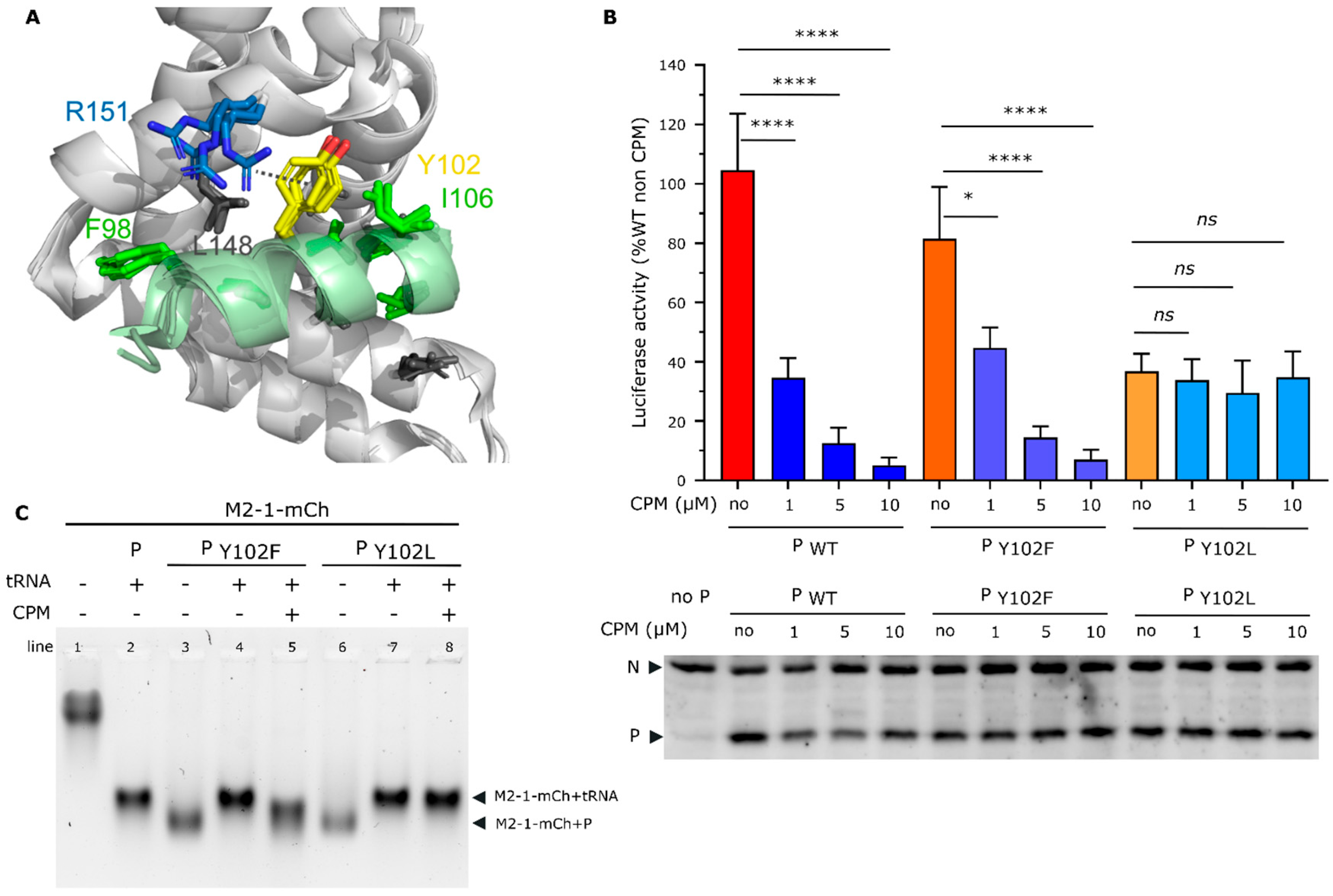
2.3. CPM Interferes with the Competitive Binding of P and RNA to M2-1
2.4. Study of the Potential Interaction of CPM with the M2-1 or M2-1-P Complex
2.5. Residue Y102 of P Is Involved in CPM Activity
3. Discussion
4. Materials and Methods
4.1. Plasmid Constructs
4.2. Cells
4.3. Time-Lapse Microscopy and Photobleaching Experiments on Pseudo-IBs
4.4. Photobleaching Experiments on In Vitro Reconstituted Pseudo-Ibs
4.5. Minigenome Assay
4.6. Expression and Purification of the Recombinant Proteins
4.7. Band Shift on Native Agarose Gels
4.8. Pulldown Assay
4.9. Docking of CPM onto M2-1
4.10. P Peptide Fold Prediction
4.11. Microscale Thermophoresis
4.12. NMR
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sagan, S.M.; Weber, S.C. Let’s phase it: Viruses are master architects of biomolecular condensates. Trends Biochem. Sci. 2023, 48, 229–243. [Google Scholar] [CrossRef]
- Li, H.; Ernst, C.; Kolonko-Adamska, M.; Greb-Markiewicz, B.; Man, J.; Parissi, V.; Ng, B.W. Phase separation in viral infections. Trends Microbiol. 2022, 30, 1217–1231. [Google Scholar] [CrossRef]
- Wei, W.; Bai, L.; Yan, B.; Meng, W.; Wang, H.; Zhai, J.; Si, F.; Zheng, C. When liquid-liquid phase separation meets viral infections. Front. Immunol. 2022, 13, 985622. [Google Scholar] [CrossRef]
- Rincheval, V.; Lelek, M.; Gault, E.; Bouillier, C.; Sitterlin, D.; Blouquit-Laye, S.; Galloux, M.; Zimmer, C.; Eleouet, J.F.; Rameix-Welti, M.A. Functional organization of cytoplasmic inclusion bodies in cells infected by respiratory syncytial virus. Nat. Commun. 2017, 8, 563. [Google Scholar] [CrossRef]
- Jobe, F.; Simpson, J.; Hawes, P.; Guzman, E.; Bailey, D. Respiratory Syncytial Virus Sequesters NF-kappaB Subunit p65 to Cytoplasmic Inclusion Bodies To Inhibit Innate Immune Signaling. J. Virol. 2020, 94, e01380-20. [Google Scholar] [CrossRef]
- Afonso, C.L.; Amarasinghe, G.K.; Banyai, K.; Bao, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef]
- Garcia, J.; Garcia-Barreno, B.; Vivo, A.; Melero, J.A. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: Formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22 K protein. Virology 1993, 195, 243–247. [Google Scholar] [CrossRef]
- Galloux, M.; Risso-Ballester, J.; Richard, C.A.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Minimal Elements Required for the Formation of Respiratory Syncytial Virus Cytoplasmic Inclusion Bodies In Vivo and In Vitro. mBio 2020, 11, e01202-20. [Google Scholar] [CrossRef]
- Shi, T.; McAllister, D.A.; O’Brien, K.L.; Simoes, E.A.F.; Madhi, S.A.; Gessner, B.D.; Polack, F.P.; Balsells, E.; Acacio, S.; Aguayo, C.; et al. Global, regional, and national disease burden estimates of acute lower respiratory infections due to respiratory syncytial virus in young children in 2015: A systematic review and modelling study. Lancet 2017, 390, 946–958. [Google Scholar] [CrossRef]
- Busack, B.; Shorr, A.F. Going Viral-RSV as the Neglected Adult Respiratory Virus. Pathogens 2022, 11, 1324. [Google Scholar] [CrossRef]
- Valarcher, J.F.; Taylor, G. Bovine respiratory syncytial virus infection. Vet. Res. 2007, 38, 153–180. [Google Scholar] [CrossRef]
- Kimman, T.G.; Westenbrink, F.; Straver, P.J. Priming for local and systemic antibody memory responses to bovine respiratory syncytial virus: Effect of amount of virus, virus replication, route of administration and maternal antibodies. Vet. Immunol. Immunopathol. 1989, 22, 145–160. [Google Scholar] [CrossRef]
- Ellis, J.A. How efficacious are vaccines against bovine respiratory syncytial virus in cattle? Vet. Microbiol. 2017, 206, 59–68. [Google Scholar] [CrossRef]
- Bailly, B.; Richard, C.A.; Sharma, G.; Wang, L.; Johansen, L.; Cao, J.; Pendharkar, V.; Sharma, D.C.; Galloux, M.; Wang, Y.; et al. Targeting human respiratory syncytial virus transcription anti-termination factor M2-1 to inhibit in vivo viral replication. Sci. Rep. 2016, 6, 25806. [Google Scholar] [CrossRef]
- Risso-Ballester, J.; Galloux, M.; Cao, J.; Le Goffic, R.; Hontonnou, F.; Jobart-Malfait, A.; Desquesnes, A.; Sake, S.M.; Haid, S.; Du, M.; et al. A condensate-hardening drug blocks RSV replication in vivo. Nature 2021, 595, 596–599. [Google Scholar] [CrossRef]
- Fix, J.; Descamps, D.; Galloux, M.; Ferret, C.; Bouguyon, E.; Zohari, S.; Naslund, K.; Hagglund, S.; Altmeyer, R.; Valarcher, J.F.; et al. Screening antivirals with a mCherry-expressing recombinant bovine respiratory syncytial virus: A proof of concept using cyclopamine. Vet. Res. 2023, 54, 36. [Google Scholar] [CrossRef]
- Richard, C.A.; Rincheval, V.; Lassoued, S.; Fix, J.; Cardone, C.; Esneau, C.; Nekhai, S.; Galloux, M.; Rameix-Welti, M.A.; Sizun, C.; et al. RSV hijacks cellular protein phosphatase 1 to regulate M2-1 phosphorylation and viral transcription. PLoS Pathog. 2018, 14, e1006920. [Google Scholar] [CrossRef]
- Cardone, C.; Caseau, C.M.; Pereira, N.; Sizun, C. Pneumoviral Phosphoprotein, a Multidomain Adaptor-Like Protein of Apparent Low Structural Complexity and High Conformational Versatility. Int. J. Mol. Sci. 2021, 22, 1537. [Google Scholar] [CrossRef]
- Tran, T.L.; Castagne, N.; Dubosclard, V.; Noinville, S.; Koch, E.; Moudjou, M.; Henry, C.; Bernard, J.; Yeo, R.P.; Eleouet, J.F. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J. Virol. 2009, 83, 6363–6374. [Google Scholar] [CrossRef]
- Esperante, S.A.; Paris, G.; de Prat-Gay, G. Modular unfolding and dissociation of the human respiratory syncytial virus phosphoprotein p and its interaction with the m(2-1) antiterminator: A singular tetramer-tetramer interface arrangement. Biochemistry 2012, 51, 8100–8110. [Google Scholar] [CrossRef]
- Tanner, S.J.; Ariza, A.; Richard, C.A.; Kyle, H.F.; Dods, R.L.; Blondot, M.L.; Wu, W.; Trincao, J.; Trinh, C.H.; Hiscox, J.A.; et al. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc. Natl. Acad. Sci. USA 2014, 111, 1580–1585. [Google Scholar] [CrossRef] [PubMed]
- Leyrat, C.; Renner, M.; Harlos, K.; Huiskonen, J.T.; Grimes, J.M. Drastic changes in conformational dynamics of the antiterminator M2-1 regulate transcription efficiency in Pneumovirinae. eLife 2014, 3, e02674. [Google Scholar] [CrossRef] [PubMed]
- Blondot, M.L.; Dubosclard, V.; Fix, J.; Lassoued, S.; Aumont-Nicaise, M.; Bontems, F.; Eleouet, J.F.; Sizun, C. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog. 2012, 8, e1002734. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, M.; Yegambaram, K.; Todd, E.; Richard, C.A.; Dods, R.L.; Pangratiou, G.M.; Trinh, C.H.; Moul, S.L.; Murphy, J.C.; Mankouri, J.; et al. The Structure of the Human Respiratory Syncytial Virus M2-1 Protein Bound to the Interaction Domain of the Phosphoprotein P Defines the Orientation of the Complex. mBio 2018, 9, e01554-18. [Google Scholar] [CrossRef]
- Gao, Y.; Cao, D.; Pawnikar, S.; John, K.P.; Ahn, H.M.; Hill, S.; Ha, J.M.; Parikh, P.; Ogilvie, C.; Swain, A.; et al. Structure of the Human Respiratory Syncytial Virus M2-1 Protein in Complex with a Short Positive-Sense Gene-End RNA. Structure 2020, 28, 979–990.e4. [Google Scholar] [CrossRef]
- Molina, I.G.; Esperante, S.A.; Marino-Buslje, C.; Chemes, L.B.; de Prat-Gay, G. Cooperative RNA Recognition by a Viral Transcription Antiterminator. J. Mol. Biol. 2018, 430, 777–792. [Google Scholar] [CrossRef]
- Brocca, S.; Grandori, R.; Longhi, S.; Uversky, V. Liquid-Liquid Phase Separation by Intrinsically Disordered Protein Regions of Viruses: Roles in Viral Life Cycle and Control of Virus-Host Interactions. Int. J. Mol. Sci. 2020, 21, 9045. [Google Scholar] [CrossRef]
- Lopez, N.; Camporeale, G.; Salgueiro, M.; Borkosky, S.S.; Visentin, A.; Peralta-Martinez, R.; Loureiro, M.E.; de Prat-Gay, G. Deconstructing virus condensation. PLoS Pathog. 2021, 17, e1009926. [Google Scholar] [CrossRef]
- Gonnin, L.; Richard, C.A.; Gutsche, I.; Chevret, D.; Troussier, J.; Vasseur, J.J.; Debart, F.; Eleouet, J.F.; Galloux, M. Importance of RNA length for in vitro encapsidation by the nucleoprotein of human respiratory syncytial virus. J. Biol. Chem. 2022, 298, 102337. [Google Scholar] [CrossRef]
- Pereira, N.; Cardone, C.; Lassoued, S.; Galloux, M.; Fix, J.; Assrir, N.; Lescop, E.; Bontems, F.; Eleouet, J.F.; Sizun, C. New Insights into Structural Disorder in Human Respiratory Syncytial Virus Phosphoprotein and Implications for Binding of Protein Partners. J. Biol. Chem. 2017, 292, 2120–2131. [Google Scholar] [CrossRef]
- Galloux, M.; Gabiane, G.; Sourimant, J.; Richard, C.A.; England, P.; Moudjou, M.; Aumont-Nicaise, M.; Fix, J.; Rameix-Welti, M.A.; Eleouet, J.F. Identification and Characterization of the Binding Site of the Respiratory Syncytial Virus Phosphoprotein to RNA-Free Nucleoprotein. J. Virol. 2015, 89, 3484–3496. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, U.J.; Finke, S.; Conzelmann, K.K. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 1999, 73, 251–259. [Google Scholar] [CrossRef] [PubMed]
- De Chaumont, F.; Dallongeville, S.; Chenouard, N.; Herve, N.; Pop, S.; Provoost, T.; Meas-Yedid, V.; Pankajakshan, P.; Lecomte, T.; Le Montagner, Y.; et al. Icy: An open bioimage informatics platform for extended reproducible research. Nat. Methods 2012, 9, 690–696. [Google Scholar] [CrossRef]
- Giakoumakis, N.N.; Rapsomaniki, M.A.; Lygerou, Z. Analysis of Protein Kinetics Using Fluorescence Recovery After Photobleaching (FRAP). Methods Mol. Biol. 2017, 1563, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Lamiable, A.; Thevenet, P.; Rey, J.; Vavrusa, M.; Derreumaux, P.; Tuffery, P. PEP-FOLD3: Faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016, 44, W449–W454. [Google Scholar] [CrossRef]
- Baaske, P.; Wienken, C.J.; Reineck, P.; Duhr, S.; Braun, D. Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew. Chem. Int. Ed. Engl. 2010, 49, 2238–2241. [Google Scholar] [CrossRef]
- Zillner, K.; Jerabek-Willemsen, M.; Duhr, S.; Braun, D.; Langst, G.; Baaske, P. Microscale thermophoresis as a sensitive method to quantify protein: Nucleic acid interactions in solution. Methods Mol. Biol. 2012, 815, 241–252. [Google Scholar] [CrossRef]
- Jerabek-Willemsen, M.; Wienken, C.J.; Braun, D.; Baaske, P.; Duhr, S. Molecular interaction studies using microscale thermophoresis. Assay. Drug Dev. Technol. 2011, 9, 342–353. [Google Scholar] [CrossRef]
- Duhr, S.; Braun, D. Why molecules move along a temperature gradient. Proc. Natl. Acad. Sci. USA 2006, 103, 19678–19682. [Google Scholar] [CrossRef]
- Skinner, S.P.; Fogh, R.H.; Boucher, W.; Ragan, T.J.; Mureddu, L.G.; Vuister, G.W. CcpNmr AnalysisAssign: A flexible platform for integrated NMR analysis. J. Biomol. NMR 2016, 66, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Dubosclard, V.; Blondot, M.L.; Eleouet, J.F.; Bontems, F.; Sizun, C. 1H, 13C, and 15N resonance assignment of the central domain of hRSV transcription antitermination factor M2-1. Biomol. NMR Assign. 2011, 5, 237–239. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diot, C.; Richard, C.-A.; Risso-Ballester, J.; Martin, D.; Fix, J.; Eléouët, J.-F.; Sizun, C.; Rameix-Welti, M.-A.; Galloux, M. Hardening of Respiratory Syncytial Virus Inclusion Bodies by Cyclopamine Proceeds through Perturbation of the Interactions of the M2-1 Protein with RNA and the P Protein. Int. J. Mol. Sci. 2023, 24, 13862. https://doi.org/10.3390/ijms241813862
Diot C, Richard C-A, Risso-Ballester J, Martin D, Fix J, Eléouët J-F, Sizun C, Rameix-Welti M-A, Galloux M. Hardening of Respiratory Syncytial Virus Inclusion Bodies by Cyclopamine Proceeds through Perturbation of the Interactions of the M2-1 Protein with RNA and the P Protein. International Journal of Molecular Sciences. 2023; 24(18):13862. https://doi.org/10.3390/ijms241813862
Chicago/Turabian StyleDiot, Cédric, Charles-Adrien Richard, Jennifer Risso-Ballester, Davy Martin, Jenna Fix, Jean-François Eléouët, Christina Sizun, Marie-Anne Rameix-Welti, and Marie Galloux. 2023. "Hardening of Respiratory Syncytial Virus Inclusion Bodies by Cyclopamine Proceeds through Perturbation of the Interactions of the M2-1 Protein with RNA and the P Protein" International Journal of Molecular Sciences 24, no. 18: 13862. https://doi.org/10.3390/ijms241813862
APA StyleDiot, C., Richard, C.-A., Risso-Ballester, J., Martin, D., Fix, J., Eléouët, J.-F., Sizun, C., Rameix-Welti, M.-A., & Galloux, M. (2023). Hardening of Respiratory Syncytial Virus Inclusion Bodies by Cyclopamine Proceeds through Perturbation of the Interactions of the M2-1 Protein with RNA and the P Protein. International Journal of Molecular Sciences, 24(18), 13862. https://doi.org/10.3390/ijms241813862