
VHL L169P Variant Does Not Alter Cellular Hypoxia Tension in Clear Cell Renal Cell Carcinoma



Abstract
:1. Introduction
2. Results
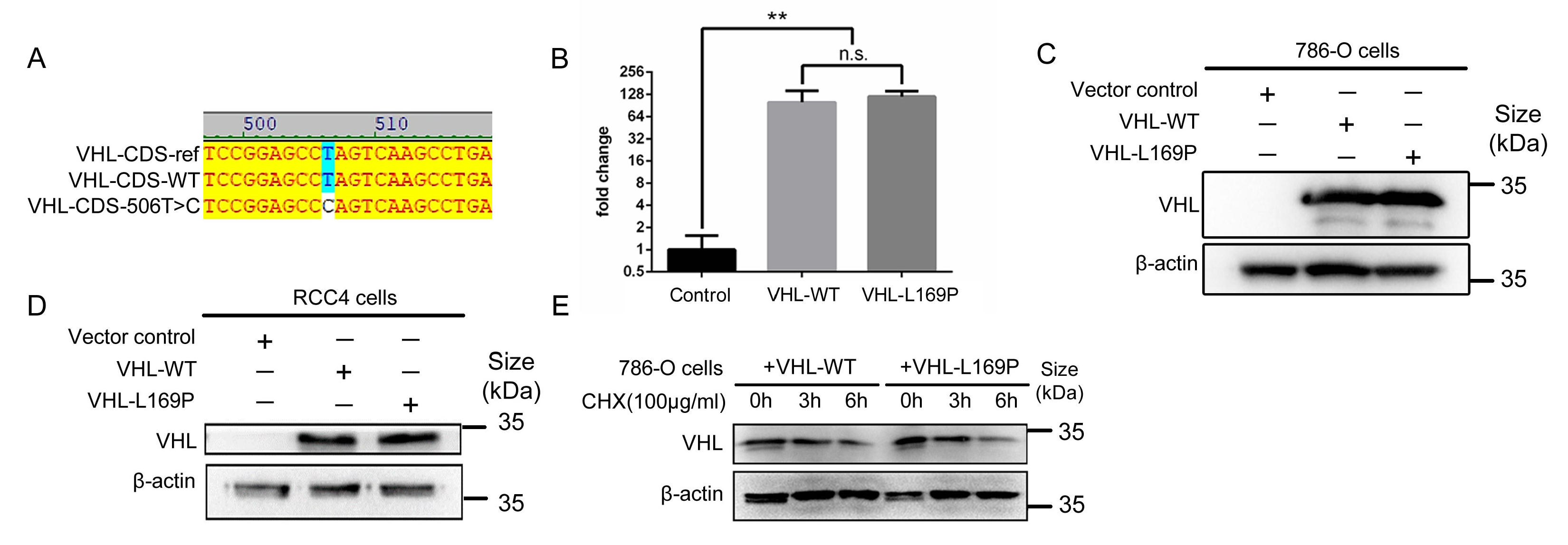
2.1. Amino Acid L169P Substitution Does Not Alter VHL Protein Stability
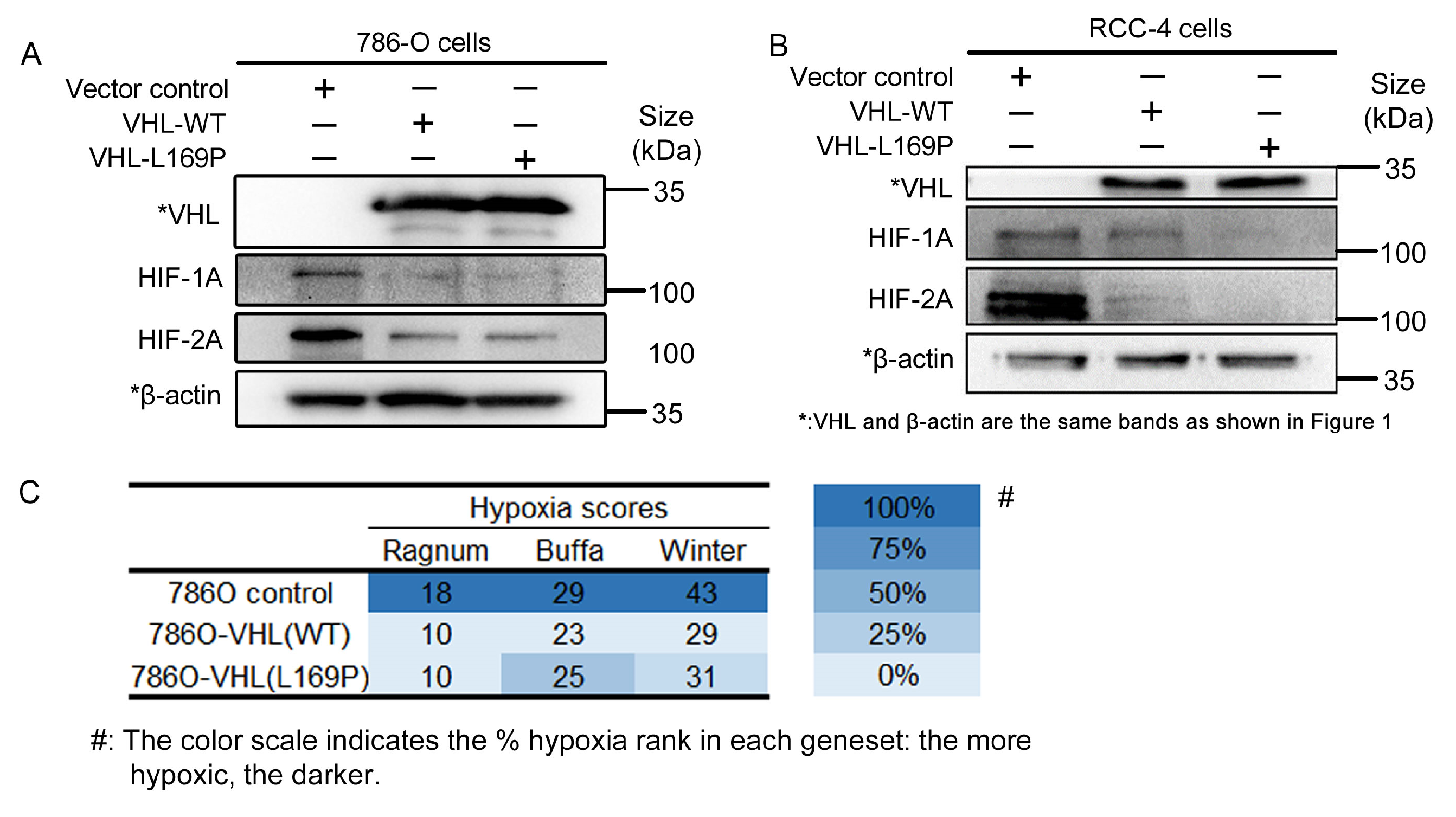
2.2. The Hypoxic Regulation of L169P VHL Is Comparable to WT VHL
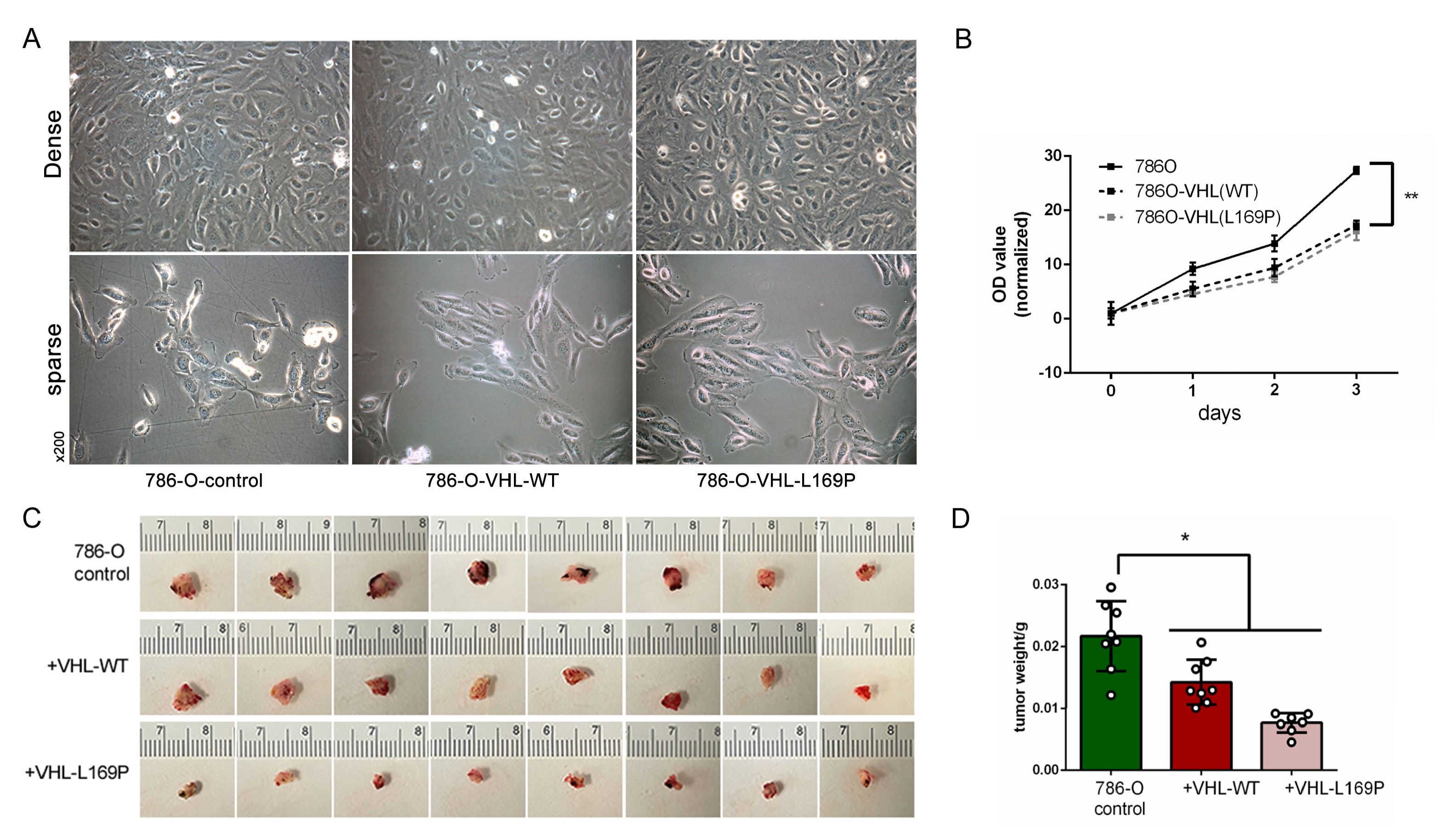
2.3. Restored Expression of WT or L169P VHL Slowed Cancer Cell Growth
3. Discussion
4. Materials and Methods
4.1. The Cell Culture, Reagents and Generation of Genetic Engineered Cell Lines
4.2. qRT-PCR, Western Blot, Cell Proliferation Assay, Chorioallantoic Membrane (CAM) Xenograft Model and VHL Stability Analysis
4.3. Bioinformatic Analysis and Hypoxic Score Calculation
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maher, E.R. Genomics and epigenomics of renal cell carcinoma. Semin. Cancer Biol. 2013, 23, 10–17. [Google Scholar] [CrossRef] [PubMed]
- The Human Gene Mutation Database; The Institute of Medical Genetics: Cardiff, UK, 2007; Available online: https://www.hgmd.cf.ac.uk/ac/index.php (accessed on 1 June 2023).
- The VHL Mutations Database. Montpellier, France. Available online: http://www.umd.be/VHL/ (accessed on 1 June 2023).
- Rechsteiner, M.P.; von Teichman, A.; Nowicka, A.; Sulser, T.; Schraml, P.; Moch, H. VHL Gene Mutations and Their Effects on Hypoxia Inducible Factor HIFα: Identification of Potential Driver and Passenger Mutations. Cancer Res. 2011, 71, 5500–5511. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Tan, P.; Ishihara, M.; Bayley, N.A.; Schokrpur, S.; Reynoso, J.G.; Zhang, Y.; Lim, R.J.; Dumitras, C.; Yang, L.; et al. Tumor heterogeneity in VHL drives metastasis in clear cell renal cell carcinoma. Signal Transduct. Target. Ther. 2023, 8, 155. [Google Scholar] [CrossRef]
- TCGA-KIRC Database: VHL L169P. Available online: https://portal.gdc.cancer.gov/ssms/49c8f652-64ce-5b88-b57d-6b00dbb6ad7f (accessed on 1 June 2023).
- Iliopoulos, O.; Ohh, M.; Kaelin, W.G., Jr. pVHL19 is a biologically active product of the von Hippel-Lindau gene arising from internal translation initiation. Proc. Natl. Acad. Sci. USA 1998, 95, 11661–11666. [Google Scholar] [CrossRef]
- Stebbins, C.E.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 1999, 284, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Gnarra, J.R.; Duan, D.R.; Weng, Y.; Humphrey, J.S.; Chen, D.Y.; Lee, S.; Pause, A.; Dudley, C.F.; Latif, F.; Kuzmin, I.; et al. Molecular cloning of the von Hippel-Lindau tumor suppressor gene and its role in renal carcinoma. Biochim. Biophys. Acta 1996, 1242, 201–210. [Google Scholar] [CrossRef]
- Kamura, T.; Koepp, D.M.; Conrad, M.N.; Skowyra, D.; Moreland, R.J.; Iliopoulos, O.; Lane, W.S.; Kaelin, W.G.; Elledge, S.J.; Conaway, R.C.; et al. Rbx1, a Component of the VHL Tumor Suppressor Complex and SCF Ubiquitin Ligase. Science 1999, 284, 657–661. [Google Scholar] [CrossRef]
- Hon, W.-C.; Wilson, M.I.; Harlos, K.; Claridge, T.D.W.; Schofield, C.J.; Pugh, C.W.; Maxwell, P.H.; Ratcliffe, P.J.; Stuart, D.I.; Jones, E.Y. Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 2002, 417, 975–978. [Google Scholar] [CrossRef]
- Min, J.H.; Yang, H.; Ivan, M.; Gertler, F.; Kaelin, W.G., Jr.; Pavletich, N.P. Structure of an HIF-1alpha-pVHL complex: Hydroxyproline recognition in signaling. Science 2002, 296, 1886–1889. [Google Scholar] [CrossRef]
- Forman, J.R.; Worth, C.L.; Bickerton, G.R.J.; Eisen, T.G.; Blundell, T.L. Structural bioinformatics mutation analysis reveals genotype–phenotype correlations in von Hippel-Lindau disease and suggests molecular mechanisms of tumorigenesis. Proteins Struct. Funct. Bioinform. 2009, 77, 84–96. [Google Scholar] [CrossRef]
- Clifford, S.C.; Cockman, M.E.; Smallwood, A.C.; Mole, D.R.; Woodward, E.R.; Maxwell, P.H.; Ratcliffe, P.J.; Maher, E.R. Contrasting effects on HIF-1alpha regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum. Mol. Genet. 2001, 10, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.A.; Ohh, M.; Yang, H.; Klco, J.M.; Ivan, M.; Kaelin, W.G., Jr. von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum. Mol. Genet. 2001, 10, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Thoma, C.R.; Frew, I.J.; Hoerner, C.R.; Montani, M.; Moch, H.; Krek, W. pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat. Cell Biol. 2007, 9, 588–595. [Google Scholar] [CrossRef] [PubMed]
- Hergovich, A.; Lisztwan, J.; Barry, R.; Ballschmieter, P.; Krek, W. Regulation of microtubule stability by the von Hippel-Lindau tumour suppressor protein pVHL. Nat. Cell Biol. 2003, 5, 64–70. [Google Scholar] [CrossRef]
- Yu, F.; White, S.B.; Zhao, Q.; Lee, F.S. HIF-1α binding to VHL is regulated by stimulus-sensitive proline hydroxylation. Proc. Natl. Acad. Sci. USA 2001, 98, 9630–9635. [Google Scholar] [CrossRef]
- Nyhan, M.J.; El Mashad, S.M.; O’Donovan, T.R.; Ahmad, S.; Collins, C.; Sweeney, P.; Rogers, E.; O’Sullivan, G.C.; McKenna, S.L. VHL genetic alteration in CCRCC does not determine de-regulation of HIF, CAIX, hnRNP A2/B1 and osteopontin. Anal. Cell. Pathol. 2010, 33, 121–132. [Google Scholar] [CrossRef]
- Schneider-Poetsch, T.; Ju, J.; Eyler, D.E.; Dang, Y.; Bhat, S.; Merrick, W.C.; Green, R.; Shen, B.; Liu, J.O. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat. Chem. Biol. 2010, 6, 209–217. [Google Scholar] [CrossRef] [PubMed]
- Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional programme associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382–390. [Google Scholar] [CrossRef] [PubMed]
- Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428–435. [Google Scholar] [CrossRef]
- Winter, S.C.; Buffa, F.M.; Silva, P.; Miller, C.; Valentine, H.R.; Turley, H.; Shah, K.A.; Cox, G.J.; Corbridge, R.J.; Homer, J.J.; et al. Relation of a Hypoxia Metagene Derived from Head and Neck Cancer to Prognosis of Multiple Cancers. Cancer Res. 2007, 67, 3441–3449. [Google Scholar] [CrossRef]
- Ishihara, M.; Hu, J.; Zhang, X.; Choi, Y.; Wong, A.; Cano-Ruiz, C.; Zhao, R.; Tan, P.; Tso, J.L.; Wu, L. Comparing Metastatic Clear Cell Renal Cell Carcinoma Model Established in Mouse Kidney and on Chicken Chorioallantoic Membrane. J. Vis. Exp. JoVE 2020, 156, e60314. [Google Scholar]
- Hu, J.; Ishihara, M.; Chin, A.I.; Wu, L. Establishment of xenografts of urological cancers on chicken chorioallantoic membrane (CAM) to study metastasis. Precis. Clin. Med. 2019, 2, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Crystallography and Bioinformatic Group. Site Directed Mutator: I-Mutant2.0: Thermodynamic Database for Protein and Mutants; University of Cambridge: Cambridge, UK, 2005. [Google Scholar]
- Crystallography and Bioinformatic Group. Site Directed Mutator: Mupro: Thermodynamic Database for Protein and Mutants; University of Cambridge: Cambridge, UK, 2005. [Google Scholar]
- Chelliah, V.; Chen, L.; Blundell, T.L.; Lovell, S.C. Distinguishing structural and functional restraints in evolution in order to identify interaction sites. J. Mol. Biol. 2004, 342, 1487–1504. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Hoey, C.; Liu, L.Y.; Lalonde, E.; Ray, J.; Livingstone, J.; Lesurf, R.; Shiah, Y.J.; Vujcic, T.; Huang, X.; et al. Molecular landmarks of tumor hypoxia across cancer types. Nat. Genet. 2019, 51, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, V.; Li, C.H.; Bristow, R.G.; Boutros, P.C.; Aaltonen, L.A.; Abascal, F.; Abeshouse, A.; Aburatani, H.; Adams, D.J.; Agrawal, N.; et al. Divergent mutational processes distinguish hypoxic and normoxic tumours. Nat. Commun. 2020, 11, 737. [Google Scholar] [CrossRef] [PubMed]
- Pause, A.; Lee, S.; Lonergan, K.M.; Klausner, R.D. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc. Natl. Acad. Sci. USA 1998, 95, 993–998. [Google Scholar] [CrossRef]
- Trotta, A.M.; Santagata, S.; Zanotta, S.; D’Alterio, C.; Napolitano, M.; Rea, G.; Camerlingo, R.; Esposito, F.; Lamantia, E.; Anniciello, A.; et al. Mutated Von Hippel-Lindau-renal cell carcinoma (RCC) promotes patients specific natural killer (NK) cytotoxicity. J. Exp. Clin. Cancer Res. 2018, 37, 297. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar] [CrossRef]
- Rankin, E.B.; Tomaszewski, J.E.; Haase, V.H. Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 2006, 66, 2576–2583. [Google Scholar] [CrossRef]
- Harlander, S.; Schönenberger, D.; Toussaint, N.C.; Prummer, M.; Catalano, A.; Brandt, L.; Moch, H.; Wild, P.J.; Frew, I.J. Combined mutation in Vhl, Trp53 and Rb1 causes clear cell renal cell carcinoma in mice. Nat. Med. 2017, 23, 869–877. [Google Scholar] [CrossRef]
- Schokrpur, S.; Hu, J.; Moughon, D.L.; Liu, P.; Lin, L.C.; Hermann, K.; Mangul, S.; Guan, W.; Pellegrini, M.; Xu, H.; et al. CRISPR-Mediated VHL Knockout Generates an Improved Model for Metastatic Renal Cell Carcinoma. Sci. Rep. 2016, 6, 29032. [Google Scholar] [CrossRef] [PubMed]
- Sharrow, A.C.; Ishihara, M.; Hu, J.; Kim, I.H.; Wu, L. Using the Chicken Chorioallantoic Membrane In Vivo Model to Study Gynecological and Urological Cancers. J. Vis. Exp. JoVE 2020, 155, e60651. [Google Scholar]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, J.; Smith, D.J.; Wu, L. VHL L169P Variant Does Not Alter Cellular Hypoxia Tension in Clear Cell Renal Cell Carcinoma. Int. J. Mol. Sci. 2023, 24, 14075. https://doi.org/10.3390/ijms241814075
Hu J, Smith DJ, Wu L. VHL L169P Variant Does Not Alter Cellular Hypoxia Tension in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences. 2023; 24(18):14075. https://doi.org/10.3390/ijms241814075
Chicago/Turabian StyleHu, Junhui, Desmond J. Smith, and Lily Wu. 2023. "VHL L169P Variant Does Not Alter Cellular Hypoxia Tension in Clear Cell Renal Cell Carcinoma" International Journal of Molecular Sciences 24, no. 18: 14075. https://doi.org/10.3390/ijms241814075
APA StyleHu, J., Smith, D. J., & Wu, L. (2023). VHL L169P Variant Does Not Alter Cellular Hypoxia Tension in Clear Cell Renal Cell Carcinoma. International Journal of Molecular Sciences, 24(18), 14075. https://doi.org/10.3390/ijms241814075