
AMMVF-DTI: A Novel Model Predicting Drug–Target Interactions Based on Attention Mechanism and Multi-View Fusion


Abstract
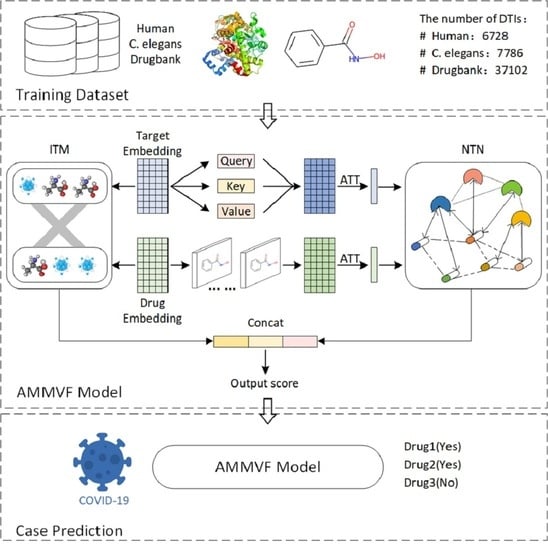
:
1. Introduction
2. Results and Discussion
2.1. Performance on Human, C. elegans, and DrugBank Baseline Datasets
2.2. Ablation Study
2.3. Case Study
2.4. Limitations
3. Materials and Methods
3.1. Datasets
3.2. Methods
3.2.1. BERT Module
3.2.2. GAT Module
3.2.3. ATT Module
3.2.4. ITM Module
3.2.5. NTN Module
3.2.6. MLP Module
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drews, J. Drug discovery: A historical perspective. Science 2000, 287, 1960–1964. [Google Scholar] [CrossRef] [PubMed]
- Hutchins, S.; Torphy, T.; Muller, C. Open partnering of integrated drug discovery: Continuing evolution of the pharmaceutical model. Drug Discov. Today 2011, 16, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Hay, M.; Thomas, D.W.; Craighead, J.L.; Economides, C.; Rosenthal, J. Clinical development success rates for investigational drugs. Nat. Biotechnol. 2014, 32, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, A.; Wu, M.; Li, X.; Kwoh, C. Computational prediction of drug-target interactions using chemogenomic approaches: An empirical survey. Brief Bioinform. 2019, 20, 1337–1357. [Google Scholar] [PubMed]
- Zhan, P.; Yu, B.; Ouyang, L. Drug repurposing: An effective strategy to accelerate contemporary drug discovery. Drug Discov. Today 2022, 27, 1785–1788. [Google Scholar] [CrossRef]
- Sakate, R.; Kimura, T. Drug repositioning trends in rare and intractable diseases. Drug Discov. Today 2022, 27, 1789–1795. [Google Scholar] [CrossRef]
- Kapetanovic, I.M. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Interact. 2008, 171, 165–176. [Google Scholar] [CrossRef]
- Zhang, R.; Wang, Z.; Wang, X.; Meng, Z.; Cui, W. MHTAN-DTI: Metapath-based hierarchical transformer and attention network for drug-target interaction prediction. Brief Bioinform. 2023, 24, 1–11. [Google Scholar] [CrossRef]
- Reddy, A.; Zhang, S. Polypharmacology: Drug discovery for the future. Expert Rev. Clin. Pharmacol. 2014, 6, 41–47. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Frantz, S. Drug discovery: Playing dirty. Nature 2005, 437, 942–943. [Google Scholar] [CrossRef] [PubMed]
- Aronson, J.K. Old drugs—New uses. Br. J. Clin. Pharmacol. 2007, 64, 563–565. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Sullivan, D.J. New uses for old drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef] [PubMed]
- Novac, N. Challenges and opportunities of drug repositioning. Trends Pharmacol. Sci. 2013, 34, 267–272. [Google Scholar] [CrossRef]
- Sharma, K.; Ahmed, F.; Sharma, T.; Grover, A.; Agarwal, M. Grover, S. Potential repurposed drug candidates for tuberculosis treatment: Progress and update of drugs identified in over a decade. ACS Omega 2023, 8, 17362–17380. [Google Scholar] [CrossRef]
- Chong, C.R.; Chen, X.; Shi, L.; Liu, J.O.; Sullivan, D.J. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2006, 2, 415–416. [Google Scholar] [CrossRef]
- Ambrosio, F.A.; Costa, G.; Romeo, I.; Esposito, F.; Alkhatib, M.; Salpini, R.; Svicher, V.; Corona, A.; Malune, P.; Tramontano, E.; et al. Targeting SARS-CoV-2 main protease: A successful story guided by an in silico drug repurposing approach. J. Chem. Inf. Model. 2023, 63, 3601–3613. [Google Scholar] [CrossRef]
- Wang, T.; Wu, M.; Zhang, R.; Chen, Z.; Hua, C.; Lin, J.; Yang, L. Advances in computational structure-based drug design and application in drug discovery. Curr. Top. Med. Chem. 2016, 16, 901–916. [Google Scholar] [CrossRef]
- Acharya, C.; Coop, A.; Polli, J.E.; Mackerell, A.D. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Aided Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Jones, L.H.; Bunnage, M.E. Applications of chemogenomic library screening in drug discovery. Nat. Rev. Drug Discov. 2017, 16, 285–296. [Google Scholar] [CrossRef]
- Wu, Z.; Li, W.; Liu, G.; Tang, Y. Network-based methods for prediction of drug-target interactions. Front. Pharmacol. 2018, 9, 1134. [Google Scholar] [CrossRef] [PubMed]
- Olayan, R.S.; Ashoor, H.; Bajic, V.B. DDR: Efficient computational method to predict drug-target interactions using graph mining and machine learning approaches. Bioinformatics 2018, 34, 1164–1173. [Google Scholar] [CrossRef] [PubMed]
- Thafar, M.A.; Olayan, R.S.; Albaradei, S.; Bajic, V.B.; Gojobori, T.; Essack, M.; Gao, X. DTi2Vec: Drug-target interaction prediction using network embedding and ensemble learning. J. Cheminform. 2021, 13, 71. [Google Scholar] [CrossRef] [PubMed]
- Bagherian, M.; Sabeti, E.; Wang, K.; Sartor, M.A.; Nikolovska-Coleska, Z.; Najarian, K. Machine learning approaches and databases for prediction of drug-target interaction: A survey paper. Brief Bioinform. 2021, 22, 247–269. [Google Scholar] [CrossRef]
- Pillai, N.; Dasgupta, A.; Sudsakorn, S.; Fretland, J.; Mavroudis, P.D. Machine learning guided early drug discovery of small molecules. Drug Discov. Today 2022, 27, 2209–2215. [Google Scholar] [CrossRef]
- Chen, R.; Liu, X.; Jin, S.; Lin, J.; Liu, J. Machine learning for drug-target interaction prediction. Molecules 2018, 23, 2208. [Google Scholar] [CrossRef]
- Yu, J.; Dai, Q.; Li, G. Deep learning in target prediction and drug repositioning: Recent advances and challenges. Drug Discov. Today 2022, 27, 1796–1814. [Google Scholar] [CrossRef]
- Liu, H.; Sun, J.; Guan, J.; Zheng, J.; Zhou, S. Improving compound-protein interaction prediction by building up highly credible negative samples. Bioinformatics 2015, 31, i221–i229. [Google Scholar] [CrossRef]
- Öztürk, H.; Özgür, A.; Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 2018, 34, i821–i829. [Google Scholar] [CrossRef]
- Karimi, M.; Wu, D.; Wang, Z.; Shen, Y. DeepAffinity: Interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks. Bioinformatics 2019, 35, 3329–3338. [Google Scholar] [CrossRef]
- Chen, L.; Tan, X.; Wang, D.; Zhong, F.; Liu, X.; Yang, T.; Luo, X.; Chen, K.; Jiang, H.; Zheng, M. TransformerCPI: Improving compound-protein interaction prediction by sequence-based deep learning with self-attention mechanism and label reversal experiments. Bioinformatics 2020, 36, 4406–4414. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Yan, C.; Wu, F.X.; Wang, J. Drug-target interaction prediction using multi-head self-attention and graph attention network. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 2208–2218. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Zeng, Y.; Liu, H.; Liu, Z.; Liu, X. DeepMHADTA: Prediction of drug-target binding affinity using multi-head self-attention and convolutional neural network. Curr. Issues Mol. Biol. 2022, 44, 2287–2299. [Google Scholar] [CrossRef]
- Wang, S.; Song, X.; Zhang, Y.; Zhang, K.; Liu, Y.; Ren, C.; Pang, S. MSGNN-DTA: Multi-Scale Topological Feature Fusion Based on Graph Neural Networks for Drug-Target Binding Affinity Prediction. Int. J. Mol. Sci. 2023, 24, 8326. [Google Scholar] [CrossRef]
- Zhao, Q.; Duan, G.; Yang, M.; Cheng, Z.; Li, Y.; Wang, J. AttentionDTA: Drug-target binding affinity prediction by sequence-based deep learning with attention mechanism. IEEE/ACM Trans. Comput. Biol. Bioinform. 2023, 20, 852–863. [Google Scholar] [CrossRef]
- Chen, W.; Chen, G.; Zhao, L.; Chen, C.Y. Predicting drug-target interactions with deep-embedding learning of graphs and sequences. J. Phys. Chem. A 2021, 125, 5633–5642. [Google Scholar] [CrossRef]
- Koshland, D.E. The key-lock theory and the induced fit theory. Angew. Chem. Int. Ed. Engl. 1994, 33, 2375–2378. [Google Scholar] [CrossRef]
- Glorot, X.; Bengio, Y. Understanding the difficulty of training deep feedforward neural networks. In Proceedings of the Thirteenth International Conference on Artificial Intelligence and Statistics, Sardinia, Italy, 13–15 May 2010; pp. 249–256. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Wei, L.; Long, W.; Wei, L. MDL-CPI: Multi-view deep learning model for compound-protein interaction prediction. Methods 2022, 204, 418–427. [Google Scholar] [CrossRef]
- Tsubaki, M.; Tomii, K.; Sese, J. Compound-protein interaction prediction with end-to-end learning of neural networks for graphs and sequences. Bioinformatics 2018, 35, 309–318. [Google Scholar] [CrossRef]
- Nguyen, T.; Le, H.; Quinn, T.P.; Nguyen, T.; Le, T.D.; Venkatesh, S. GraphDTA: Predicting drug-target binding affinity with graph neural networks. Bioinformatics 2021, 37, 1140–1147. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Li, Y.; Chen, S.; Xu, J.; Yang, Y. Predicting drug-protein interaction using quasi-visual question answering system. Nat. Mach. Intell. 2020, 2, 134–140. [Google Scholar] [CrossRef]
- Lee, I.; Nam, H. Identification of drug-target interaction by a random walk with restart method on an interactome network. BMC Bioinf. 2018, 19, 208. [Google Scholar] [CrossRef]
- Yuan, Q.; Gao, J.; Wu, D.; Zhang, S.; Mamitsuka, H.; Zhu, S. DrugE-rank: Improving drug-target interaction prediction of new candidate drugs or targets by ensemble learning to rank. Bioinformatics 2016, 32, i18–i27. [Google Scholar] [CrossRef] [PubMed]
- Wan, F.; Zhu, Y.; Hu, H.; Dai, A.; Cai, X.; Chen, L.; Gong, H.; Xia, T.; Yang, D.; Wang, M.; et al. DeepCPI: A deep learning-based framework for large-scale in silico drug screening. Genom. Proteom. Bioinf. 2019, 17, 478–495. [Google Scholar] [CrossRef]
- Lee, I.; Keum, J.; Nam, H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS Comput. Biol. 2019, 15, e1007129. [Google Scholar] [CrossRef]
- Qian, Y.; Wu, J.; Zhang, Q. CAT-CPI: Combining CNN and transformer to learn compound image features for predicting compound-protein interactions. Front. Mol. Biosci. 2022, 9, 963912. [Google Scholar] [CrossRef]
- Zhan, Y.; Chen, B. Drug target identification and drug repurposing in Psoriasis through systems biology approach, DNN-based DTI model and genome-wide microarray data. Int. J. Mol. Sci. 2023, 24, 10033. [Google Scholar] [CrossRef]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2008, 36, D901–D906. [Google Scholar] [CrossRef]
- Genovese, M.C.; Kremer, J.; Zamani, O.; Ludivico, C.; Krogulec, M.; Xie, L.; Beattie, S.D.; Koch, A.E.; Cardillo, T.E.; Rooney, T.P.; et al. Baricitinib in patients with refractory rheumatoid arthritis. N. Engl. J. Med. 2016, 374, 1243–1252. [Google Scholar] [CrossRef]
- Kuriya, B.; Cohen, M.D.; Keystone, E. Baricitinib in rheumatoid arthritis: Evidence-to-date and clinical potential. Ther. Adv. Musculoskelet Dis. 2017, 9, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Mayence, A.; Vanden Eynde, J.J. Baricitinib: A 2018 novel FDA-approved small molecule inhibiting janus kinases. Pharmaceuticals 2019, 12, 37. [Google Scholar] [CrossRef]
- Yang, Z.; Zhong, W.; Zhao, L.; Chen, C.Y. MGraphDTA: Deep multiscale graph neural network for explainable drug-target binding affinity prediction. Chem. Sci. 2022, 13, 816–833. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Zhong, W.; Lv, Q.; Dong, T.; Chen, C.Y. Geometric interaction graph neural network for predicting protein-ligand binding affinities from 3D structures (GIGN). J. Phys. Chem. Lett. 2023, 14, 2020–2033. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Takigawa, I.; Mamitsuka, H.; Zhu, S. Similarity-based machine learning methods for predicting drug-target interactions: A brief review. Brief Bioinform. 2013, 15, 734–747. [Google Scholar] [CrossRef]
- Günther, S.; Kuhn, M.; Dunkel, M.; Campillos, M.; Senger, C.; Petsalaki, E.; Ahmed, J.; Urdiales, E.G.; Gewiess, A.; Jensen, L.J.; et al. SuperTarget and Matador: Resources for exploring drug-target relationships. Nucleic Acids Res. 2008, 36, D919–D922. [Google Scholar] [CrossRef]
- Bai, Y.; Ding, H.; Bian, S.; Chen, T.; Sun, Y.; Wang, W. SimGNN: A neural network approach to fast graph similarity computation. In Proceedings of the twelfth ACM International Conference on Web Search and Data Mining, WSDM ’19, Melbourne, VIC, Australia, 11–15 February 2019; pp. 384–392. [Google Scholar]
- Mikolov, T.; Chen, K.; Corrado, G.; Dean, J. Efficient estimation of word representations in vector space. In Proceedings of the 1st International Conference on Learning Representations, ICLR 2013, Scottsdale, AZ, USA, 2–4 May 2013. [Google Scholar]
- Devlin, J.; Chang, M.W.; Lee, K.; Toutanova, K. BERT: Pre-training of deep bidirectional transformers for language understanding. In Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Volume 1 (Long and Short Papers), Minneapolis, MN, USA, 2–7 June 2019; Association for Computational Linguistics: Minneapolis, MN, USA, 2019; pp. 4171–4186. [Google Scholar]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł; Polosukhin, I. Attention is all you need. In Proceedings of the 31st Conference on Neural Information Processing Systems, NIPS 2017, Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Kang, H.; Goo, S.; Lee, H.; Chae, J.; Yun, H.; Jung, S. Fine-tuning of BERT model to accurately predict drug-target interactions. Pharmaceutics 2022, 14, 1710. [Google Scholar] [CrossRef]
- Kipf, T.N.; Welling, M. Semi-supervised classification with graph convolutional networks. arXiv 2016, arXiv:1609.02907. [Google Scholar]
- Klicpera, J.; Bojchevski, A.; Günnemann, S. Predict then propagate: Graph neural networks meet personalized pagerank. arXiv 2018, arXiv:1810.05997. [Google Scholar]
- Xu, K.; Hu, W.; Leskovec, J.; Jegelka, S. How powerful are graph neural networks. arXiv 2018, arXiv:1810.00826. [Google Scholar]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Delving deep into rectifiers: Surpassing human-level performance on imagenet classification. In Proceedings of the IEEE International Conference on Computer Vision, Santiago, Chile, 7–13 December 2015; pp. 1026–1034. [Google Scholar]
- Li, F.; Zhang, Z.; Guan, J.; Zhou, S. Effective drug-target interaction prediction with mutual interaction neural network. Bioinformatics 2022, 38, 3582–3589. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Fan, Q.; Panda, R. CrossViT: Cross-attention multi-scale vision transformer for image classification. In Proceedings of the IEEE/CVF International Conference on Computer Vision (ICCV), Montreal, QC, Canada, 10–17 October 2021; pp. 347–356. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Hyperparameter | Value |
|---|---|
| Epoch | 40 |
| Dropout | 0.1 |
| Learning rate | 1 × 10−3 |
| Regularization coefficient | 1 × 10−4 |
| The radius | 2 |
| The n-gram | 3 |
| The number of major potential associations K | 16 |
| The dimensions of the hidden layer | 64 |
| The number of GAT layers | 3 |
| The number of multi-head self-attention | 8 |
| Model | AUC | Precision | Recall |
|---|---|---|---|
| KNN | 0.860 | 0.927 | 0.798 |
| RF | 0.940 | 0.897 | 0.861 |
| L2 | 0.911 | 0.913 | 0.867 |
| SVM | 0.910 | 0.966 | 0.969 |
| MDL-CPI | 0.959 | 0.924 | 0.905 |
| GNN | 0.970 | 0.918 | 0.923 |
| GCN | 0.956 ± 0.004 | 0.862 ± 0.006 | 0.928 ± 0.010 |
| GraphDTA | 0.960 ± 0.005 | 0.882 ± 0.040 | 0.912 ± 0.040 |
| DrugVQA (VQA-seq) | 0.964 ± 0.005 | 0.897 ± 0.004 | 0.948 ± 0.003 |
| TransformerCPI | 0.973 ± 0.002 | 0.916 ± 0.006 | 0.925 ± 0.006 |
| AMMVF-DTI (this work) | 0.986 | 0.976 | 0.938 |
| Model | AUC | Precision | Recall |
|---|---|---|---|
| KNN | 0.858 | 0.801 | 0.827 |
| RF | 0.902 | 0.821 | 0.844 |
| L2 | 0.892 | 0.890 | 0.877 |
| SVM | 0.894 | 0.785 | 0.818 |
| MDL-CPI | 0.975 | 0.943 | 0.923 |
| GNN | 0.978 | 0.938 | 0.929 |
| GCN | 0.975 ± 0.004 | 0.921 ± 0.008 | 0.927 ± 0.006 |
| GraphDTA | 0.974 ± 0.004 | 0.927 ± 0.015 | 0.912 ± 0.023 |
| TransformerCPI | 0.988 ± 0.002 | 0.952 ± 0.006 | 0.953 ± 0.005 |
| AMMVF-DTI (this work) | 0.990 | 0.962 | 0.960 |
| Model | AUC | Precision | Recall |
|---|---|---|---|
| RWR | 0.7595 | 0.7046 | 0.6511 |
| DrugE-Rank | 0.7591 | 0.7070 | 0.6289 |
| DeepConv-DTI | 0.8531 | 0.7891 | 0.7385 |
| DeepCPI | 0.7003 | 0.7006 | 0.5563 |
| MHSADTI | 0.8628 | 0.7706 | 0.7918 |
| AMMVF-DTI (this work) | 0.9570 | 0.9034 | 0.9084 |
| Human | C. elegans | DrugBank | |
|---|---|---|---|
| Number of drugs | 1052 | 1434 | 6707 |
| Number of target proteins | 852 | 2504 | 4794 |
| Number of total samples | 6728 | 7786 | 37,102 |
| Number of positive interactions | 3364 | 3893 | 18,398 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Zhou, Y.; Chen, Q. AMMVF-DTI: A Novel Model Predicting Drug–Target Interactions Based on Attention Mechanism and Multi-View Fusion. Int. J. Mol. Sci. 2023, 24, 14142. https://doi.org/10.3390/ijms241814142
Wang L, Zhou Y, Chen Q. AMMVF-DTI: A Novel Model Predicting Drug–Target Interactions Based on Attention Mechanism and Multi-View Fusion. International Journal of Molecular Sciences. 2023; 24(18):14142. https://doi.org/10.3390/ijms241814142
Chicago/Turabian StyleWang, Lu, Yifeng Zhou, and Qu Chen. 2023. "AMMVF-DTI: A Novel Model Predicting Drug–Target Interactions Based on Attention Mechanism and Multi-View Fusion" International Journal of Molecular Sciences 24, no. 18: 14142. https://doi.org/10.3390/ijms241814142
APA StyleWang, L., Zhou, Y., & Chen, Q. (2023). AMMVF-DTI: A Novel Model Predicting Drug–Target Interactions Based on Attention Mechanism and Multi-View Fusion. International Journal of Molecular Sciences, 24(18), 14142. https://doi.org/10.3390/ijms241814142