
Identification of Two Flip-Over Genes in Grass Family as Potential Signature of C4 Photosynthesis Evolution


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
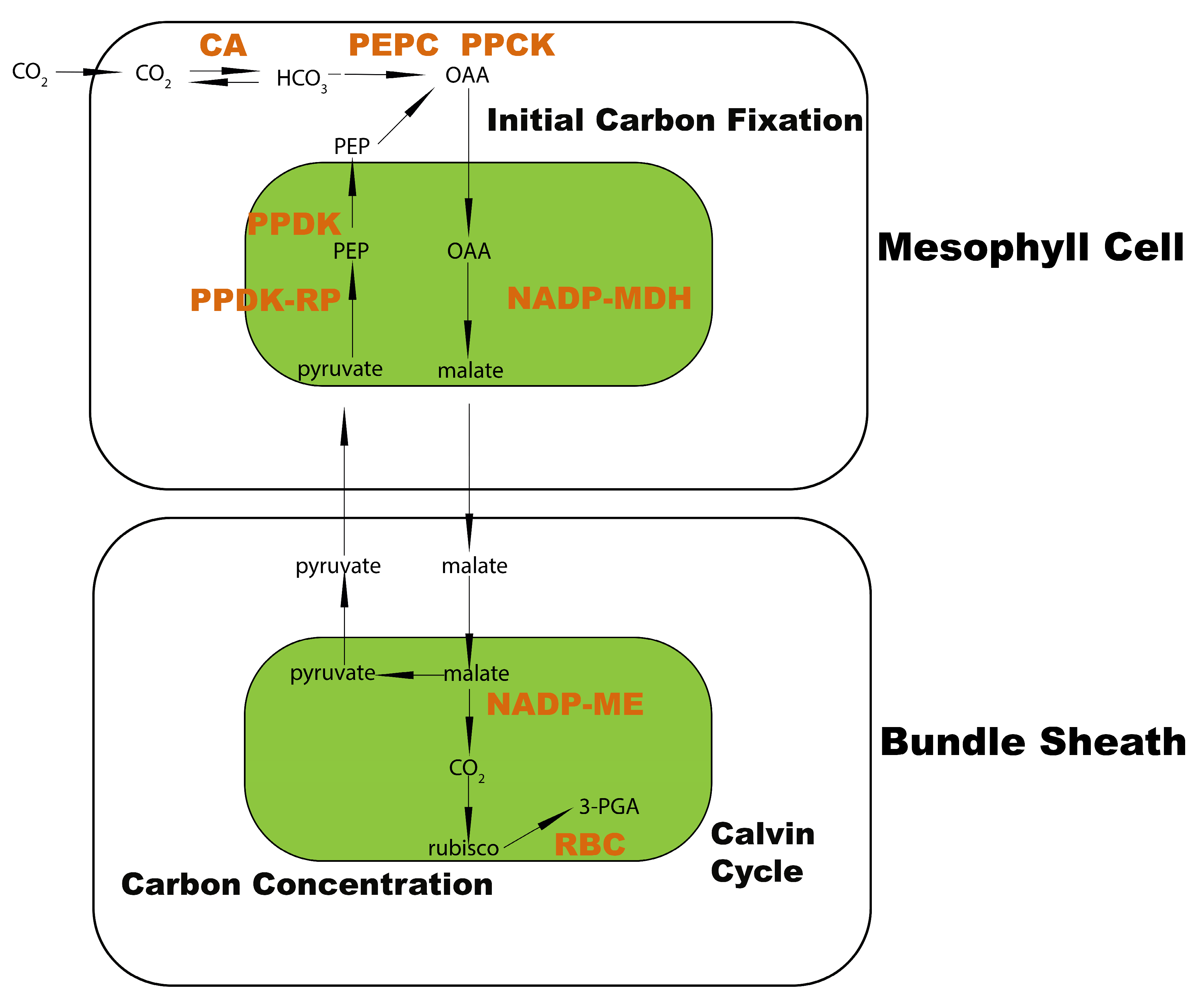
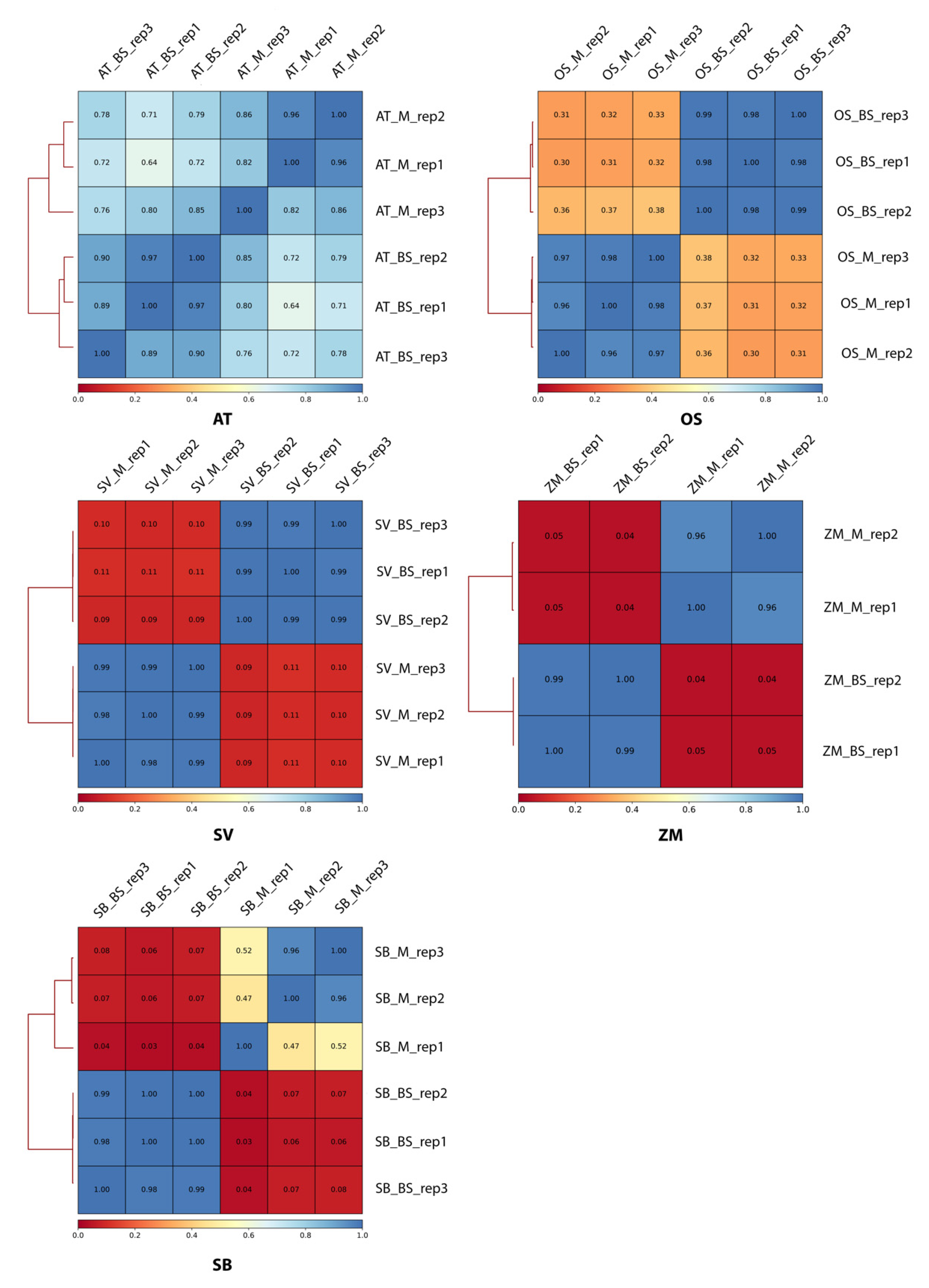
2.1. Transcriptome Divergence between C3 and C4 Plants
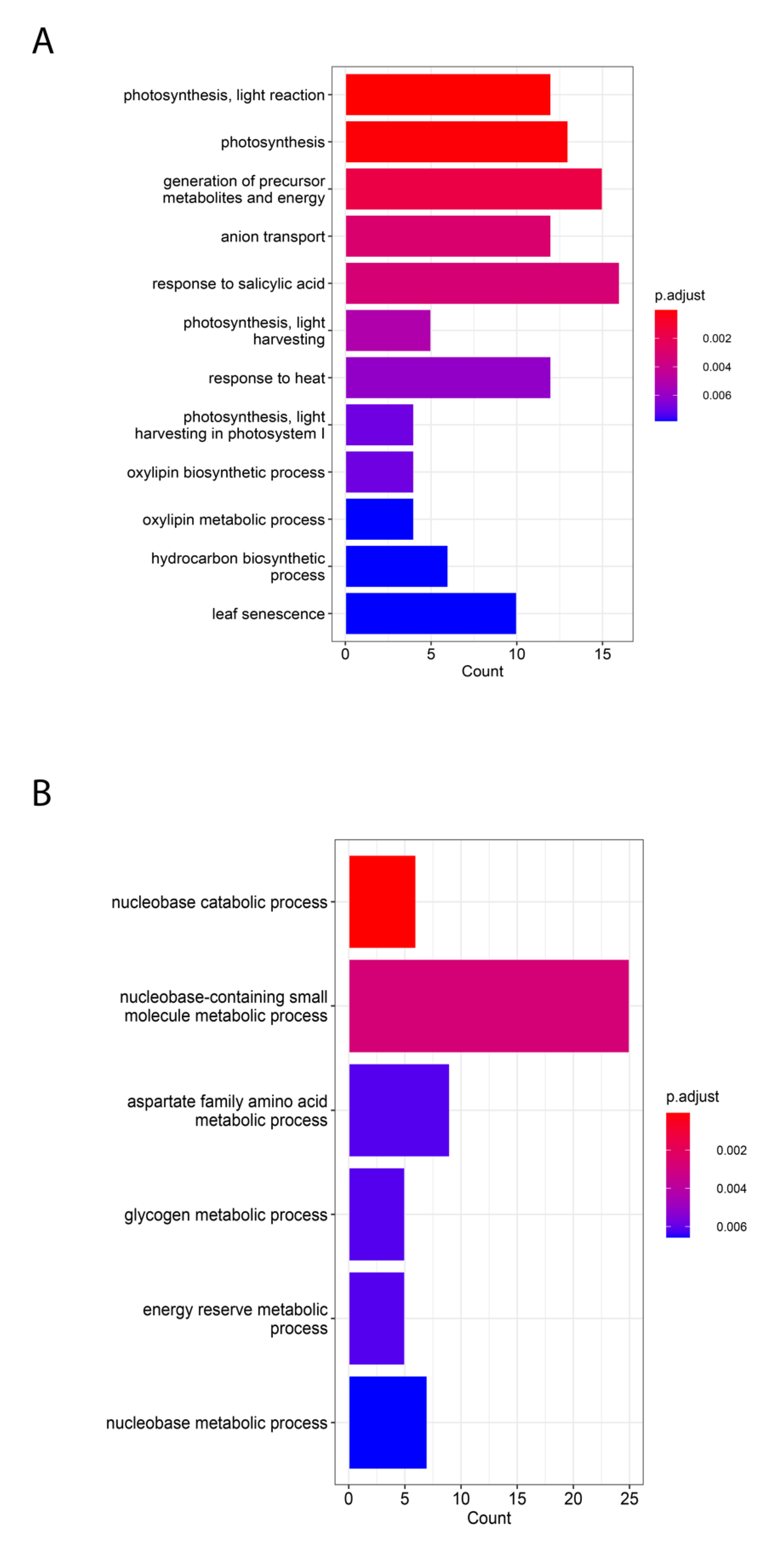
2.2. Common and Specific Pathways Enriched in C3 and C4 Plants
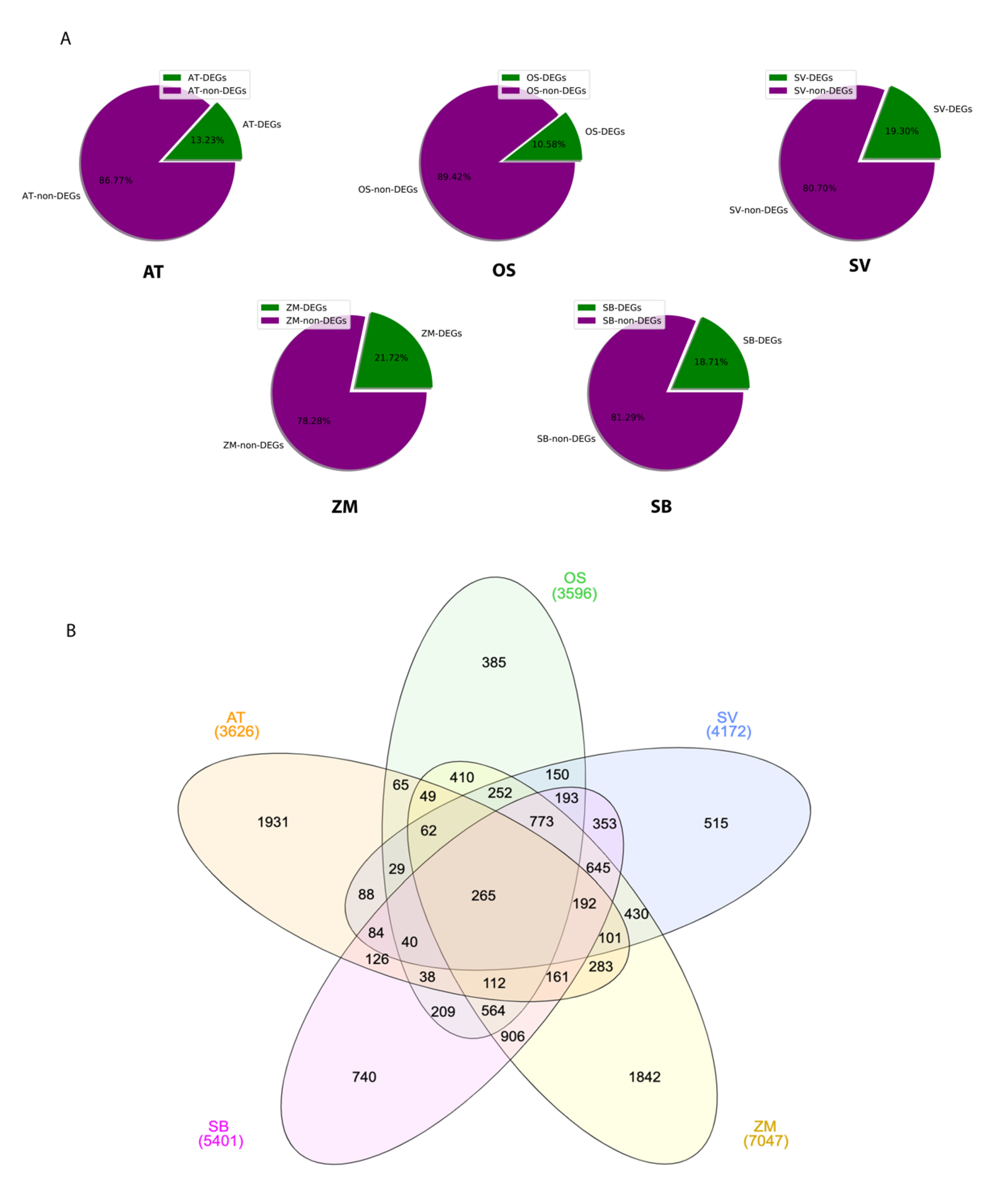
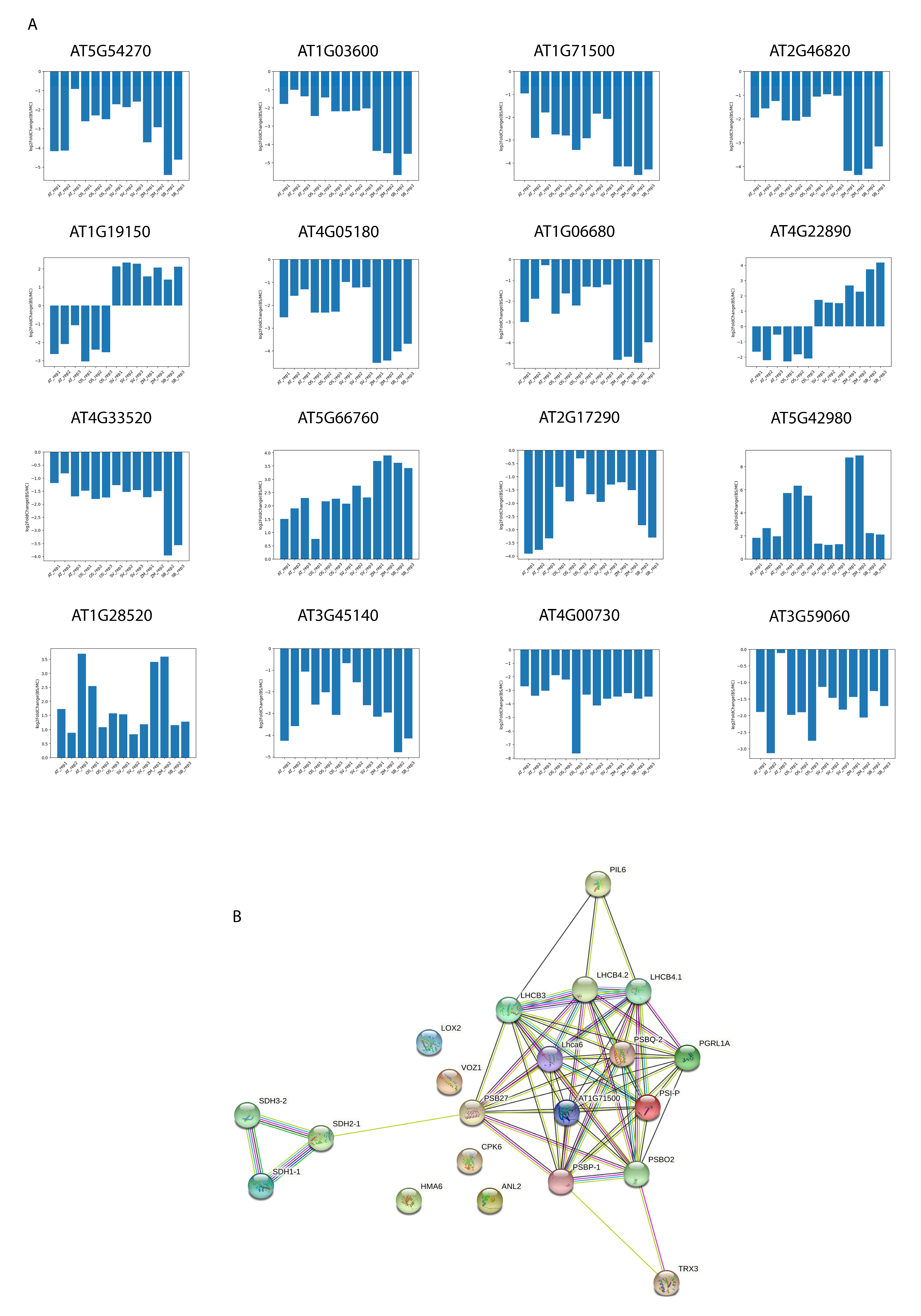
2.3. Transcriptome Signature Discovery in C3 and C4 Plants
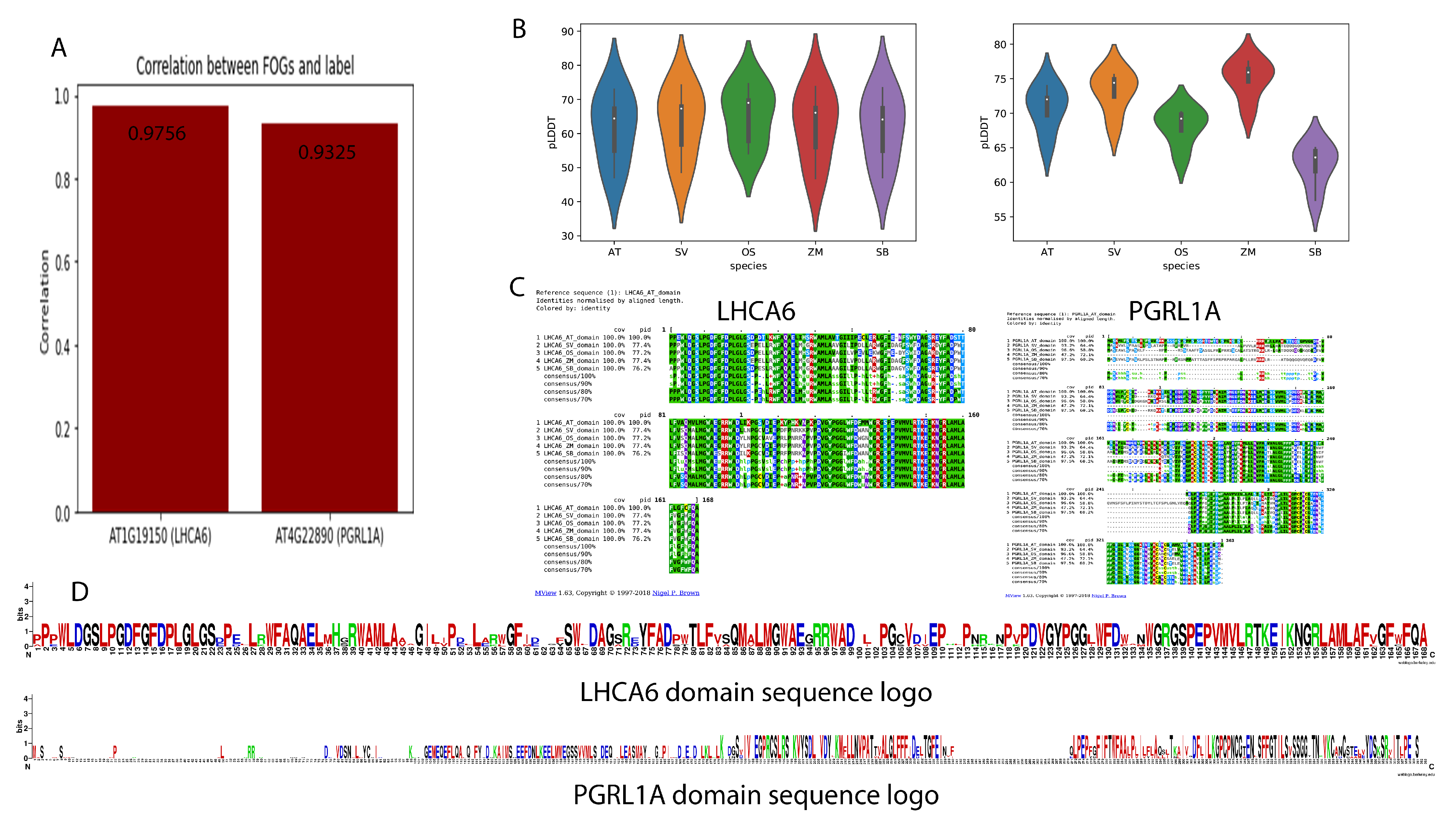
2.4. Domain Analysis and Protein Folding Prediction of Two FOGs
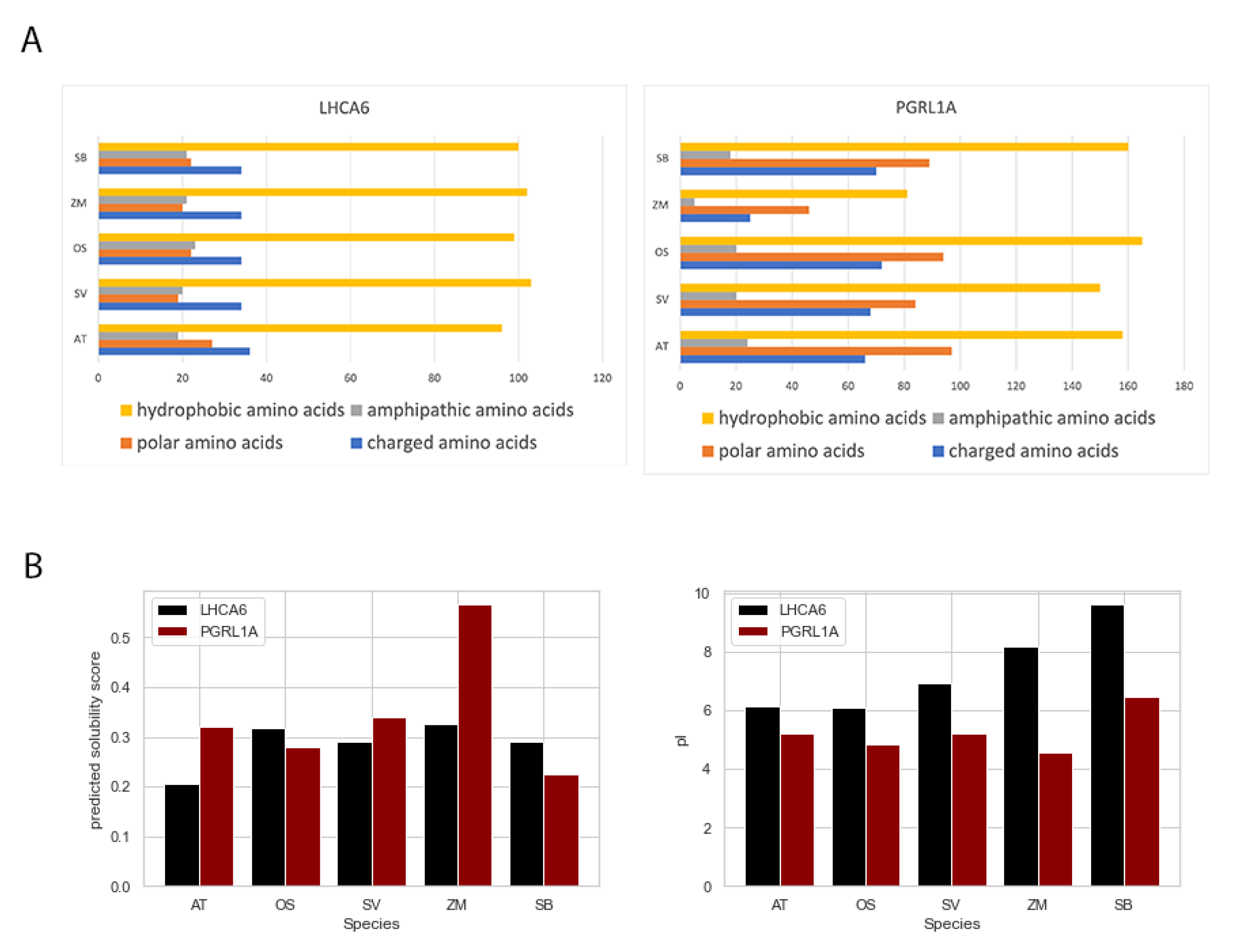
2.5. Amino Acids Composition Analysis and Protein Solubility Prediction of FOGs
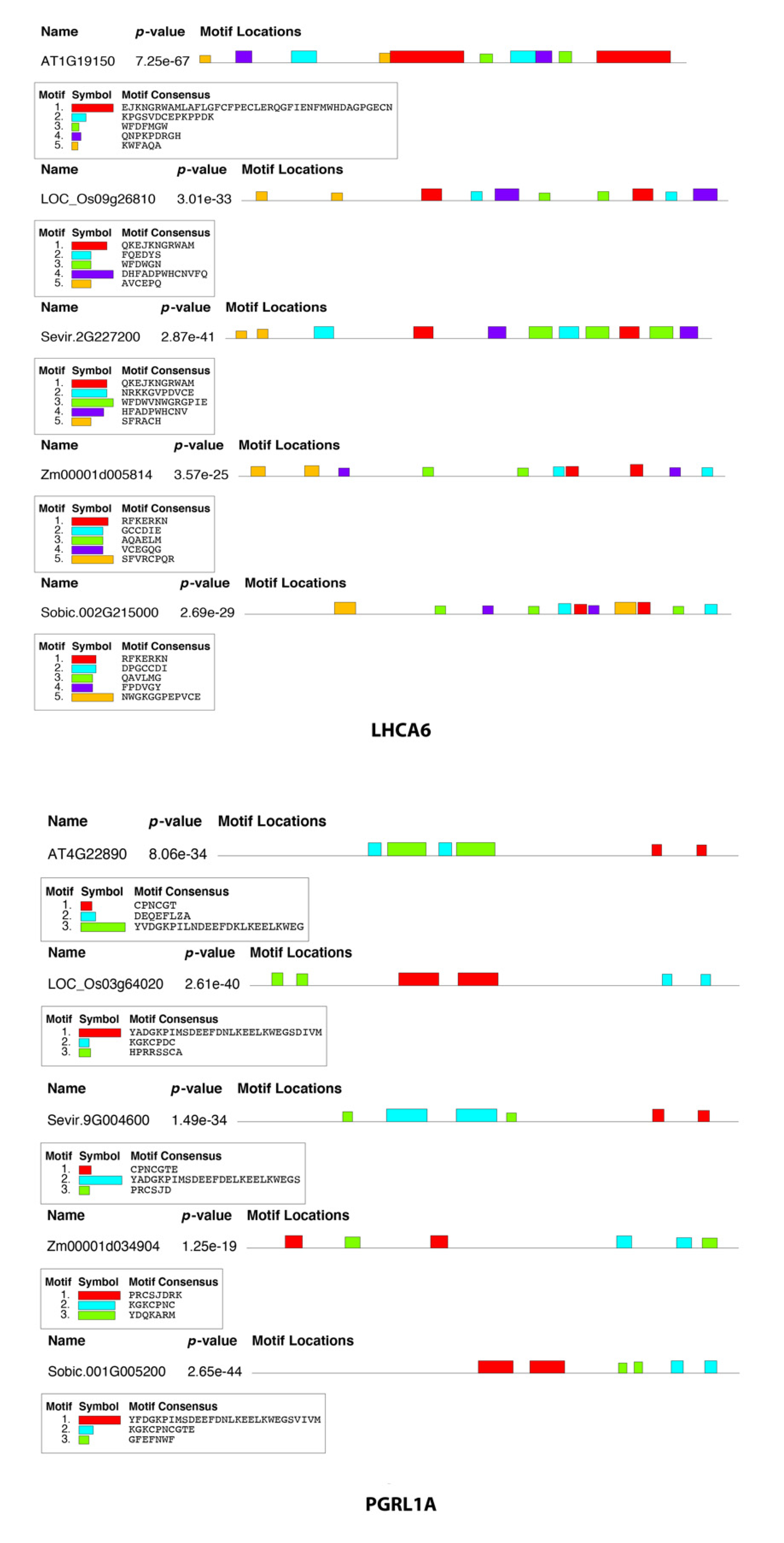
2.6. Sequence Motif Discovery in FOGs
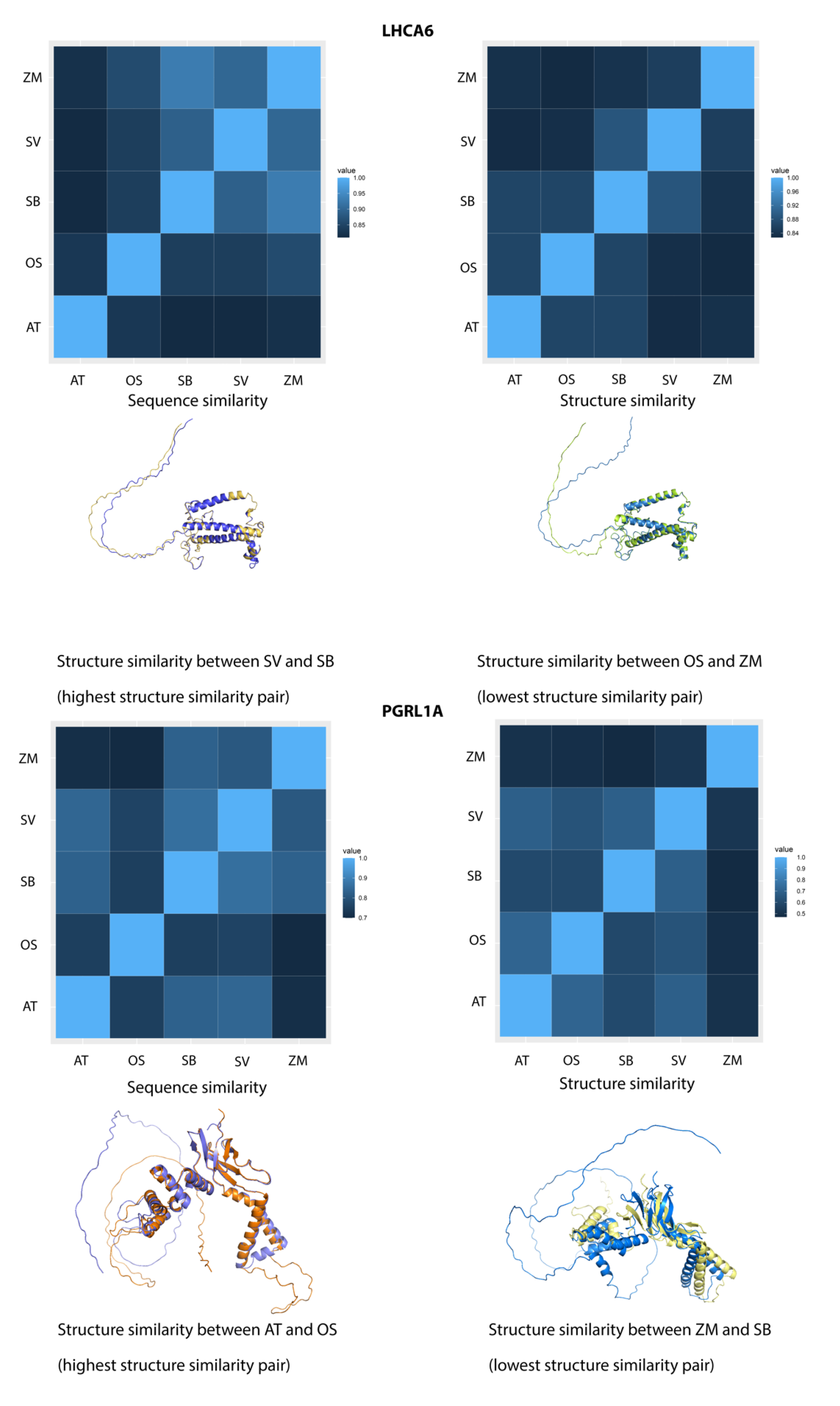
2.7. Sequence and Structure Similarities Comparison of FOG Proteins
3. Discussion
4. Perspective
5. Materials and Methods
5.1. Comparative Transcriptome Analysis between C3 and C4 Plants
5.2. Protein-Protein Interaction Prediction of C3/C4 Transcriptome Signature Genes
5.3. Multiple Sequence Alignment for Specific Domains and FOGs Expression Pattern
5.4. Domain Amino Acids Composition Analysis, Protein Solubility Prediction, and Global Motif Discovery
5.5. Three-Dimensional Structure Prediction and Sequence Structure Similarity Comparison
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sage, R.F. The evolution of C 4 photosynthesis. New Phytol. 2004, 161, 341–370. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Czedik-Eysenberg, A.; Mertz, R.A.; Si, Y.; Tohge, T.; Nunes-Nesi, A.; Arrivault, S.; Dedow, L.K.; Bryant, D.W.; Zhou, W.; et al. Comparative analyses of C4 and C3 photosynthesis in developing leaves of maize and rice. Nat. Biotechnol. 2014, 32, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Way, D.A.; Katul, G.G.; Manzoni, S.; Vico, G. Increasing water use efficiency along the C3 to C4 evolutionary pathway: A stomatal optimization perspective. J. Exp. Bot. 2014, 65, 3683–3693. [Google Scholar] [CrossRef]
- Killi, D.; Bussotti, F.; Raschi, A.; Haworth, M. Adaptation to high temperature mitigates the impact of water deficit during combined heat and drought stress in C3 sunflower and C4 maize varieties with contrasting drought tolerance. Physiol. Plant 2017, 159, 130–147. [Google Scholar] [CrossRef]
- Lopez-Hernandez, F.; Cortes, A.J. Last-Generation Genome-Environment Associations Reveal the Genetic Basis of Heat Tolerance in Common Bean (Phaseolus vulgaris L.). Front. Genet. 2019, 10, 954. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.J.; Blair, M.W. Genotyping by Sequencing and Genome-Environment Associations in Wild Common Bean Predict Widespread Divergent Adaptation to Drought. Front. Plant Sci. 2018, 9, 128. [Google Scholar] [CrossRef]
- Cortes, A.J.; Skeen, P.; Blair, M.W.; Chacon-Sanchez, M.I. Does the Genomic Landscape of Species Divergence in Phaseolus Beans Coerce Parallel Signatures of Adaptation and Domestication? Front. Plant Sci. 2018, 9, 1816. [Google Scholar] [CrossRef]
- Blair, M.W.; Cortes, A.J.; This, D. Identification of an ERECTA gene and its drought adaptation associations with wild and cultivated common bean. Plant Sci. 2016, 242, 250–259. [Google Scholar] [CrossRef]
- Cortés, A.J.; Chavarro, M.C.; Madriñán, S.; This, D.; Blair, M.W. Molecular ecology and selection in the drought-related Asr gene polymorphisms in wild and cultivated common bean (Phaseolus vulgaris L.). BMC Genom. Data 2012, 13, 58. [Google Scholar] [CrossRef]
- Cortes, A.J.; This, D.; Chavarro, C.; Madrinan, S.; Blair, M.W. Nucleotide diversity patterns at the drought-related DREB2 encoding genes in wild and cultivated common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 2012, 125, 1069–1085. [Google Scholar] [CrossRef]
- Buitrago-Bitar, M.A.; Cortes, A.J.; Lopez-Hernandez, F.; Londono-Caicedo, J.M.; Munoz-Florez, J.E.; Munoz, L.C.; Blair, M.W. Allelic Diversity at Abiotic Stress Responsive Genes in Relationship to Ecological Drought Indices for Cultivated Tepary Bean, Phaseolus acutifolius A. Gray, and Its Wild Relatives. Genes 2021, 12, 556. [Google Scholar] [CrossRef] [PubMed]
- Brutnell, T.P.; Wang, L.; Swartwood, K.; Goldschmidt, A.; Jackson, D.; Zhu, X.-G.; Kellogg, E.; Van Eck, J. Setaria viridis: A Model for C4 Photosynthesis. Plant Cell 2010, 22, 2537–2544. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, P.; Li, J.; Wei, S.; Yan, Y.; Yang, J.; Zhao, M.; Langdale, J.A.; Zhou, W. Maize GOLDEN2-LIKE genes enhance biomass and grain yields in rice by improving photosynthesis and reducing photoinhibition. Commun. Biol. 2020, 3, 151. [Google Scholar] [CrossRef]
- Wang, P.; Fouracre, J.; Kelly, S.; Karki, S.; Gowik, U.; Aubry, S.; Shaw, M.K.; Westhoff, P.; Slamet-Loedin, I.H.; Quick, W.P.; et al. Evolution of GOLDEN2-LIKE gene function in C(3) and C(4) plants. Planta 2013, 237, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Reeves, G.; Grange-Guermente, M.J.; Hibberd, J.M. Regulatory gateways for cell-specific gene expression in C4 leaves with Kranz anatomy. J. Exp. Bot. 2017, 68, 107–116. [Google Scholar] [CrossRef]
- Majeran, W.; Cai, Y.; Sun, Q.; van Wijk, K.J. Functional differentiation of bundle sheath and mesophyll maize chloroplasts determined by comparative proteomics. Plant Cell 2005, 17, 3111–3140. [Google Scholar] [CrossRef]
- Leegood, R.C. Roles of the bundle sheath cells in leaves of C3 plants. J. Exp. Bot. 2007, 59, 1663–1673. [Google Scholar] [CrossRef]
- Sinha, N.R.; Kellogg, E.A. Parallelism and diversity in multiple origins of c4photosynthesis in the grass family. Am. J. Bot. 1996, 83, 1458–1470. [Google Scholar] [CrossRef]
- Sawers, R.J.; Liu, P.; Anufrikova, K.; Hwang, J.T.; Brutnell, T.P. A multi-treatment experimental system to examine photosynthetic differentiation in the maize leaf. BMC Genom. 2007, 8, 12. [Google Scholar] [CrossRef]
- Li, P.; Ponnala, L.; Gandotra, N.; Wang, L.; Si, Y.; Tausta, S.L.; Kebrom, T.H.; Provart, N.; Patel, R.; Myers, C.R.; et al. The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 2010, 42, 1060–1067. [Google Scholar] [CrossRef]
- Ding, Z.; Weissmann, S.; Wang, M.; Du, B.; Huang, L.; Wang, L.; Tu, X.; Zhong, S.; Myers, C.; Brutnell, T.P.; et al. Identification of Photosynthesis-Associated C4 Candidate Genes through Comparative Leaf Gradient Transcriptome in Multiple Lineages of C3 and C4 Species. PLoS ONE 2015, 10, e0140629. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Brutnell, T.P. A synthesis of transcriptomic surveys to dissect the genetic basis of C4 photosynthesis. Curr. Opin. Plant Biol. 2016, 31, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Guo, D. Computational Docking Reveals Co-Evolution of C4 Carbon Delivery Enzymes in Diverse Plants. Int. J. Mol. Sci. 2022, 23, 12688. [Google Scholar] [CrossRef] [PubMed]
- Berkowitz, O.; Xu, Y.; Liew, L.C.; Wang, Y.; Zhu, Y.; Hurgobin, B.; Lewsey, M.G.; Whelan, J. RNA-seq analysis of laser microdissected Arabidopsis thaliana leaf epidermis, mesophyll and vasculature defines tissue-specific transcriptional responses to multiple stress treatments. Plant J. 2021, 107, 938–955. [Google Scholar] [CrossRef]
- Hua, L.; Stevenson, S.R.; Reyna-Llorens, I.; Xiong, H.; Kopriva, S.; Hibberd, J.M. The bundle sheath of rice is conditioned to play an active role in water transport as well as sulfur assimilation and jasmonic acid synthesis. Plant J. 2021, 107, 268–286. [Google Scholar] [CrossRef]
- Aubry, S.; Kelly, S.; Kumpers, B.M.; Smith-Unna, R.D.; Hibberd, J.M. Deep evolutionary comparison of gene expression identifies parallel recruitment of trans-factors in two independent origins of C4 photosynthesis. PLoS Genet. 2014, 10, e1004365. [Google Scholar] [CrossRef]
- Madhok, A.; Bhat, S.A.; Philip, C.S.; Sureshbabu, S.K.; Chiplunkar, S.; Galande, S. Transcriptome Signature of Vgamma9Vdelta2 T Cells Treated With Phosphoantigens and Notch Inhibitor Reveals Interplay Between TCR and Notch Signaling Pathways. Front. Immunol. 2021, 12, 660361. [Google Scholar] [CrossRef]
- Casella, G.; Munk, R.; Kim, K.M.; Piao, Y.; De, S.; Abdelmohsen, K.; Gorospe, M. Transcriptome signature of cellular senescence. Nucleic Acids Res. 2019, 47, 7294–7305. [Google Scholar] [CrossRef]
- Liang, P.; Yang, W.; Chen, X.; Long, C.; Zheng, L.; Li, H.; Zuo, Y. Machine Learning of Single-Cell Transcriptome Highly Identifies mRNA Signature by Comparing F-Score Selection with DGE Analysis. Mol. Ther. Nucleic Acids 2020, 20, 155–163. [Google Scholar] [CrossRef]
- Li, R.; Zhu, J.; Zhong, W.D.; Jia, Z. Comprehensive Evaluation of Machine Learning Models and Gene Expression Signatures for Prostate Cancer Prognosis Using Large Population Cohorts. Cancer Res. 2022, 82, 1832–1843. [Google Scholar] [CrossRef]
- Kong, S.W.; Collins, C.D.; Shimizu-Motohashi, Y.; Holm, I.A.; Campbell, M.G.; Lee, I.H.; Brewster, S.J.; Hanson, E.; Harris, H.K.; Lowe, K.R.; et al. Characteristics and predictive value of blood transcriptome signature in males with autism spectrum disorders. PLoS ONE 2012, 7, e49475. [Google Scholar] [CrossRef] [PubMed]
- Aubry, S.; Aresheva, O.; Reyna-Llorens, I.; Smith-Unna, R.D.; Hibberd, J.M.; Genty, B. A Specific Transcriptome Signature for Guard Cells from the C4 Plant Gynandropsis gynandra. Plant Physiol. 2016, 170, 1345–1357. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, R.N., Jr.; Poelwijk, F.J.; Raman, A.; Gosal, W.S.; Ranganathan, R. The spatial architecture of protein function and adaptation. Nature 2012, 491, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V. Fold change in evolution of protein structures. J. Struct. Biol. 2001, 134, 167–185. [Google Scholar] [CrossRef]
- Shi, Y. A glimpse of structural biology through X-ray crystallography. Cell 2014, 159, 995–1014. [Google Scholar] [CrossRef]
- Fowler, N.J.; Sljoka, A.; Williamson, M.P. A method for validating the accuracy of NMR protein structures. Nat. Commun. 2020, 11, 6321. [Google Scholar] [CrossRef]
- Nogales, E.; Scheres, S.H. Cryo-EM: A Unique Tool for the Visualization of Macromolecular Complexity. Mol. Cell 2015, 58, 677–689. [Google Scholar] [CrossRef]
- Todd, A.E.; Orengo, C.A.; Thornton, J.M. Evolution of function in protein superfamilies, from a structural perspective. J. Mol. Biol. 2001, 307, 1113–1143. [Google Scholar] [CrossRef]
- Yang, J.; Anishchenko, I.; Park, H.; Peng, Z.; Ovchinnikov, S.; Baker, D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. USA 2020, 117, 1496–1503. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Rives, A.; Meier, J.; Sercu, T.; Goyal, S.; Lin, Z.; Liu, J.; Guo, D.; Ott, M.; Zitnick, C.L.; Ma, J.; et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. USA 2021, 118, e2016239118. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Goulet, A.; Cambillau, C. Structure and Topology Prediction of Phage Adhesion Devices Using AlphaFold2: The Case of Two Oenococcus oeni Phages. Microorganisms 2021, 9, 2151. [Google Scholar] [CrossRef]
- Tsaban, T.; Varga, J.K.; Avraham, O.; Ben-Aharon, Z.; Khramushin, A.; Schueler-Furman, O. Harnessing protein folding neural networks for peptide-protein docking. Nat. Commun. 2022, 13, 176. [Google Scholar] [CrossRef]
- Krissinel, E. On the relationship between sequence and structure similarities in proteomics. Bioinformatics 2007, 23, 717–723. [Google Scholar] [CrossRef]
- Chothia, C.; Lesk, A.M. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986, 5, 823–826. [Google Scholar] [CrossRef]
- Murzin, A.G. How far divergent evolution goes in proteins. Curr. Opin. Struct. Biol. 1998, 8, 380–387. [Google Scholar] [CrossRef]
- Todd, C.; Wood, W.R.P. Evolution of Protein Sequences and Structures. J. Mol. Biol. 1999, 291, 977–995. [Google Scholar]
- Koehl, P.; Levitt, M. Sequence variations within protein families are linearly related to structural variations. J. Mol. Biol. 2002, 323, 551–562. [Google Scholar] [CrossRef]
- He, Y.; Maisuradze, G.G.; Yin, Y.; Kachlishvili, K.; Rackovsky, S.; Scheraga, H.A. Sequence-, structure-, and dynamics-based comparisons of structurally homologous CheY-like proteins. Proc. Natl. Acad. Sci. USA 2017, 114, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Kosloff, M.; Kolodny, R. Sequence-similar, structure-dissimilar protein pairs in the PDB. Proteins 2008, 71, 891–902. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Hebditch, M.; Carballo-Amador, M.A.; Charonis, S.; Curtis, R.; Warwicker, J. Protein-Sol: A web tool for predicting protein solubility from sequence. Bioinformatics 2017, 33, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Tholen, D.; Zhu, X.G. C(4) photosynthesis in C(3) rice: A theoretical analysis of biochemical and anatomical factors. Plant Cell Environ. 2017, 40, 80–94. [Google Scholar] [CrossRef]
- Hughes, T.E.; Sedelnikova, O.V.; Wu, H.; Becraft, P.W.; Langdale, J.A. Redundant SCARECROW genes pattern distinct cell layers in roots and leaves of maize. Development 2019, 146, dev177543. [Google Scholar] [CrossRef]
- di Iulio, J.; Bartha, I.; Spreafico, R.; Virgin, H.W.; Telenti, A. Transfer transcriptomic signatures for infectious diseases. Proc. Natl. Acad. Sci. USA 2021, 118, e2022486118. [Google Scholar] [CrossRef]
- Hibberd, J.M.; Sheehy, J.E.; Langdale, J.A. Using C4 photosynthesis to increase the yield of rice—Rationale and feasibility. Curr. Opin. Plant Biol. 2008, 11, 228–231. [Google Scholar] [CrossRef]
- Ermakova, M.; Danila, F.R.; Furbank, R.T.; von Caemmerer, S. On the road to C4 rice: Advances and perspectives. Plant J. 2020, 101, 940–950. [Google Scholar] [CrossRef]
- Eckardt, N.A. Oxylipin signaling in plant stress responses. Plant Cell 2008, 20, 495–497. [Google Scholar] [CrossRef]
- Barros, T.; Kuhlbrandt, W. Crystallisation, structure and function of plant light-harvesting Complex II. Biochim. Biophys. Acta 2009, 1787, 753–772. [Google Scholar] [CrossRef] [PubMed]
- Araujo, W.L.; Nunes-Nesi, A.; Nikoloski, Z.; Sweetlove, L.J.; Fernie, A.R. Metabolic control and regulation of the tricarboxylic acid cycle in photosynthetic and heterotrophic plant tissues. Plant Cell Environ. 2012, 35, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Theobald, D.L.; Wuttke, D.S. Divergent evolution within protein superfolds inferred from profile-based phylogenetics. J. Mol. Biol. 2005, 354, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Gilson, A.I.; Marshall-Christensen, A.; Choi, J.M.; Shakhnovich, E.I. The Role of Evolutionary Selection in the Dynamics of Protein Structure Evolution. Biophys. J. 2017, 112, 1350–1365. [Google Scholar] [CrossRef] [PubMed]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold: Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef] [PubMed]
- DalCorso, G.; Pesaresi, P.; Masiero, S.; Aseeva, E.; Schunemann, D.; Finazzi, G.; Joliot, P.; Barbato, R.; Leister, D. A complex containing PGRL1 and PGR5 is involved in the switch between linear and cyclic electron flow in Arabidopsis. Cell 2008, 132, 273–285. [Google Scholar] [CrossRef]
- Hertle, A.P.; Blunder, T.; Wunder, T.; Pesaresi, P.; Pribil, M.; Armbruster, U.; Leister, D. PGRL1 is the elusive ferredoxin-plastoquinone reductase in photosynthetic cyclic electron flow. Mol. Cell 2013, 49, 511–523. [Google Scholar] [CrossRef]
- Peng, L.; Fukao, Y.; Fujiwara, M.; Takami, T.; Shikanai, T. Efficient operation of NAD(P)H dehydrogenase requires supercomplex formation with photosystem I via minor LHCI in Arabidopsis. Plant Cell 2009, 21, 3623–3640. [Google Scholar] [CrossRef]
- Peng, L.; Shikanai, T. Supercomplex formation with photosystem I is required for the stabilization of the chloroplast NADH dehydrogenase-like complex in Arabidopsis. Plant Physiol. 2011, 155, 1629–1639. [Google Scholar] [CrossRef]
- Liao, B.Y.; Weng, M.P. Unraveling the association between mRNA expressions and mutant phenotypes in a genome-wide assessment of mice. Proc. Natl. Acad. Sci. USA 2015, 112, 4707–4712. [Google Scholar] [CrossRef]
- Yerramsetty, P.; Agar, E.M.; Yim, W.C.; Cushman, J.C.; Berry, J.O. An rbcL mRNA-binding protein is associated with C3 to C4 evolution and light-induced production of Rubisco in Flaveria. J. Exp. Bot. 2017, 68, 4635–4649. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cortes, A.J.; Restrepo-Montoya, M.; Bedoya-Canas, L.E. Modern Strategies to Assess and Breed Forest Tree Adaptation to Changing Climate. Front. Plant Sci. 2020, 11, 583323. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.J.; Lopez-Hernandez, F.; Osorio-Rodriguez, D. Predicting Thermal Adaptation by Looking Into Populations’ Genomic Past. Front. Genet. 2020, 11, 564515. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.J.; Lopez-Hernandez, F.; Blair, M.W. Genome-Environment Associations, an Innovative Tool for Studying Heritable Evolutionary Adaptation in Orphan Crops and Wild Relatives. Front. Genet. 2022, 13, 910386. [Google Scholar] [CrossRef]
- John, C.R.; Smith-Unna, R.D.; Woodfield, H.; Covshoff, S.; Hibberd, J.M. Evolutionary convergence of cell-specific gene expression in independent lineages of C4 grasses. Plant Physiol. 2014, 165, 62–75. [Google Scholar] [CrossRef]
- Chang, Y.M.; Liu, W.Y.; Shih, A.C.; Shen, M.N.; Lu, C.H.; Lu, M.Y.; Yang, H.W.; Wang, T.Y.; Chen, S.C.; Chen, S.M.; et al. Characterizing regulatory and functional differentiation between maize mesophyll and bundle sheath cells by transcriptomic analysis. Plant Physiol. 2012, 160, 165–177. [Google Scholar] [CrossRef]
- Doring, F.; Streubel, M.; Brautigam, A.; Gowik, U. Most photorespiratory genes are preferentially expressed in the bundle sheath cells of the C4 grass Sorghum bicolor. J. Exp. Bot. 2016, 67, 3053–3064. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Ouyang, S.; Zhu, W.; Hamilton, J.; Lin, H.; Campbell, M.; Childs, K.; Thibaud-Nissen, F.; Malek, R.L.; Lee, Y.; Zheng, L.; et al. The TIGR Rice Genome Annotation Resource: Improvements and new features. Nucleic Acids Res. 2007, 35, D883–D887. [Google Scholar] [CrossRef]
- Mamidi, S.; Healey, A.; Huang, P.; Grimwood, J.; Jenkins, J.; Barry, K.; Sreedasyam, A.; Shu, S.; Lovell, J.T.; Feldman, M.; et al. A genome resource for green millet Setaria viridis enables discovery of agronomically valuable loci. Nat. Biotechnol. 2020, 38, 1203–1210. [Google Scholar] [CrossRef] [PubMed]
- McCormick, R.F.; Truong, S.K.; Sreedasyam, A.; Jenkins, J.; Shu, S.; Sims, D.; Kennedy, M.; Amirebrahimi, M.; Weers, B.D.; McKinley, B.; et al. The Sorghum bicolor reference genome: Improved assembly, gene annotations, a transcriptome atlas, and signatures of genome organization. Plant J. 2018, 93, 338–354. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Genome Project Data Processing, S. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Ramirez, F.; Dundar, F.; Diehl, S.; Gruning, B.A.; Manke, T. deepTools: A flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 2014, 42, W187–W191. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Heberle, H.; Meirelles, G.V.; da Silva, F.R.; Telles, G.P.; Minghim, R. InteractiVenn: A web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 2015, 16, 169. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hunter, S.; Apweiler, R.; Attwood, T.K.; Bairoch, A.; Bateman, A.; Binns, D.; Bork, P.; Das, U.; Daugherty, L.; Duquenne, L.; et al. InterPro: The integrative protein signature database. Nucleic Acids Res. 2009, 37, D211–D215. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How significant is a protein structure similarity with TM-score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Sadreyev, R.I.; Pei, J.; Kinch, L.N.; Grishin, N.V. Using homology relations within a database markedly boosts protein sequence similarity search. Proc. Natl. Acad. Sci. USA 2015, 112, 7003–7008. [Google Scholar] [CrossRef]
- Sadowski, M.I.; Jones, D.T. The sequence-structure relationship and protein function prediction. Curr. Opin. Struct. Biol. 2009, 19, 357–362. [Google Scholar] [CrossRef]









Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, C.; Guo, D. Identification of Two Flip-Over Genes in Grass Family as Potential Signature of C4 Photosynthesis Evolution. Int. J. Mol. Sci. 2023, 24, 14165. https://doi.org/10.3390/ijms241814165
Wu C, Guo D. Identification of Two Flip-Over Genes in Grass Family as Potential Signature of C4 Photosynthesis Evolution. International Journal of Molecular Sciences. 2023; 24(18):14165. https://doi.org/10.3390/ijms241814165
Chicago/Turabian StyleWu, Chao, and Dianjing Guo. 2023. "Identification of Two Flip-Over Genes in Grass Family as Potential Signature of C4 Photosynthesis Evolution" International Journal of Molecular Sciences 24, no. 18: 14165. https://doi.org/10.3390/ijms241814165
APA StyleWu, C., & Guo, D. (2023). Identification of Two Flip-Over Genes in Grass Family as Potential Signature of C4 Photosynthesis Evolution. International Journal of Molecular Sciences, 24(18), 14165. https://doi.org/10.3390/ijms241814165