Abstract
Rhabdomyosarcoma (RMS) is the most common pediatric soft-tissue cancer with a survival rate below 27% for high-risk children despite aggressive multi-modal therapeutic interventions. After decades of research, no targeted therapies are currently available. Therapeutically targeting actin-binding proteins, although promising, has historically been challenging. Recent advances have made this possibility more salient, including our lab’s identification of advillin (AVIL), a novel oncogenic actin-binding protein that plays a role in many cytoskeletal functions. AVIL is overexpressed in many RMS cell lines, patient-derived xenograft models, and a cohort of 30 clinical samples of both the alveolar (ARMS) and embryonal (ERMS) subtypes. Overexpression of AVIL in mesenchymal stem cells induces neoplastic transformation both in vitro and in vivo, and reversing overexpression through genetic modulation reverses the transformation. This suggests a critical role of AVIL in RMS tumorigenesis and maintenance. As an actin-binding protein, AVIL would not traditionally be considered a druggable target. This perspective will address the feasibility of targeting differentially expressed actin-binding proteins such as AVIL therapeutically, and how critical cell infrastructure can be damaged in a cancer-specific manner.
1. Introduction
Rhabdomyosarcoma (RMS) is the most common cancer of soft tissue in children, accounting for at least half of all pediatric sarcomas [1]. Approximately 90% of all RMS cases occur in patients under 25 years of age, with the majority occurring in children under 10 [2]. RMS has significant variability in outcomes. Low-risk patients with localized disease face a survival rate of 90%, while high risk patients with metastatic or recurrent disease face dismal survival rates of 21% and 30%, respectively [3,4]. These high-risk groups have seen no improvement in outcomes in the last 30 years, making the lack of novel targeted treatments a major roadblock to durable cures.
The major anatomic sites of RMS include the head, neck, and genitourinary tract. Previously, the classification of RMS was primarily based upon histology to delineate alveolar RMS (ARMS) and embryonal RMS (ERMS). The most recent World Health Organization (WHO) classification of RMS consists of four groups: alveolar, embryonal, pleiomorphic, and spindle cell/sclerosing, with new subdivisions of spindle cell/sclerosing tumors based upon molecular alterations (Table 1) [5,6,7]. These alterations differentially regulate the pathways involved in promoting tumorigenesis. For instance, PAX3/7 and FOXO1 function as transcriptional activators of genes that regulate muscle differentiation, cell lineage, proliferation, cell migration, muscle growth, and metabolism [8,9]. Chromosomal translocations can lead to PAX3-FOXO1 or PAX7-FOXO1 gene fusions, and the translated fusion proteins have enhanced transcriptional activity in contrast to the parent proteins [10]. In ERMS, the RAS genes encode small GTPase transductor domains involved in regulating cell proliferation, migration, and growth by cycling between GTP-bound active and GDP-bound inactive forms. Mutations in these genes lead to a gain-of-function in the protein with a constitutive GTP-bound active form, promoting carcinogenesis [11]. Additionally, p53 is a tumor suppressor protein that functions to negatively regulate cell cycle progression and is frequently mutated in ERMS [12]. Most ERMS present with loss-of-function mutations in p53, which allows for uncontrolled cell cycle progression [12]. Several other functional genetic aberrations that promote RMS were recently reviewed [13]. Table 1 provides an overview of the different genetic profiles, histopathologies, and common sites associated with the four groups of RMS based on the WHO classifications.

Table 1.
Overview of the classification of rhabdomyosarcoma subtypes. The genetic alterations and histopathological characteristics of each of the four types are described.
Frontline therapy is the same for all RMS risk groups and is generally a combination of surgery, radiotherapy, and a three-drug cytotoxic chemotherapy regimen consisting of vincristine sulfate, dactinomycin (Actinomycin-D), and cyclophosphamide (VAC) in the USA, with ifosfamide replacing cyclophosphamide (IVA or VAI) in Europe [14,15]. The clinical trial results for traditional chemotherapy regiments are mixed, with some showing marginal increases in overall survival (OS) and disease/event free survival (DFS/EVS), and some showing no statistically significant difference between the two groups (Table 2). Despite some positive outcomes, traditional chemotherapy creates numerous deleterious side effects due to its non-specific cytotoxicity. For low-risk patients with localized disease, attempts to decrease the dosing of radiotherapy and chemotherapy, specifically cyclophosphamide, have been made to reduce deleterious toxicities such as lifetime infertility and myelosuppression [16,17]. With no targeted therapies approved, high-risk children face lifelong consequences even if a durable cure is achieved.
Table 2.
Overview of several recent clinical trials for traditional chemotherapy and targeted molecular therapy treatments for RMS.
Efforts have been made to pharmacologically inhibit molecular targets in RMS, such as platelet-derived growth factor receptors (PDGFRs) and vascular endothelial growth factor receptors (VEGFRs), but phase II clinical trials have indicated no improved outcomes (Table 2). The lack of approved targeted therapies is a major unmet need in the field, and novel therapeutic targets may be necessary to decrease cytotoxic chemotherapy dosing and reduce long-lasting toxicities. Our lab has recently discovered AVIL, an actin-binding protein, as an oncogenic driver of RMS. This review summarizes the current literature on research into targeting actin-binding proteins including AVIL and its family members, and explores the therapeutic potential of drugging actin-binding proteins such as AVIL for precision oncology applications in RMS.
2. Improving VAC Chemotherapy and Molecular Targeted Therapies
The standard management of RMS includes chemotherapy, radiation therapy, and tumor resection. In patients with metastatic RMS, efforts toward complete remission with high-dose chemotherapy resulted in treatment-related adverse effects. Several phase III clinical trials have been conducted to improve disease-free survival, including the addition of low-dose maintenance chemotherapy after standard chemotherapy with IVA, which showed a modest increase in the 5-year disease-free survival rate of 8% [18] (Table 2). The addition of doxorubicin, a drug widely used to treat soft-tissue sarcoma, to IVA chemotherapy failed to improve the 3-year event-free survival and also resulted in treatment-related adverse effects such as infections, anemia, leukopenia, gastrointestinal disorder, and even death [19]. Another phase III clinical trial attempted to modify a similar standard therapy, VAC, to reduce toxicity by substituting half of the VAC course with vincristine and irinotecan; this resulted in no significant difference in the oncological outcome [20]. Efforts have been made to increase complete remission and disease-free survival; however, no recent trials have significantly prolonged patient survival and remission. High dose chemotherapy, low-dose maintenance chemo following standard chemo, and the addition of doxorubicin and irinotecan were all evaluated in clinical trials and found to be ineffective.
Molecular targeted therapies have also been investigated, but their efficacy is still in question. A phase II clinical trial demonstrated that patients receiving vinorelvine (V), cyclophosphamide (C), and bevacizumab experienced a response rate of 32%, while patients receiving VC and temsirolumus experienced an increased response rate of 47% [24]. Despite promising preclinical evidence in their efficacy against RMS, sorafenib and crizotinib both proved inactive against RMS in phase II clinical trials [21,22,25]. Vismodegib trials in advanced chrondrosarcoma showed some efficacy, a 25.6% clinical benefit in patients, which was short of the 40% clinical benefit goal [26,27]. Overall, there is immense genetic, biological, and clinical response heterogeneity common to RMS, underscoring the dire need for some form of targeted therapy to open new therapeutic avenues and reduce the toxicities seen in the current standard of care.
3. Targeting Actin and Actin-Binding Proteins
Eukaryotic actin is a 375 amino acid polypeptide that folds into four subdomains, with an ATP-binding cleft important for regulating the dynamic switch between its two forms: globular actin (G-actin) and filamentous actin (F-actin). These processes are critical to the physiological functions of cells including cell division, maintaining structural integrity, cell migration, vesicular trafficking, cell signaling, cell adhesions, and tight junction formation. While healthy cells depend on the controlled regulation of actin to maintain cellular function, cancer cells harness these same mechanisms to promote migration, invasion, and metastasis by dysregulating actin-binding proteins (ABPs) and disrupting the balance in actin dynamics, thereby facilitating the formation of invasive structures like lamellipodia and filopodia [28]. As such, efforts to therapeutically target ABPs in cancer have been an attractive but challenging field of research over recent decades.
Although targeting ABPs confers an advantage to targeting cancer cells, normal cells become vulnerable to high and unbearable toxicity. For example, while cytochalasins have exhibited promising effects on breast, lung, and prostate cancers, congestion necrosis in rats has been reported at the edge of the liver, as well as negative affects on cardiac contractility [29,30,31,32]. Also, chaetoglobosin has been shown to be lethal at a dose of 2 mg/kg in rats and induce spermatocyte degeneration in mice [33,34]. Furthermore, Jasplakinolide, latrunculin, and MKT-077 are other inhibitors targeting the actin cytoskeleton with anti-cancer effects and have been shown to induce cardiac toxicity, chronic seizures in rats, and retinal toxicity in humans [35,36,37,38]. ABP targeting as a treatment option has struggled as a result of this widespread toxicity.
4. Targeting Nucleation Factors
ABPs that nucleate and mediate branching include the Arp2/3 complex with nuclear-promoting factors, the formin family of proteins, and the tandem monomer binding nucleators. Arp2/3 has been shown to be overexpressed in several cancers including gastric, glioma, breast, lung, and colorectal cancer, where it facilitates cancer pathogenesis, growth, and invasion [39,40,41,42,43].
A major activator of Arp2/3 is the Wiskott–Aldrich Syndrome protein (WASp) nuclear-promoting factor expressed in hematopoietic stem cells. WASp has been targeted with small molecule compound #13 (SMC #13), which has a bioavailability score of 0.5, a molecular weight of 461.6 g/mol, four hydrogen bond acceptors, high gastrointestinal absorption, and drug-likeness potential [44]. Investigators have shown that SMC #13 directly interacts with WASp to promote its degradation via ubiquitination, which significantly attenuates WASp-dependent actin dynamics in SMC #13 treated hematopoietic malignancies with fewer toxicities in healthy naïve cells. Nolen and colleagues have also drugged Arp2/3 using small molecule inhibitors CK-0944636 and CK-0993548. Their experiments suggest that CK-0944636 binds between Arp2 and Arp3, potentially inhibiting the movement of the complex into their active conformation, while CK-0993548 modifies the confirmation of Arp3 by binding in the hydrophobic domain [45], thereby inhibiting actin polymerization.
Formins have also been of interest as a potential target in cancer. These proteins contain the highly conserved formin homology 2 (FH2) domain for facilitating actin assembly. Formins have been shown to be overexpressed in colorectal cancer [46], induce an epithelial-mesenchymal transition to facilitate colorectal carcinoma invasion [47], and regulate cell migration and metastasis in colorectal carcinoma [48]. Silencing the formin like 2 (FMNL2) gene has been shown to slow the growth of gastric cancer cells, demonstrating its therapeutic potential [49]. A small molecule inhibitor of the FH2 domain (SMIFH2) was the first to be discovered to drug formins over a decade ago, both in vitro and in vivo. SMIFH2 disrupts formin-dependent actin dynamics and has no effect on Arp2/3-dependent actin dynamics [50].
5. Targeting Actin Polymerization and Depolymerization
Several drugs have been described to stabilize F-actin and thereby inhibit depolymerization. While these drugs may have some anti-tumorigenic activities [51], their inability to selectively target cancerous cells limits their usefulness to research purposes only. Jasplakinolide is a membrane-permeable cyclo-depsipeptide isolated from the marine sponge Jaspis sp. [52,53]. It binds to F-actin to stabilize polymerization, which impairs cell migration and the protrusion of lamellipodia [51]. Jasplakinolide competes with phalloidin, another depolymerization-inhibiting compound, for F-actin binding and stabilization [54]. Phalloidin, however, is membrane impermeable.
Other F-actin stabilizing drugs include doliculide [55], chondramides, and dollastin 11 [56]. While these drugs enhance actin polymerization, other drugs inhibit polymerization. Cytochalasins are membrane-permeable fungal metabolites that bind to the barbed ends of F-actin to inhibit actin polymerization [57]. Cytochalasin D has been shown to induce the hydrolysis of ATP in G-actin dimers to inhibit F-actin assembly, and, eventually, cell migration and proliferation. While the potential benefits of cytochalasins as a supplement to improve chemotherapies have been exploited [58] and reviewed [59], their safety in patients remains to be seen.
Furthermore, there are several other small molecule inhibitors targeting other actin-binding proteins that have been extensively reviewed [60]. These have shown promising effects in some actin-binding targets traditionally considered “undruggable”, such as Rho-GTPases. The ABP AVIL is of particular interest among actin-binding proteins because it is significantly overexpressed in RMS cancer cells while showing low level expression in very few normal cells [61]. Amplification at the AVIL locus has been shown not only in RMS but in other sarcomas as well, suggesting broader oncogenic properties. This selective overexpression of AVIL in RMS makes cancer cells vulnerable to targeting of the cytoskeleton while sparing actively dividing but healthy cells.
6. Advillin Background
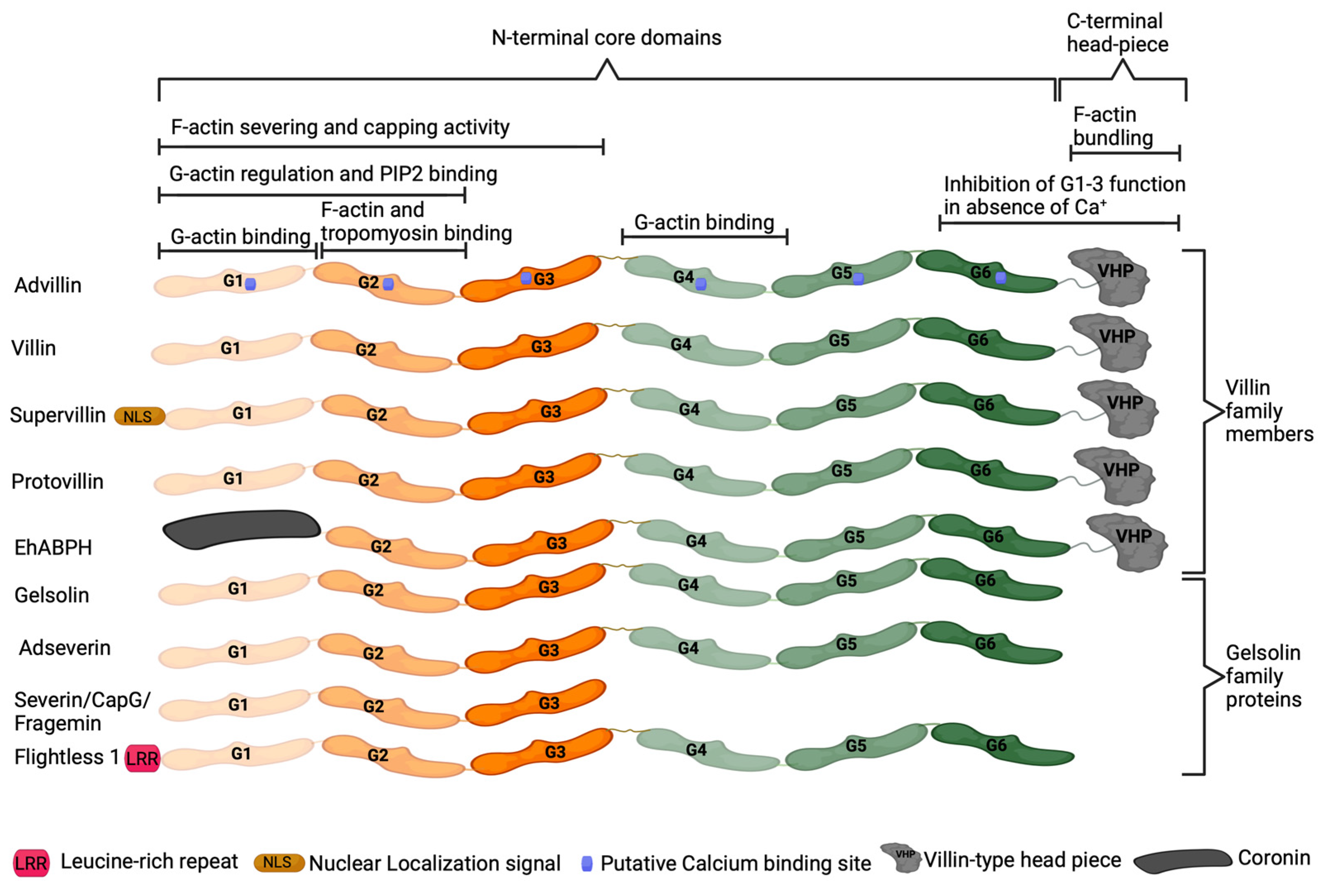
Advillin (AVIL), encoded by the AVIL gene, is a calcium-regulated actin-binding protein originally identified in the adult murine brain [62,63]. Advillin is a member of the gelsolin protein superfamily, sharing 65 to 75% homology with adseverin [62]. Proteins of this superfamily, which also includes villin, gelsolin, and adseverin, regulate actin organization [62,63,64]. Advillin contains six homologous domains termed gelsolin-like (G1–G6) which are conserved within gelsolin superfamily members (Figure 1) [65]. The G1 and G2 domains allow for binding to phosphatidylinositol 4,5-bisphosphate (PIP2) and regulation of monomeric actin (G-actin), while G1, G2, and G3 domains can also function in severing and capping actin filaments. The G1 and G4 domains bind actin monomers, while the G2 domain is responsible for filamentous actin (F-actin) and tropomyosin binding [64,65]. The carboxy-terminus headpiece domain present in advillin, villin, villin-like protein, supervillin, and flightless 1 enables actin filament bundling. In the absence of calcium ions, the G6 and HP domains inhibit the function of G1–G3 domains [64,65].
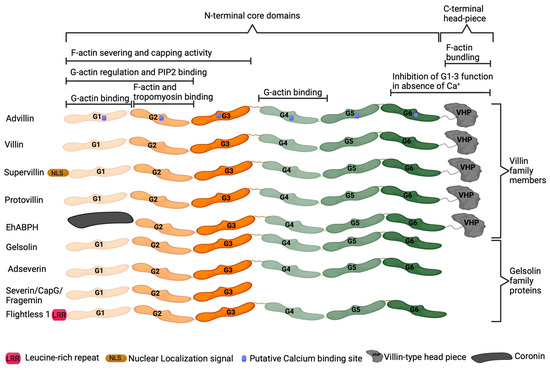
Figure 1.
Structure of gelsolin superfamily proteins. The gelsolin-like (G1–G6) domains are largely conserved within gelsolin superfamily proteins, with domain function conserved between proteins.
Although the precise function and impact on signal transduction remains unclear, advillin seems to be involved in many processes, playing a vital role in neurite outgrowth and the development of neuronal cells that form ganglia [62,66]. Moreover, in normal physiology, advillin is rarely expressed outside of sensory neurons during development, or non-peptidergic nociceptors in dorsal root ganglia, the Merkel cells of the skin, and tuft cells in the gastrointestinal and biliary tracts in adulthood [67,68]. Expression in the dorsal root ganglia is restricted to isolectin B4-positive neurons, and may be involved in growth cone formation, axonal regeneration, and neuropathic pain [69].
Due to its key role in the organization of the actin cytoskeleton, which affects polarity, movement, cell division, and trafficking, it is not surprising that advillin appears to play a role in many cancers [70]. Figure 2 summarizes the general interactions between AVIL and actin, demonstrating that AVIL can modulate many actin-based processes. The AVIL gene is overexpressed in nearly 100% of glioblastomas and was identified as a bona fide oncogene that is crucial for glioblastoma tumorigenesis [71]. In recent studies, we have shown that AVIL expression is abnormally upregulated in RMS, where silencing this gene results in a dramatic reduction in proliferation and migration, killing cancer cells and preventing tumor formation [61]. Therefore, AVIL seems to be a viable therapeutic target in glioblastoma and RMS.
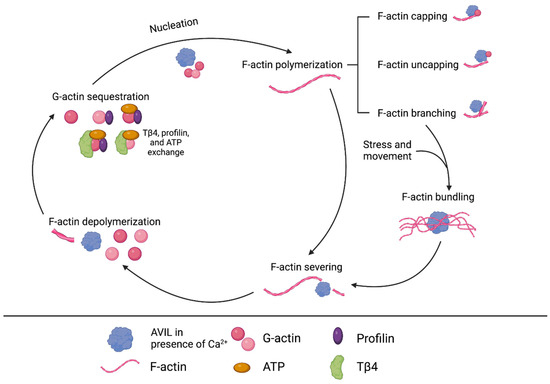
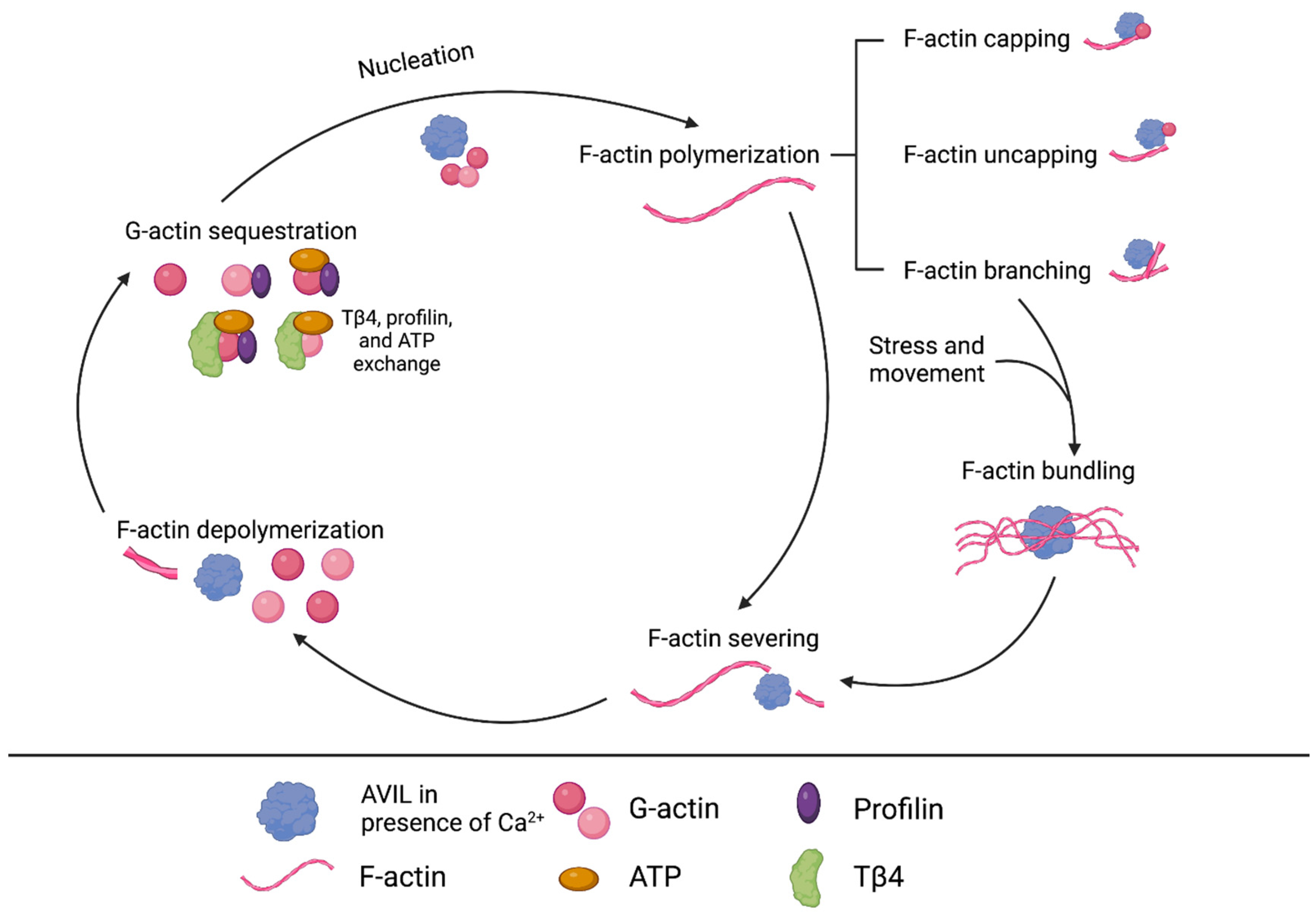
Figure 2.
Overview of known interactions between AVIL and actin, including nucleation, capping, uncapping, branching, bundling, severing, and depolymerization. Actin is a key protein of the cytoskeleton, and its dynamic qualities enable cells to respond to change. It exists in globular (G) and filamentous (F) forms and switches between the two via polymerization or depolymerization, affecting cytoskeletal structure and cellular processes. AVIL can cause changes in actin structure and composition, affecting cellular functions like movement and division.
7. AVIL Functionality in ERMS and ARMS Subtypes
Embryonal RMS (ERMS) affects mostly children, with the most common sites of presentation being the head, neck, and genitourinary tract [9]. ERMS presents varying degrees of differentiation, from well-differentiated neoplasms to poorly differentiated tumors [72]. Anaplastic cells in some cases of RMS have been characterized by a significant hyperchromasia. Several genetic alterations have been suggested to drive the pathogenesis of ERMS, including enhanced RAS signaling with mutations in KRAS, NRAS, and HRAS; activation of Hedgehog (Hh) signaling; and inactivation of p53 and Rb pathways [73]. Alteration in each of these pathways allows the cells to evade growth suppressors and apoptosis, promoting proliferation. Copy number alterations have also been observed, including gain of chromosome 8 and loss of chromosomes 10 and 15 [72].
Alveolar RMS (ARMS) affects mostly adolescents and young adults. While ARMS can arise from any part of the body, it is commonly observed in perineal and paraspinal regions as well as the extremities. Pathological features include poorly differentiated rhabdomyoblasts with enlarged nuclei and scant cytoplasm [2]. Tumor cells are usually nested in fibrovascular septa and have loosened intercellular connections, creating alveolar or slit-like spaces. ARMS is characterized by the expression of diffused MYOD1 and myogenin. About 80–90% of ARMS are associated with recurrent Forkhead Box O1 (FOXO1) fusions. FOXO1 forms a fusion with PAX3 or PAX7, resulting in altered expression, localization, and function compared to wild-type FOXO1 [74]. Functionally, the PAX3–FOXO1 fusion joins the DNA-binding region of PAX3 with the transactivation domain of FOXO1, creating a novel transcription factor that can activate cellular pathways that promote oncogeneic hallmarks such as rampant proliferation and evasion of apoptosis [74]. The PAX7–FOXO1 fusion product exhibits similar functionality in ARMS [75].
AVIL is thought to activate the RAS signaling pathway, which is a major cell proliferation pathway and a hallmark of ERMS. Interestingly, an oncogenic cooperativity assay found no difference in foci formation between overexpressing AVIL alone, RAS alone, or both [61]. Furthermore, AVIL overexpression in mesenchymal stem cells is sufficient to differentially express many RAS targets, increase p-MEK1/2 and p-ERK1/2, and mimic published gene signatures for RAS signaling. AVIL overexpression also appears to mimic PAX3–FOXO1 fusion signaling in ARMS, with AVIL leading to the differential expression of PAX3–FOXO1 fusion targets. AVIL is reported to be significantly expressed in many cell lines of both ARMS and ERMS, mimicking pathways upstream of both RMS subtypes including PAX3–FOXO1 and RAS. It may, therefore, serve as a connecting node for the major pathways associated with ARMS and ERMS [61]. Given its differential overexpression in numerous cancers including RMS, and its ability to activate both RAS and PAX3–FOXO1 signaling pathways, AVIL demonstrates promising therapeutic potential as a target for both ERMS and ARMS.
8. Perspective
Cytotoxic chemotherapies targeting the cytoskeleton are some of the most potent therapeutics for most cancers, with almost all approved therapies targeting tubulin [76,77,78]. These cytotoxic agents also come with a large list of deleterious toxicities given that they target all dividing cells, both normal and malignant. Identifying a cancer-specific cytoskeletal protein that is differentially targetable, especially in rarer malignancies, would be a major step forward in treating these diseases. AVIL appears to be a relatively cancer-specific cytoskeletal protein overexpressed in RMS, a pediatric cancer with limited treatment options. Targeting AVIL sits at the crossroads of two therapeutic philosophies: broad range cytotoxic agents that target critical cellular infrastructure common to all dividing cells, and a targeted cancer-specific therapy that limits toxicities in normal cell populations. It is this unique combination that positions AVIL as an attractive druggable target for RMS therapies.
Author Contributions
Conceptualization, H.L. and R.C.; writing—original draft preparation, L.M., S.L., M.G., A.F. and A.T.; writing—review and editing, L.M., S.L. and C.P.; visualization, L.M., S.L. and A.F.; supervision, L.M. and R.C.; funding acquisition, H.L. and L.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Cancer Institute (NCI) Grant R01CA269594. L.M. was supported by an Ingrassia Family Echols Scholars Research Grant.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We acknowledge BioRender software for use in figure creation.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the writing of the manuscript.
References
- Dasgupta, R.; Fuchs, J.; Rodeberg, D. Rhabdomyosarcoma. Semin. Pediatr. Surg. 2016, 25, 276–283. [Google Scholar] [CrossRef] [PubMed]
- Dziuba, I.; Kurzawa, P.; Dopierała, M.; Larque, A.B.; Januszkiewicz-Lewandowska, D. Rhabdomyosarcoma in children—Current pathologic and molecular classification. Pol. J. Pathol. 2018, 69, 20–32. [Google Scholar] [CrossRef]
- Rudzinski, E.R.; Kelsey, A.; Vokuhl, C.; Linardic, C.M.; Shipley, J.; Hettmer, S.; Koscielniak, E.; Hawkins, D.S.; Bisogno, G. Pathology of childhood rhabdomyosarcoma: A consensus opinion document from the Children’s Oncology Group, European Paediatric Soft Tissue Sarcoma Study Group, and the Cooperative Weichteilsarkom Studiengruppe. Pediatr. Blood Cancer 2020, 68, e28798. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Garcia, H.D.; Scheer, M.; Henssen, A.G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef]
- Agaram, N.P. Evolving classification of rhabdomyosarcoma. Histopathology 2021, 80, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Ro, J.Y. The 2020 WHO Classification of Tumors of Soft Tissue: Selected Changes and New Entities. Adv. Anat. Pathol. 2021, 28, 44–58. [Google Scholar] [CrossRef]
- Noujaim, J.; Thway, K.; Jones, R.L.; Miah, A.; Khabra, K.; Langer, R.; Kasper, B.; Judson, I.; Benson, C.; Kollàr, A. Adult Pleomorphic Rhabdomyosarcoma: A Multicentre Retrospective Study. Anticancer Res. 2015, 35, 6213. [Google Scholar]
- Xu, M.; Chen, X.; Chen, D.; Yu, B.; Huang, Z. FoxO1: A novel insight into its molecular mechanisms in the regulation of skeletal muscle differentiation and fiber type specification. Oncotarget 2017, 8, 10662–10674. [Google Scholar] [CrossRef]
- Skapek, S.X.; Ferrari, A.; Gupta, A.A.; Lupo, P.J.; Butler, E.; Shipley, J.; Barr, F.G.; Hawkins, D.S. Rhabdomyosarcoma. Nat. Rev. Dis. Primers 2019, 5, 1. [Google Scholar] [CrossRef]
- Bennicelli, J.L.; Advani, S.; Schäfer, B.W.; Barr, F.G. PAX3 and PAX7 exhibit conserved cis-acting transcription repression domains and utilize a common gain of function mechanism in alveolar rhabdomyosarcoma. Oncogene 1999, 18, 4348–4356. [Google Scholar] [CrossRef]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Diller, L.; Sexsmith, E.; Gottlieb, A.; Li, F.P.; Malkin, D. Germline p53 mutations are frequently detected in young children with rhabdomyosarcoma. J. Clin. Investig. 1995, 95, 1606–1611. [Google Scholar] [CrossRef] [PubMed]
- Martin-Giacalone, B.A.; Weinstein, P.A.; Plon, S.E.; Lupo, P.J. Pediatric rhabdomyosarcoma: Epidemiology and genetic susceptibility. J. Clin. Med. 2021, 10, 2028. [Google Scholar] [CrossRef]
- Halstead, N.V.; Cost, N.G.; Hecht, S.L.; Walker, J.P. Neurofibromatosis-1 and Rhabdomyosarcoma: An Unusual Recurrence. Urology 2020, 137, 168–172. [Google Scholar] [CrossRef] [PubMed]
- Steenman, M.; Westerveld, A.; Mannens, M. Genetics of Beckwith-Wiedemann syndrome-associated tumors: Common genetic pathways. Genes Chromosomes Cancer 2000, 28, 1–13. [Google Scholar] [CrossRef]
- Doros, L.; Yang, J.; Dehner, L.; Rossi, C.T.; Skiver, K.; Jarzembowski, J.A.; Messinger, Y.; Schultz, K.A.; Williams, G.; André, N.; et al. DICER1 Mutations in embryonal rhabdomyosarcomas from children with and without familial PPB-tumor predisposition syndrome. Pediatr. Blood Cancer 2012, 59, 558–560. [Google Scholar] [CrossRef]
- Shern, J.F.; Yohe, M.E.; Khan, J. Pediatric rhabdomyosarcoma. Crit. Rev. Oncog. 2015, 20, 227–243. [Google Scholar] [CrossRef]
- Bisogno, G.; De Salvo, G.L.; Bergeron, C.; Melcón, S.G.; Merks, J.H.; Kelsey, A.; Martelli, H.; Minard-Colin, V.; Orbach, D.; Glosli, H.; et al. Vinorelbine and continuous low-dose cyclophosphamide as maintenance chemotherapy in patients with high-risk rhabdomyosarcoma (RMS 2005): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2019, 20, 1566–1575. [Google Scholar] [CrossRef]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Melcón, S.G.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A.; et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): A multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Hawkins, D.S.; Chi, Y.-Y.; Anderson, J.R.; Tian, J.; Arndt, C.A.; Bomgaars, L.; Donaldson, S.S.; Hayes-Jordan, A.; Mascarenhas, L.; McCarville, M.B.; et al. Addition of vincristine and irinotecan to vincristine, dactinomycin, and cyclophosphamide does not improve outcome for intermediate-risk rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2018, 36, 2770–2777. [Google Scholar] [CrossRef]
- Kim, A.; Widemann, B.C.; Krailo, M.; Jayaprakash, N.; Fox, E.; Weigel, B.; Blaney, S.M. Phase 2 trial of sorafenib in children and young adults with refractory solid tumors: A report from the Children’s Oncology Group. Pediatr. Blood Cancer 2015, 62, 1562–1566. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Geoerger, B.; Kieran, M.W.; Grupp, S.; Perek, D.; Clancy, J.; Krygowski, M.; Ananthakrishnan, R.; Boni, J.P.; Berkenblit, A.; Spunt, S.L. Phase II trial of temsirolimus in children with high-grade glioma, neuroblastoma and rhabdomyosarcoma. Eur. J. Cancer 2012, 48, 253–262. [Google Scholar] [CrossRef]
- Mascarenhas, L.; Chi, Y.-Y.; Hingorani, P.; Anderson, J.R.; Lyden, E.R.; Rodeberg, D.A.; Indelicato, D.J.; Kao, S.C.; Dasgupta, R.; Spunt, S.L.; et al. Randomized Phase II Trial of Bevacizumab or Temsirolimus in Combination With Chemotherapy for First Relapse Rhabdomyosarcoma: A Report From the Children’s Oncology Group. J. Clin. Oncol. 2019, 37, 2866–2874. [Google Scholar] [CrossRef] [PubMed]
- Miwa, S.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Igarashi, K.; Tsuchiya, H. Recent Advances and Challenges in the Treatment of Rhabdomyosarcoma. Cancers 2020, 12, 1758. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French Sarcoma Group/US and French National Cancer Institute Single-Arm Phase II Collaborative Study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef] [PubMed]
- Gounder, M.M.; Rosenbaum, E.; Wu, N.; Dickson, M.A.; Sheikh, T.N.; D’Angelo, S.P.; Chi, P.; Keohan, M.L.; Erinjeri, J.P.; Antonescu, C.R.; et al. A Phase Ib/II Randomized Study of RO4929097, a Gamma-Secretase or Notch Inhibitor with or without Vismodegib, a Hedgehog Inhibitor, in Advanced Sarcoma. Clin. Cancer Res. 2022, 28, 1586–1594. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.G.; Niu, J.-T.; Wu, H.-W.; Si, X.-L.; Zhang, S.-J.; Li, D.-H.; Bian, T.-T.; Li, Y.-F.; Yan, X.-K. Actin-Binding Proteins as Potential Biomarkers for Chronic Inflammation-Induced Cancer Diagnosis and Therapy. Anal. Cell. Pathol. 2021, 2021, 6692811. [Google Scholar] [CrossRef]
- Calaghan, S.C.; White, E.; Bedut, S.; Guennec, J.-Y. Cytochalasin D reduces Ca2+ sensitivity and maximum tension via interactions with myofilaments in skinned rat cardiac myocytes. J. Physiol. 2000, 529, 405–411. [Google Scholar] [CrossRef]
- Chao, J.I.; Liu, H.F. The blockage of survivin and securin expression increases the cytochalasin B-induced cell death and growth inhibition in human cancer cells. Mol. Pharmacol. 2006, 69, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Glinsukon, T.; Lekutai, S. Comparative toxicity in the rat of cytochalasins B and E. Toxicon 1979, 17, 137–144. [Google Scholar] [CrossRef]
- Van Goietsenoven, G.; Mathieu, V.; Andolfi, A.; Cimmino, A.; Lefranc, F.; Kiss, R.; Evidente, A. In vitro growth inhibitory effects of cytochalasins and derivatives in cancer cells. Planta Medica 2011, 77, 711–717. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Saito, M.; Sekita, S.; Yoshihira, K.; Natori, S. Acute toxic effects of chaetoglobosin A, a new cytochalasan compound produced by Chaetomium globosum, on mice and rats. Jpn. J. Exp. Med. 1978, 48, 105–110. [Google Scholar] [PubMed]
- Knudsen, P.B.; Hanna, B.; Ohl, S.; Sellner, L.; Zenz, T.; Döhner, H.; Stilgenbauer, S.; Larsen, T.O.; Lichter, P.; Seiffert, M. Chaetoglobosin A preferentially induces apoptosis in chronic lymphocytic leukemia cells by targeting the cytoskeleton. Leukemia 2014, 28, 1289–1298. [Google Scholar] [CrossRef] [PubMed]
- Schweikart, K.; Guo, L.; Shuler, Z.; Abrams, R.; Chiao, E.T.; Kolaja, K.L.; Davis, M. The effects of jaspamide on human cardiomyocyte function and cardiac ion channel activity. Toxicol. Vitr. 2013, 27, 745–751. [Google Scholar] [CrossRef]
- Sierra-Paredes, G.; Oreiro-García, T.; Núñez-Rodriguez, A.; Vázquez-López, A.; Sierra-Marcuño, G. Seizures induced by in vivo latrunculin A and jasplakinolide microperfusion in the rat hippocampus. J. Mol. Neurosci. 2006, 28, 151–160. [Google Scholar] [CrossRef]
- Konishi, H.; Kikuchi, S.; Ochiai, T.; Ikoma, H.; Kubota, T.; Ichikawa, D.; Fujiwara, H.; Okamoto, K.; Sakakura, C.; Sonoyama, T.; et al. Latrunculin A has a strong anticancer effect in a peritoneal dissemination model of human gastric cancer in mice. Anticancer Res. 2009, 29, 2091–2097. [Google Scholar]
- Propper, D.J.; Braybrooke, J.P.; Taylor, D.J.; Lodi, R.; Styles, P.; Cramer, J.A.; Collins, W.C.J.; Levitt, N.C.; Talbot, D.C.; Ganesan, T.S.; et al. Phase I trial of the selective mitochondrial toxin MKT 077 in chemo- resistant solid tumours. Ann. Oncol. 1999, 10, 923–927. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yang, X.; Chen, C.; Liu, B.; Ren, B.; Wang, L.; Zhao, K.; Yu, S.; Ming, H. Expression of the Arp2/3 complex in human gliomas and its role in the migration and invasion of glioma cells. Oncol. Rep. 2013, 30, 2127–2136. [Google Scholar] [CrossRef]
- Georgopoulou, M.P.; Tosios, K.I.; Goutas, N.; Kouloukoussa, M. Arp2/3 Complex Is Expressed in Oral Squamous Cell Carcinoma: An Immunohistochemical Study of 88 Cases. Open J. Stomatol. 2019, 09, 29–38. [Google Scholar] [CrossRef]
- Semba, S.; Iwaya, K.; Matsubayashi, J.; Serizawa, H.; Kataba, H.; Hirano, T.; Kato, H.; Matsuoka, T.; Mukai, K. Coexpression of actin-related protein 2 and Wiskott-Aldrich syndrome family verproline-homologous protein 2 in adenocarcinoma of the lung. Clin. Cancer Res. 2006, 12, 2449–2454. [Google Scholar] [CrossRef]
- Iwaya, K.; Norio, K.; Mukai, K. Coexpression of Arp2 and WAVE2 predicts poor outcome in invasive breast carcinoma. Mod. Pathol. 2007, 20, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C.; Zheng, Y.-S.; Li, X.-H.; Takahashi, H.; Hara, T.; Masuda, S.; Yang, X.-H.; Guan, Y.-F.; Takano, Y. Arp2/3 overexpression contributed to pathogenesis, growth and invasion of gastric carcinoma. Anticancer. Res. 2008, 28, 2225–2232. [Google Scholar]
- Biber, G.; Ben-Shmuel, A.; Noy, E.; Joseph, N.; Puthenveetil, A.; Reiss, N.; Levy, O.; Lazar, I.; Feiglin, A.; Ofran, Y.; et al. Targeting the actin nucleation promoting factor WASp provides a therapeutic approach for hematopoietic malignancies. Nat. Commun. 2021, 12, 5581. [Google Scholar] [CrossRef] [PubMed]
- Nolen, B.J.; Tomasevic, N.; Russell, A.; Pierce, D.W.; Jia, Z.; McCormick, C.D.; Hartman, J.; Sakowicz, R.; Pollard, T.D. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature 2009, 460, 1031–1034. [Google Scholar] [CrossRef]
- Zhu, X.L.; Liang, L.; Ding, Y.Q. Overexpression of FMNL2 is closely related to metastasis of colorectal cancer. Int. J. Color. Dis. 2008, 23, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhu, X.; Zeng, Y.; Wang, J.; Zhang, X.; Ding, Y.-Q.; Liang, L. FMNL2 enhances invasion of colorectal carcinoma by inducing epithelial-mesenchymal transition. Mol. Cancer Res. 2010, 8, 1579–1590. [Google Scholar] [CrossRef]
- Zhu, X.L.; Zeng, Y.-F.; Guan, J.; Li, Y.-F.; Deng, Y.-J.; Bian, X.-W.; Ding, Y.-Q.; Liang, L. FMNL2 is a positive regulator of cell motility and metastasis in colorectal carcinoma. J. Pathol. 2011, 224, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Wang, K.; Xu, H.; Kong, F. Silencing Formin-like 2 inhibits growth and metastasis of gastric cancer cells through suppressing internalization of integrins. Cancer Cell Int. 2018, 18, 79. [Google Scholar] [CrossRef]
- Rizvi, S.A.; Neidt, E.M.; Cui, J.; Feiger, Z.; Skau, C.T.; Gardel, M.L.; Kozmin, S.A.; Kovar, D.R. Identification and Characterization of a Small Molecule Inhibitor of Formin-Mediated Actin Assembly. Chem. Biol. 2009, 16, 1158–1168. [Google Scholar] [CrossRef]
- Takeuchi, H.; Ara, G.; Sausville, E.A.; Teicher, B. Jasplakinolide: Interaction with radiation and hyperthermia in human prostate carcinoma and Lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998, 42, 491–496. [Google Scholar] [CrossRef] [PubMed]
- Sasse, F.; Kunze, B.; Gronewold, T.M.A.; Reichenbach, H. The chondramides: Cytostatic agents from myxobacteria acting on the actin cytoskeleton. J. Natl. Cancer Inst. 1998, 90, 1559–1563. [Google Scholar] [CrossRef]
- Crews, P.; Manes, L.V.; Boehler, M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis Sp. Tetrahedron Lett. 1986, 27, 2797–2800. [Google Scholar] [CrossRef]
- Bubb, M.R.; Senderowicz, A.M.J.; Sausville, E.A.; Duncan, K.L.K.; Korn, E.D. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994, 269, 14869–14871. [Google Scholar] [CrossRef] [PubMed]
- Bai, R.; Verdier-Pinard, P.; Gangwar, S.; Stessman, C.C.; McClure, K.J.; Sausville, E.A.; Pettit, G.R.; Bates, R.B.; Hamel, E. Dolastatin 11, a marine depsipeptide, arrests cells at cytokinesis and induces hyperpolymerization of purified actin. Mol. Pharmacol. 2001, 59, 462–469. [Google Scholar] [CrossRef]
- Bai, R.; Covell, D.G.; Liu, C.; Ghosh, A.K.; Hamel, E. (-)-doliculide, a new macrocyclic depsipeptide enhancer of actin assembly. J. Biol. Chem. 2002, 277, 32165–32171. [Google Scholar] [CrossRef]
- Cooper, J.A. Effects of cytochalasin and phalloidin on actin. J. Cell Biol. 1987, 105, 1473–1478. [Google Scholar] [CrossRef]
- Trendowski, M.; Mitchell, J.M.; Corsette, C.M.; Acquafondata, C.; Fondy, T.P. Chemotherapy with cytochalasin congeners in vitro and in vivo against murine models. Investig. New Drugs 2015, 33, 290–299. [Google Scholar] [CrossRef]
- Trendowski, M. Using Cytochalasins to Improve Current Chemotherapeutic Approaches. Anti-Cancer Agents Med. Chem. 2015, 15, 327–335. [Google Scholar] [CrossRef]
- Ruggiero, C.; Lalli, E. Targeting the cytoskeleton against metastatic dissemination. Cancer Metastasis Rev. 2021, 40, 89–140. [Google Scholar] [CrossRef]
- Xie, Z.; Janczyk, P.L.; Shi, X.; Wang, Q.; Singh, S.; Cornelison, R.; Xu, J.; Mandell, J.W.; Barr, F.G.; Li, H. Rhabdomyosarcomas are oncogene addicted to the activation of AVIL. Proc. Natl. Acad. Sci. USA 2022, 119, e2118048119. [Google Scholar] [CrossRef] [PubMed]
- Marks, P.W.; Arai, M.; Bandura, J.L.; Kwiatkowski, D.J. Advillin (p92): A new member of the gelsolin/villin family of actin regulatory proteins. J. Cell Sci. 1998, 111, 2129–2136. [Google Scholar] [CrossRef] [PubMed]
- Silacci, P.; Mazzolai, L.; Gauci, C.; Stergiopulos, N.; Yin, H.L.; Hayoz, D. Gelsolin superfamily proteins: Key regulators of cellular functions. Cell Mol. Life Sci. 2004, 61, 2614–2623. [Google Scholar] [CrossRef]
- Nag, S.; Larsson, M.; Robinson, R.C.; Burtnick, L.D. Gelsolin: The tail of a molecular gymnast. Cytoskeleton 2013, 70, 360–384. [Google Scholar] [CrossRef]
- George, S.P.; Esmaeilniakooshkghazi, A.; Roy, S.; Khurana, S. F-actin-bundling sites are conserved in proteins with villin-type headpiece domains. Mol. Biol. Cell 2020, 31, 1857–1866. [Google Scholar] [CrossRef]
- Ravenall, S.J.; Gavazzi, I.; Wood, J.N.; Akopian, A.N. A peripheral nervous system actin-binding protein regulates neurite outgrowth. Eur. J. Neurosci. 2002, 15, 281–290. [Google Scholar] [CrossRef]
- Ruppert, A.L.; Keshavarz, M.; Winterberg, S.; Oberwinkler, J.; Kummer, W.; Schütz, B. Advillin is a tuft cell marker in the mouse alimentary tract. J. Mol. Histol. 2020, 51, 421–435. [Google Scholar] [CrossRef]
- Hunter, D.V.; Smaila, B.D.; Lopes, D.M.; Takatoh, J.; Denk, F.; Ramer, M.S. Advillin Is Expressed in All Adult Neural Crest-Derived Neurons. eNeuro 2018, 5, ENEURO.0077-18.2018. [Google Scholar] [CrossRef]
- Chuang, Y.-C.; Lee, C.-H.; Sun, W.-H.; Chen, C.-C. Involvement of advillin in somatosensory neuron subtype-specific axon regeneration and neuropathic pain. Proc. Natl. Acad. Sci. USA 2018, 115, E8557–E8566. [Google Scholar] [CrossRef]
- Izdebska, M.; Zielińska, W.; Grzanka, D.; Gagat, M. The Role of Actin Dynamics and Actin-Binding Proteins Expression in Epithelial-to-Mesenchymal Transition and Its Association with Cancer Progression and Evaluation of Possible Therapeutic Targets. BioMed Res. Int. 2018, 2018, 4578373. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Janczyk, P.; Zhang, Y.; Liu, A.; Shi, X.; Singh, S.; Facemire, L.; Kubow, K.; Li, Z.; Jia, Y.; et al. A cytoskeleton regulator AVIL drives tumorigenesis in glioblastoma. Nat. Commun. 2020, 11, 3457. [Google Scholar] [CrossRef]
- Leiner, J.; Le Loarer, F. The current landscape of rhabdomyosarcomas: An update. Virchows Arch. 2020, 476, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Paulson, V.; Chandler, G.; Rakheja, D.; Galindo, R.L.; Wilson, K.; Amatruda, J.F.; Cameron, S. High-resolution array CGH identifies common mechanisms that drive embryonal rhabdomyosarcoma pathogenesis. Genes Chromosomes Cancer 2011, 50, 397–408. [Google Scholar] [CrossRef]
- Linardic, C.M. PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 2008, 270, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.D.; Grosveld, G.C. Alveolar rhabdomyosarcoma—The molecular drivers of PAX3/7-FOXO1-induced tumorigenesis. Skelet. Muscle 2012, 2, 25. [Google Scholar] [CrossRef]
- Jordan, M.A. Mechanism of action of antitumor drugs that interact with microtubules and tubulin. Curr. Med. Chem.-Anti-Cancer Agents 2002, 2, 1–17. [Google Scholar] [CrossRef]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).