
Ginsenoside F1-Mediated Telomere Preservation Delays Cellular Senescence

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
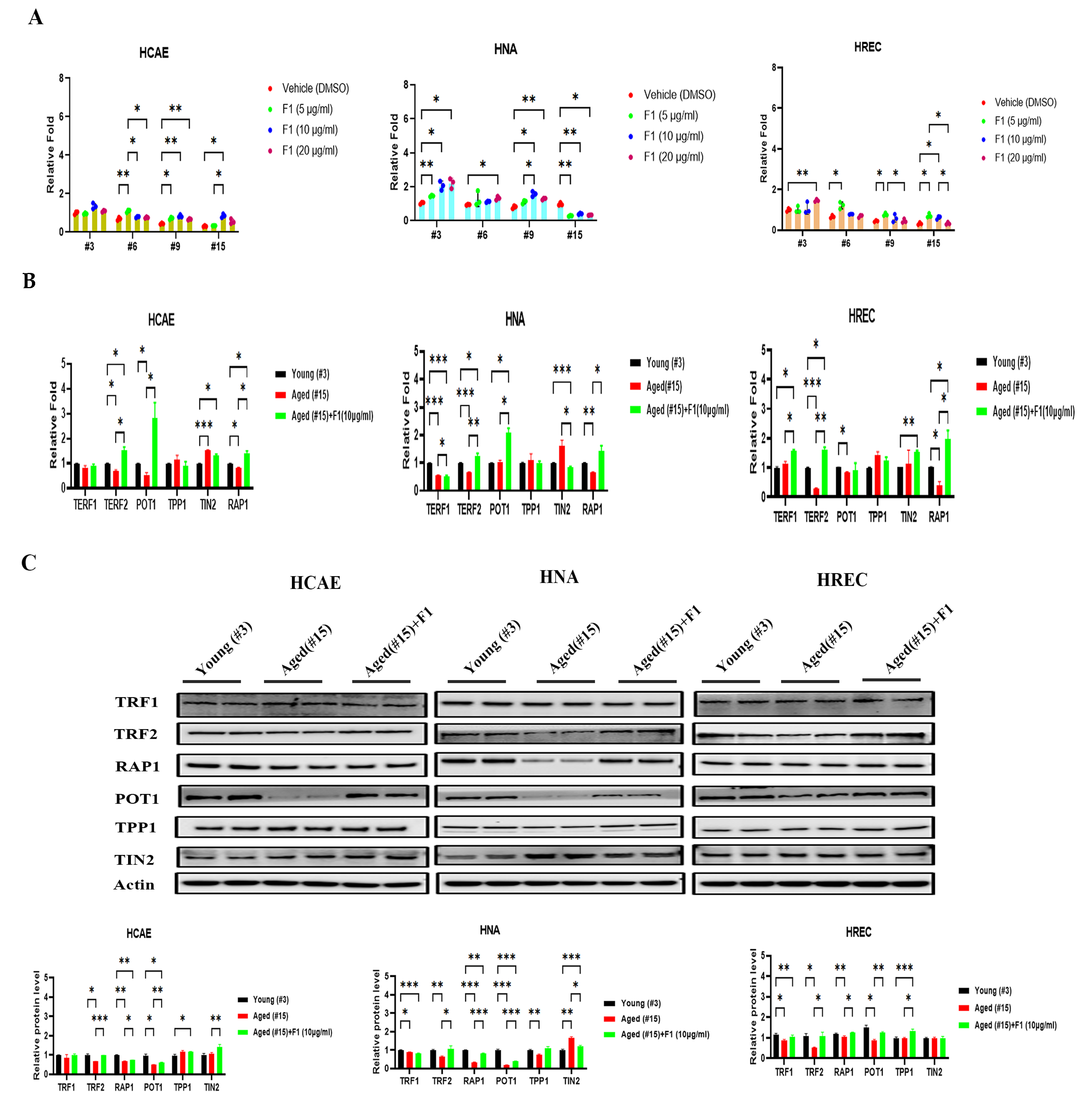
2.1. Ginsenoside F1 Preserved the Telomere Length in Aged Somatic Cells
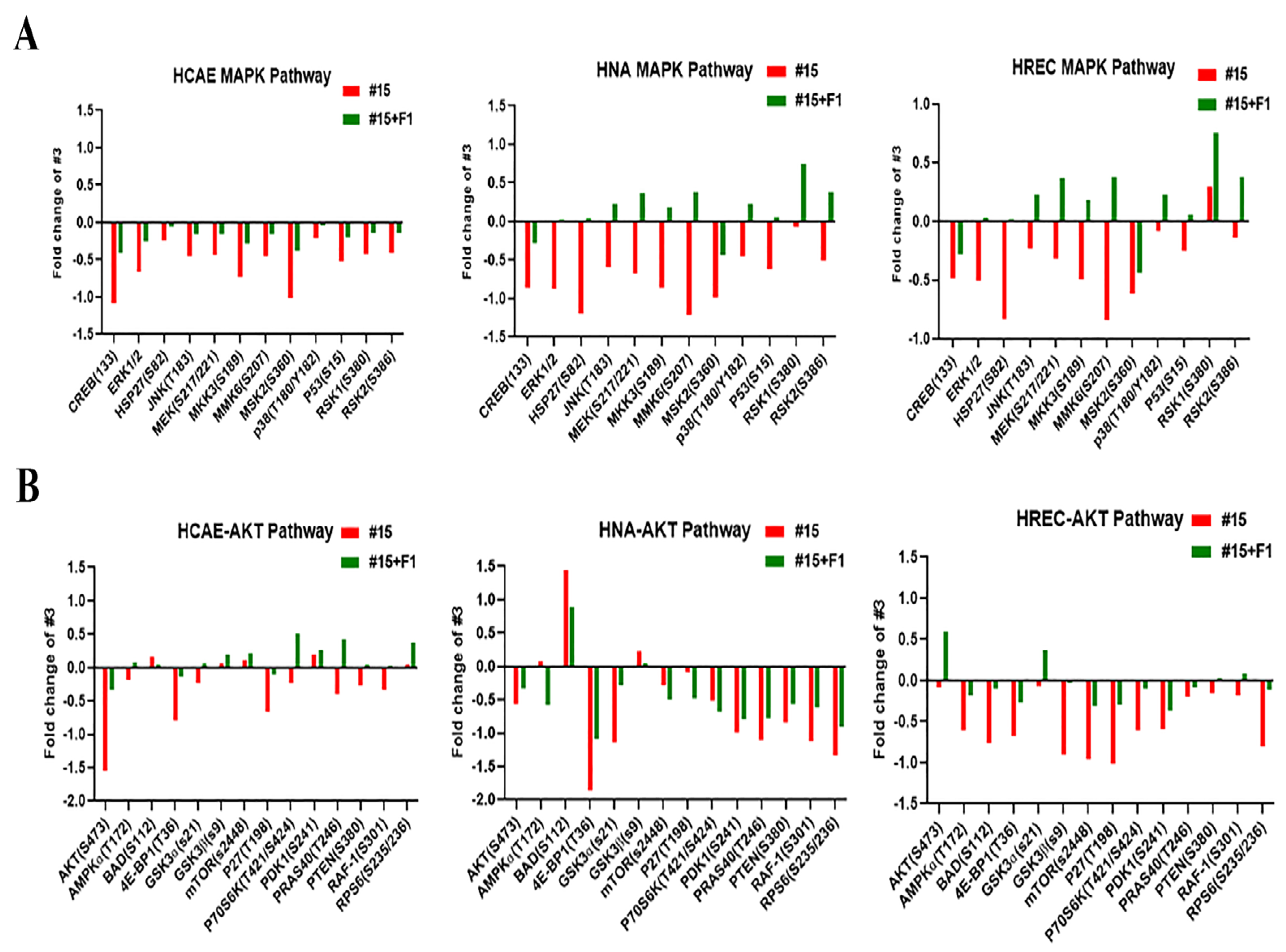
2.2. Ginsenoside F1 Modulated the Protein Kinase B (AKT) and Mitogen-Activated Protein Kinase (MAPK) Pathways in Aged Somatic Cells
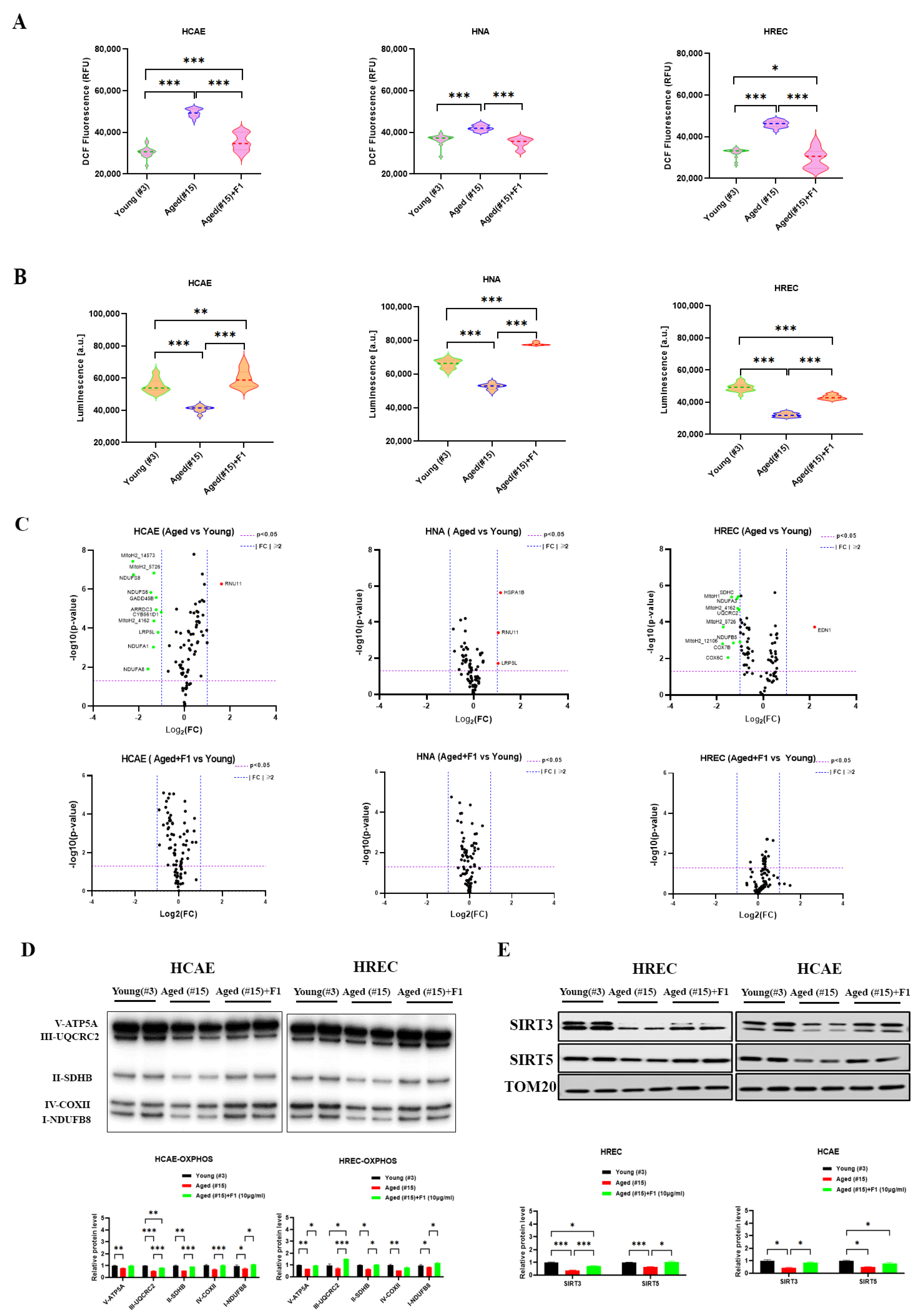
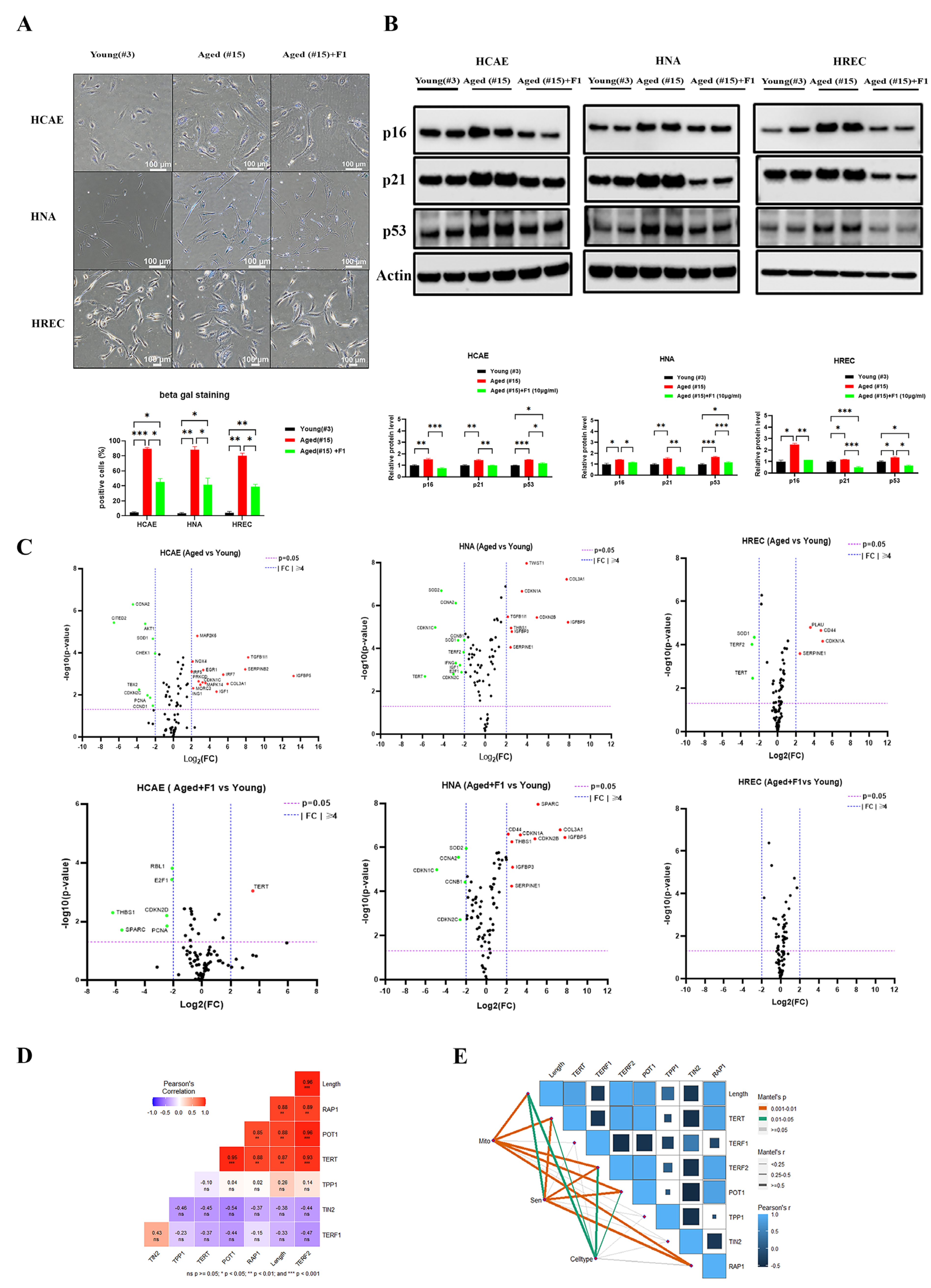
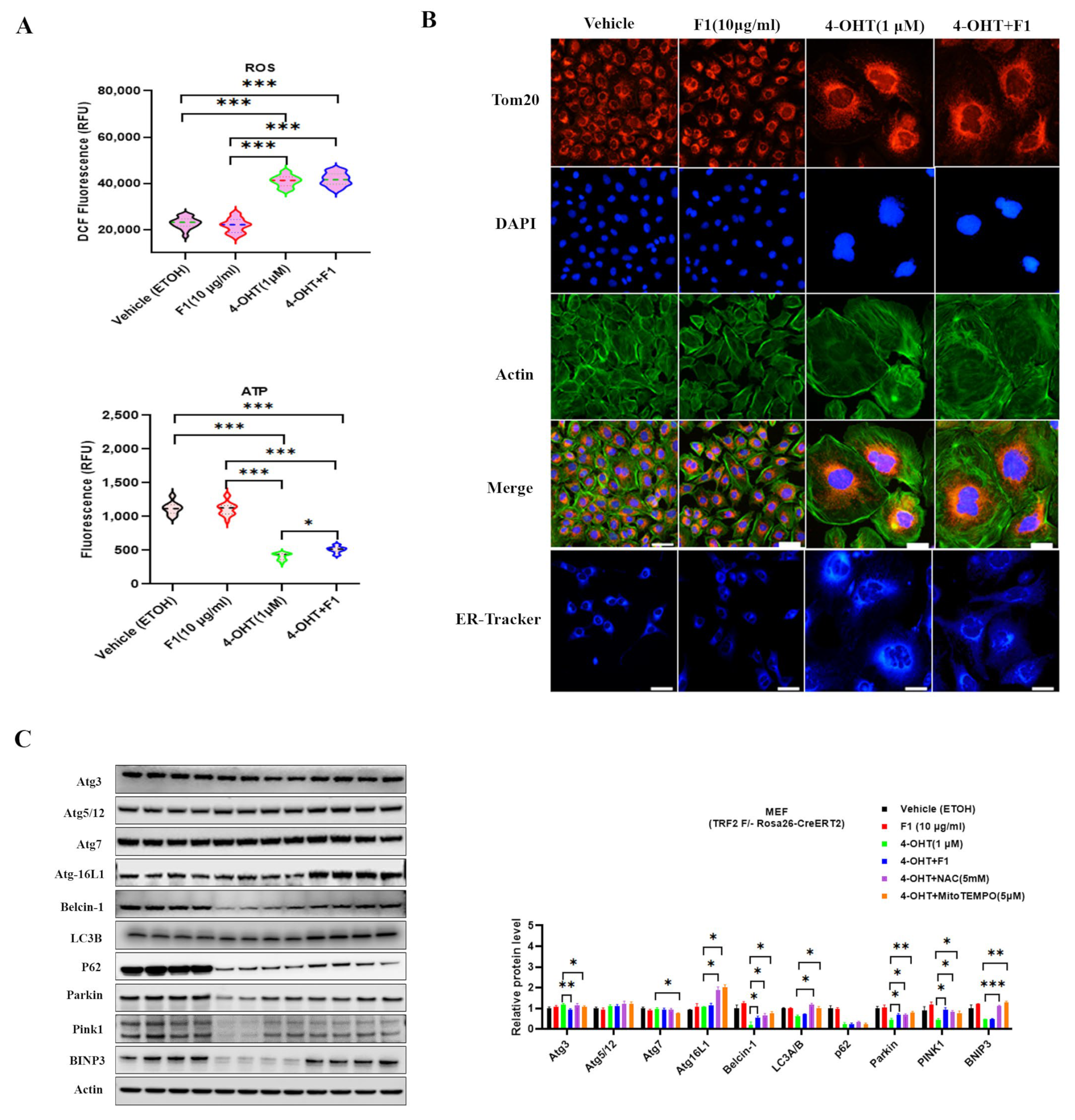
2.3. Ginsenoside F1 Ameliorated Mitochondrial Dysfunction and Cellular Senescence in Aged Somatic Cells
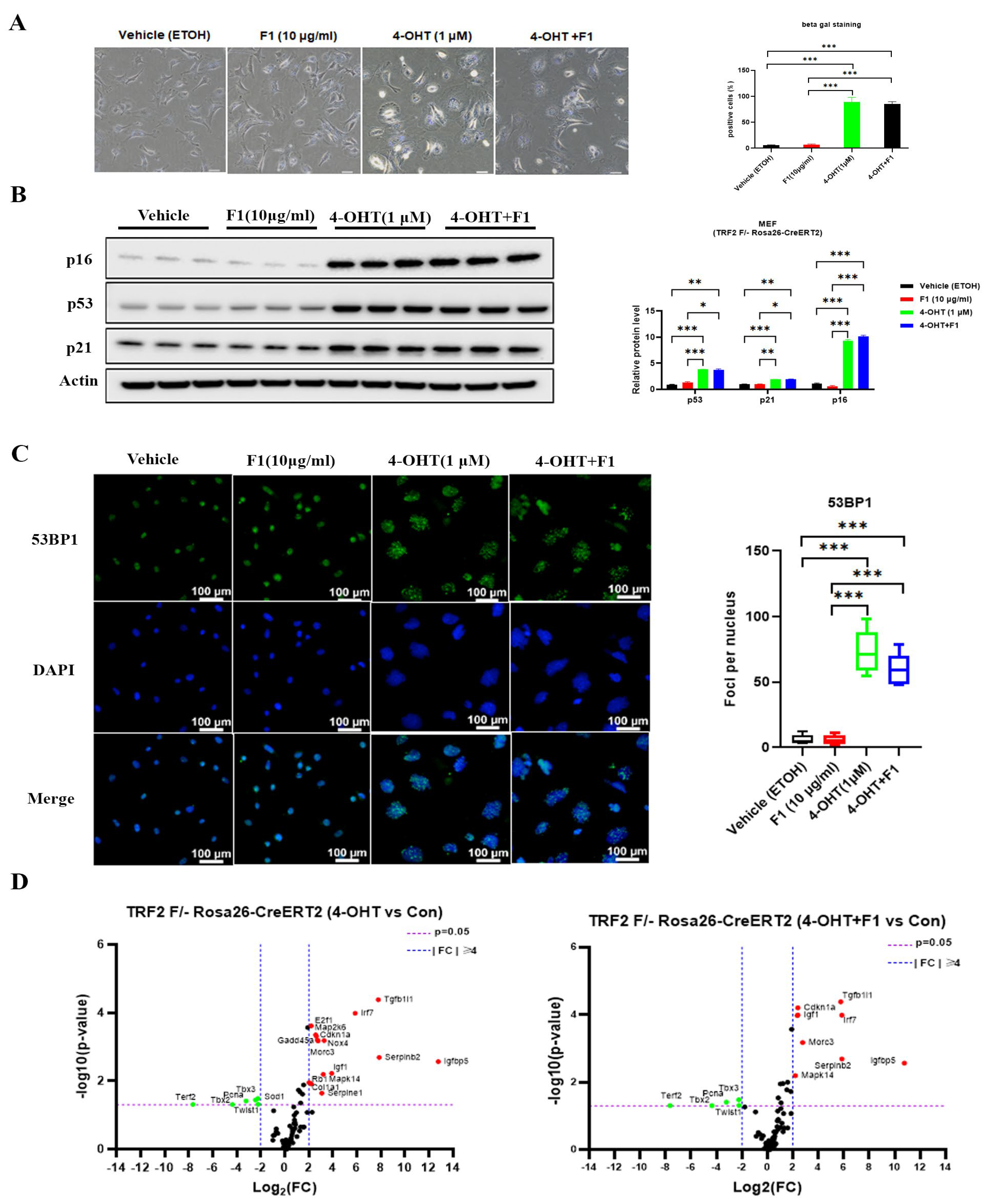
2.4. TRF2 Depletion-Induced Cellular Senescence
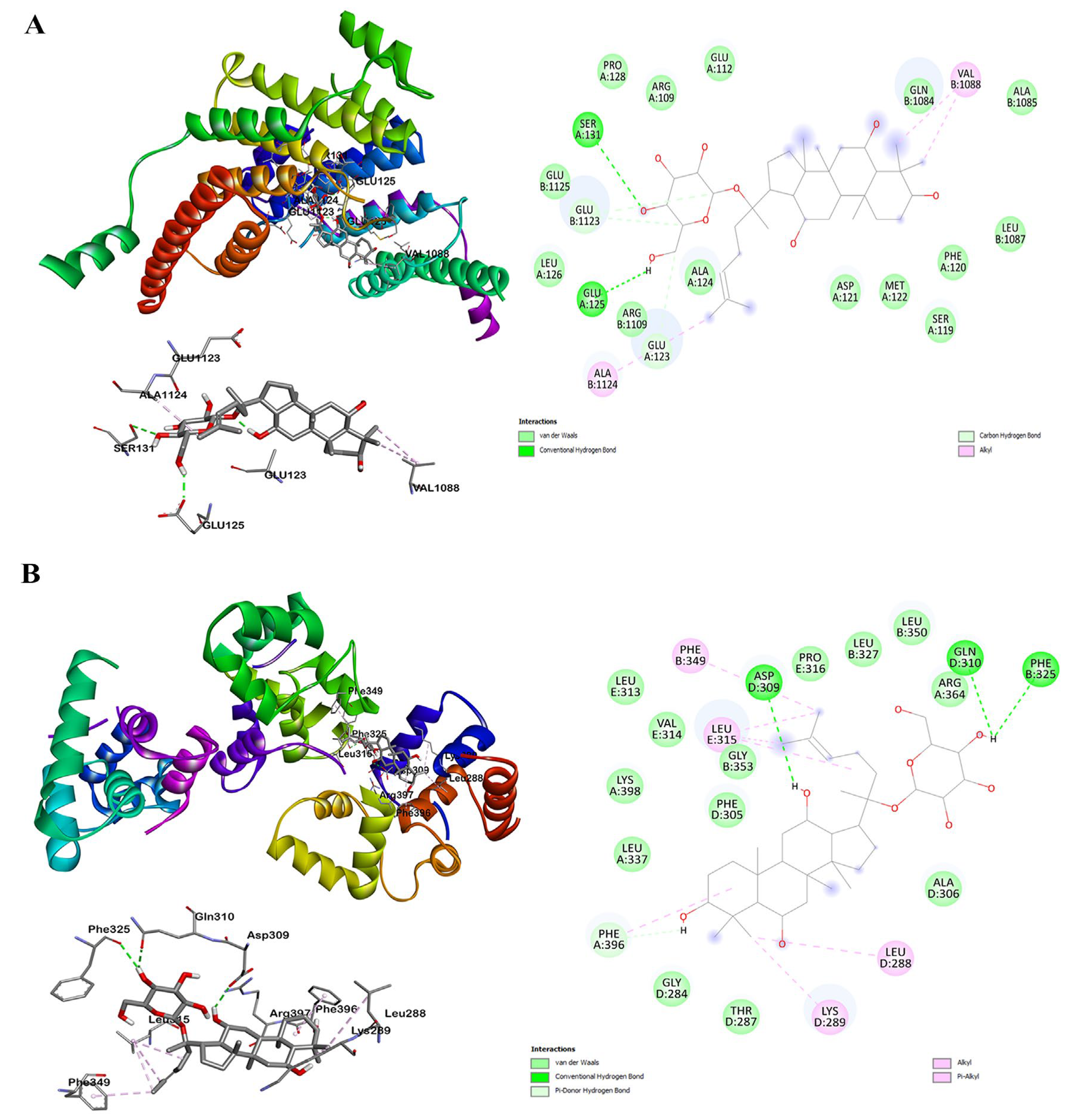
2.5. Molecular Docking Simulation in F1 and TRF2
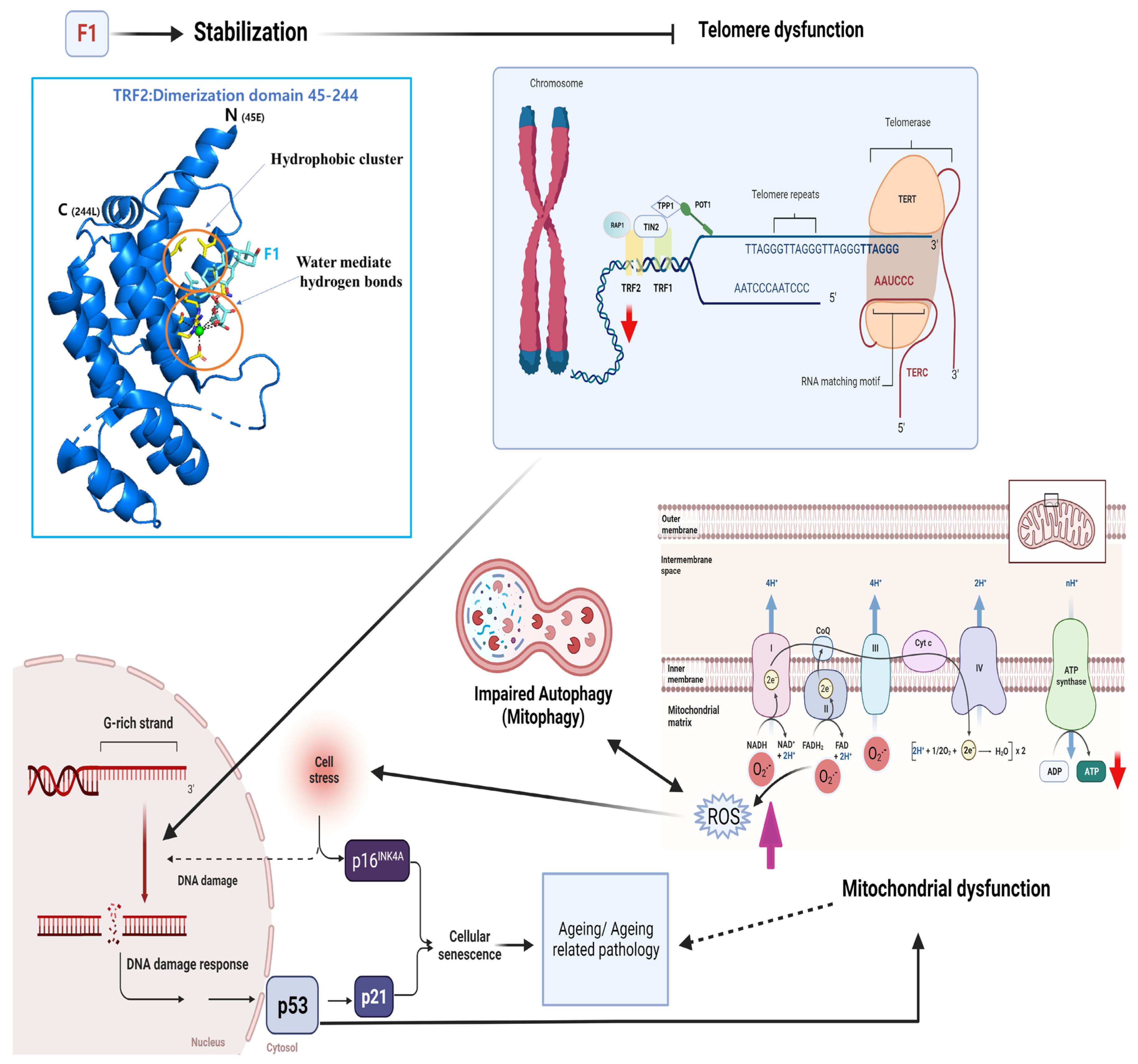
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cells and Cell Culture
4.3. Cell Aging
4.4. Telomere Length Assay
4.5. PCR Array
4.6. Antibody Array
4.7. Western Blot
4.8. Intracellular Superoxide Detection
4.9. ATP Assay
4.10. Senescence-Associated β-Galactosidase (SA-βG) Staining
4.11. Real-Time qPCR
4.12. Immunofluorescence
4.13. Molecular Docking
4.14. Statistical Analysis
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Chakravarti, D.; Hu, B.; Mao, X.; Rashid, A.; Li, J.; Li, J.; Liao, W.-T.; Whitley, E.M.; Dey, P.; Hou, P. Telomere dysfunction activates YAP1 to drive tissue inflammation. Nat. Commun. 2020, 11, 4766. [Google Scholar] [CrossRef]
- Yildirim, E.; Yaba, A. Determination of c-Abl tyrosine kinase and mTERT catalytic subunit of telomerase expression level during prenatal-postnatal mouse ovary-testis development. Reprod. Biol. 2020, 20, 555–567. [Google Scholar] [CrossRef]
- Niewisch, M.R.; Savage, S.A. An update on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev. Hematol. 2019, 12, 1037–1052. [Google Scholar] [CrossRef]
- Povedano, J.M.; Martinez, P.; Flores, J.M.; Mulero, F.; Blasco, M.A. Mice with pulmonary fibrosis driven by telomere dysfunction. Cell Rep. 2015, 12, 286–299. [Google Scholar] [CrossRef] [PubMed]
- Bär, C.; Povedano, J.M.; Serrano, R.; Benitez-Buelga, C.; Popkes, M.; Formentini, I.; Bobadilla, M.; Bosch, F.; Blasco, M.A. Telomerase gene therapy rescues telomere length, bone marrow aplasia, and survival in mice with aplastic anemia. Blood J. Am. Soc. Hematol. 2016, 127, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Daniali, L.; Benetos, A.; Susser, E.; Kark, J.D.; Labat, C.; Kimura, M.; Desai, K.K.; Granick, M.; Aviv, A. Telomeres shorten at equivalent rates in somatic tissues of adults. Nat. Commun. 2013, 4, 1597. [Google Scholar] [CrossRef] [PubMed]
- Blasco, M.A. Telomere length, stem cells and aging. Nat. Chem. Biol. 2007, 3, 640–649. [Google Scholar] [CrossRef]
- Feldser, D.M.; Hackett, J.A.; Greider, C.W. Telomere dysfunction and the initiation of genome instability. Nat. Rev. Cancer 2003, 3, 623–627. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Aging: Ros or tor. Cell Cycle 2008, 7, 3344–3354. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; Kotanko, P.; Schols, A.M.W.J.; Shiels, P.G.; Stenvinkel, P. Chronic kidney disease and premature ageing. Nat. Rev. Nephrol. 2014, 10, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Lee, S.; Kim, Y.; Cho, S.; Kim, K.; Kim, Y.C.; Han, S.S.; Lee, H.; Lee, J.P.; Joo, K.W. A Mendelian randomization study found causal linkage between telomere attrition and chronic kidney disease. Kidney Int. 2021, 100, 1063–1070. [Google Scholar] [CrossRef]
- Gao, K.; Wei, C.; Zhu, J.; Wang, X.; Chen, G.; Luo, Y.; Zhang, D.; Yue, W.; Yu, H. Exploring the causal pathway from telomere length to Alzheimer’s disease: An update Mendelian randomization study. Front. Psychiatry 2019, 10, 843. [Google Scholar] [CrossRef] [PubMed]
- Rolyan, H.; Scheffold, A.; Heinrich, A.; Begus-Nahrmann, Y.; Langkopf, B.H.; Hölter, S.M.; Vogt-Weisenhorn, D.M.; Liss, B.; Wurst, W.; Lie, D.C. Telomere shortening reduces Alzheimer’s disease amyloid pathology in mice. Brain 2011, 134, 2044–2056. [Google Scholar] [CrossRef] [PubMed]
- Fyhrquist, F.; Saijonmaa, O.; Strandberg, T. The roles of senescence and telomere shortening in cardiovascular disease. Nat. Rev. Cardiol. 2013, 10, 274–283. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Feng, X.; Wang, H.; Xu, W.; Zhao, Y.; Ma, W.; Jiang, S.; Liu, D.; Huang, J.; Songyang, Z. Mir-23a induces telomere dysfunction and cellular senescence by inhibiting TRF 2 expression. Aging Cell 2015, 14, 391–399. [Google Scholar] [CrossRef]
- Fujita, K.; Horikawa, I.; Mondal, A.M.; Jenkins, L.M.M.; Appella, E.; Vojtesek, B.; Bourdon, J.-C.; Lane, D.P.; Harris, C.C. Positive feedback between p53 and TRF2 during telomere-damage signalling and cellular senescence. Nat. Cell Biol. 2010, 12, 1205–1212. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Oh, J.-P.; Yoo, M.; Cui, C.-H.; Jeon, B.-M.; Kim, S.-C.; Han, J.-H. Minor ginsenoside F1 improves memory in APP/PS1 mice. Mol. Brain 2019, 12, 77. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Cui, C.; Kim, S.; Sung, C.; Choi, C. Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 2018, 283, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Jeon, B.; Baek, J.; Yun, Y.; Kim, D.; Chang, B.; Kim, S.; Kim, S. High fat diet-induced brain damaging effects through autophagy-mediated senescence, inflammation and apoptosis mitigated by ginsenoside F1-enhanced mixture. J. Ginseng Res. 2022, 46, 79–90. [Google Scholar] [CrossRef]
- Yun, Y.-J.; Park, B.-H.; Hou, J.; Oh, J.-P.; Han, J.-H.; Kim, S.-C. Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP. Life 2022, 12, 58. [Google Scholar] [CrossRef]
- Chakravarti, D.; LaBella, K.A.; DePinho, R.A. Telomeres: History, health, and hallmarks of aging. Cell 2021, 184, 306–322. [Google Scholar] [CrossRef]
- Lanza, R.P.; Cibelli, J.B.; Blackwell, C.; Cristofalo, V.J.; Francis, M.K.; Baerlocher, G.M.; Mak, J.; Schertzer, M.; Chavez, E.A.; Sawyer, N. Extension of cell life-span and telomere length in animals cloned from senescent somatic cells. Science 2000, 288, 665–669. [Google Scholar] [CrossRef]
- Erdel, F.; Kratz, K.; Willcox, S.; Griffith, J.D.; Greene, E.C.; de Lange, T. Telomere Recognition and Assembly Mechanism of Mammalian Shelterin. Cell Rep. 2017, 18, 41–53. [Google Scholar] [CrossRef]
- Markiewicz-Potoczny, M.; Lobanova, A.; Loeb, A.M.; Kirak, O.; Olbrich, T.; Ruiz, S.; Lazzerini Denchi, E. TRF2-mediated telomere protection is dispensable in pluripotent stem cells. Nature 2021, 589, 110–115. [Google Scholar] [CrossRef]
- Ye, J.; Lenain, C.; Bauwens, S.; Rizzo, A.; Saint-Léger, A.; Poulet, A.; Benarroch, D.; Magdinier, F.; Morere, J.; Amiard, S.; et al. TRF2 and Apollo Cooperate with Topoisomerase 2α to Protect Human Telomeres from Replicative Damage. Cell 2010, 142, 230–242. [Google Scholar] [CrossRef]
- Méndez-Pertuz, M.; Martínez, P.; Blanco-Aparicio, C.; Gómez-Casero, E.; Belen García, A.; Martínez-Torrecuadrada, J.; Palafox, M.; Cortés, J.; Serra, V.; Pastor, J. Modulation of telomere protection by the PI3K/AKT pathway. Nat. Commun. 2017, 8, 1278. [Google Scholar] [CrossRef]
- Sahin, E.; Colla, S.; Liesa, M.; Moslehi, J.; Müller, F.L.; Guo, M.; Cooper, M.; Kotton, D.; Fabian, A.J.; Walkey, C.; et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature 2011, 470, 359–365. [Google Scholar] [CrossRef]
- Schmutz, I.; Timashev, L.; Xie, W.; Patel, D.J.; de Lange, T. TRF2 binds branched DNA to safeguard telomere integrity. Nat. Struct. Mol. Biol. 2017, 24, 734–742. [Google Scholar] [CrossRef]
- Reinhardt, H.C.; Schumacher, B. The p53 network: Cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012, 28, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Fairall, L.; Chapman, L.; Moss, H.; de Lange, T.; Rhodes, D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol. Cell 2001, 8, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Asghar, M.; Odeh, A.; Fattahi, A.J.; Henriksson, A.E.; Miglar, A.; Khosousi, S.; Svenningsson, P. Mitochondrial biogenesis, telomere length and cellular senescence in Parkinson’s disease and Lewy body dementia. Sci. Rep. 2022, 12, 17578. [Google Scholar] [CrossRef] [PubMed]
- An, D.-S.; Cui, C.-H.; Siddiqi, M.Z.; Yu, H.S.; Jin, F.-X.; Kim, S.-G.; Im, W.-T. Gram-scale production of ginsenoside F1 using a recombinant bacterial β-glucosidase. J. Microbiol. Biotechnol. 2017, 27, 1559–1565. [Google Scholar] [CrossRef]
- Sunagawa, S.; Coelho, L.P.; Chaffron, S.; Kultima, J.R.; Labadie, K.; Salazar, G.; Djahanschiri, B.; Zeller, G.; Mende, D.R.; Alberti, A.; et al. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hou, J.; Yun, Y.; Jeon, B.; Baek, J.; Kim, S. Ginsenoside F1-Mediated Telomere Preservation Delays Cellular Senescence. Int. J. Mol. Sci. 2023, 24, 14241. https://doi.org/10.3390/ijms241814241
Hou J, Yun Y, Jeon B, Baek J, Kim S. Ginsenoside F1-Mediated Telomere Preservation Delays Cellular Senescence. International Journal of Molecular Sciences. 2023; 24(18):14241. https://doi.org/10.3390/ijms241814241
Chicago/Turabian StyleHou, Jingang, Yeejin Yun, Byeongmin Jeon, Jongin Baek, and Sunchang Kim. 2023. "Ginsenoside F1-Mediated Telomere Preservation Delays Cellular Senescence" International Journal of Molecular Sciences 24, no. 18: 14241. https://doi.org/10.3390/ijms241814241
APA StyleHou, J., Yun, Y., Jeon, B., Baek, J., & Kim, S. (2023). Ginsenoside F1-Mediated Telomere Preservation Delays Cellular Senescence. International Journal of Molecular Sciences, 24(18), 14241. https://doi.org/10.3390/ijms241814241