Abstract
The spectral and dynamic properties of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) in a series of 1-alkanols ranging from methanol to 1-decanol over a temperature range 100–300 K were investigated by electron spin resonance (ESR). The main characteristic ESR temperatures connected with slow to fast motion regime transition; T50G ‘s and TX1fast ‘s are situated above the corresponding glass temperatures, Tg, and for the shorter members, the T50G ‘s lie above or close to melting point, Tm, while the longer ones the T50G < Tm relationship indicates that the TEMPO molecules are in the local disordered regions of the crystalline media. The T50G ‘s and especially TX1fast ‘s are compared with the dynamic crossover temperatures, TXVISC = 8.72M0.66, as obtained by fitting the viscosity data in the liquid n-alkanols with the empirical power law. In particular, for NC > 6, the TX1fast ‘s lie rather close to the TXVISC resembling apolar n-alkanes [PCCP 2018,20,11145-11151], while for NC < 6, they are situated in the vicinity of Tm. The absence of a coincidence for lower1-alkanols indicates that the T50G is significantly influenced by the mutual interaction between the polar TEMPO and the protic polar medium due to the increased polarity and proticity destroyed by the larger-scale melting transition.
1. Introduction
In general, the dynamics of glass-forming liquids, i.e., organics and inorganics forming the supercooled liquid by their cooling below the melting temperature, Tm, and finished via a liquid−to−glass transition to a glass below the glass temperature, Tg, is non-monotonous and exhibits a change at the so-called dynamic crossover temperature, Tcross = TB or TX, lying between Tm and Tg [1,2,3,4,5,6,7,8,9,10,11,12]. This dynamic crossover phenomenon between the relatively weakly and strongly changing supercooled liquid dynamics is observed using experimental techniques, such as viscosity (VISC) at TB,η or TX [1,2], and dielectric spectroscopy (DS) at TB,DSST [3,4] or TB,DSMG [5], as well as TB,DSKWW [6] and TB,DSSCH [7].
Usually, the crossover temperatures are determined by fitting the supercooled liquid dynamics using a combination of classic phenomenological expressions for viscosity, η, or structural relaxation time, τ, such as the Vogel–Fulcher–Tamman–Hesse (VFTH) equation [1], or using the power law (PL) equation [2]. Lately, a special evaluation method using Stickel’s temperature-derivative analysis or the more general Martinez–Garcia apparent enthalpy analysis of the relevant dynamic quantities, giving TB,DS ST and TB,DS MG, respectively, was proposed [3,4,5]. Other ways of determination of the crossover temperature are based on the onset of the increasing broadening of the frequency dispersion of the structural relaxation time distribution, and on the change in structural relaxation strength, Δεα, leading to TB,DS KWW or TB,DS SCH, respectively [6,7]. In the case of the PL eq. with TX [2,9], this expression is rationalized theoretically using the idealized mode coupling theory (I−MCT) of liquid dynamics [11] via the derivation of the same form of temperature dependence for viscosity and relaxation time using the so-called critical temperature, Tc ≈ TX.
The crossover transition is a very significant feature of the supercooled liquid’s behavior, as has been demonstrated by the findings of several empirical correlations of TB or TX with various characteristic temperatures of a variety of structural–dynamic phenomena, such as the decoupling or bifurcation of the primary α relaxation and the secondary β process, Tαβ, from DS or dynamic light scattering (DLS) [8]. Moreover, the crossover phenomenon is also illustrated by various extrinsic probe techniques, such as FS, ESR and PALS. These have revealed the decoupling of the translation from the rotation of molecular probes and medium dynamics at Tdecoup, either for the relatively large fluorescence probes via fluorescence spectroscopy (FS) [13] or the decoupling of rotation of the spin probes from the medium dynamics, using electron spin resonance (ESR) [14]. Finally, crossover in the supercooled liquid state is also manifested by a bend effect in ortho-positronium lifetime τ3 vs. T dependence, as detected by positron annihilation lifetime (PALS). This slope change reflects a change in the free volume expansion at the characteristic PALS temperature Tb1L above Tg in amorphous glass−formers [15]. Evidently, it is increasingly recognized that the dynamic crossover before glass transition temperature plays an essential if not fundamental role in our understanding of the glass transition phenomenon [9].
In contrast to the aforementioned cases of amorphous glass-formers, observations of crossover transition in strongly crystallizing, i.e., relatively hardy supercooled apolar and polar organics, is substantially more difficult. This is connected with the problem of the formation of sufficiently large amorphous domains in the otherwise dominantly ordered material, and subsequently, with their characterization by suitable experimental techniques. Recently, we have proposed one special method for creating such amorphous domains in crystalline materials, consisting of the introduction of an appropriate molecular probe disordering the immediate surroundings of the otherwise ordered medium. This includes the spin probe (2,2,6,6−tetramethyl piperidin−1−yl) oxyl (TEMPO) with VTEMPOvdW = 170 Å3 in a series of apolar n−alkanes ranging from n-hexane to n−nonadecane using the ESR technique [16]. On the basis of the close correlation between one of the characteristic ESR temperatures, namely, TX1fast, lying a bit above the main slow−to−fast transition at T50G, and marking the onset of the pure fast motion regime of TEMPO and the crossover temperatures, TX VISC, as obtained from fitting the corresponding viscosity data for a series of n-alkanes using the PL eq., dynamic crossovers in the local disordered regions around the probe molecules in the otherwise dominantly crystalline organics were detected.
One of the most important aspects of extrinsic probe techniques such as ESR is the potential interaction between the probe used and the medium’s constituents, which can to a greater or lesser extent influence the corresponding probe response to the investigated organic matrix. In our previous work on a series of apolar crystallizing n-alkanes, we used one of the smallest polar spin probes, TEMPO, where in this interaction aspect is supposed to be small [16]. The aim of this work was to test other types of strongly crystallizing organic media consisting of protic polar compounds, such as 1−alkanols, with the potential for an intermolecular H−bonding interaction, not only between its own polar molecules but also between these polar molecules and polar spin probe TEMPO. The spectral and dynamic data obtained for the TEMPO on the family of aliphatic monoalcohols or 1−alkanols H(CH2)N OH with NC = 1–10, i.e., ranging from methanol to 1−decanol, are interpreted using the newly analyzed viscosity data in the literature in order to reveal the roles of the thermodynamic and dynamic transitions, as well as of the interaction aspect, in the main slow-to-fast transition behavior of the spin probe TEMPO used.
2. Results and Discussion
2.1. Thermodynamic and Crossover Transition Behaviors in 1−alkanols
It is well known that 1−alkanols, similarly to n−alkanes, belong to the class of relatively easily crystalizing organic compounds. For this strong ordering tendency, they must have special means of preparation of the totally or partially amorphous samples, with the one exception of 1−propanol (C3OH), which is a very good glass−former [17].
2.1.1. Thermodynamic Transitions in 1−alkanols
Figure 1 and Figure 2 and Table 1 summarize the data from the literature on the three basic thermodynamic transitions of condensed materials, i.e., the glass−to−liquid (devitrification) transition of the amorphous phase, the solid-to-liquid (melting) transition of the crystalline phase, as well as the liquid−to−gas (evaporation) transition of the liquid phase to gas. In general, the corresponding transition temperatures, i.e., glass temperature Tg and melting temperature Tm, for 1−alkanols, exhibit a non-monotonous characteristic as a function of their molecular, size expressed by the number of carbon atoms in the chain, Nc, or molecular weight, M, while the boiling temperature, Tb, shows a monotonous type of dependence over the whole molecular size interval. In contrast to the melting with well-defined Tm values [18], the Tg values measured so far exhibit a scatter up to 10 K, which depends on both the preparation procedure and the measuring technique, such as dynamic–mechanical spectroscopy (DMS), differential thermal analysis (DTA) or differential scanning calorimetry (DSC), and dielectric spectroscopy (DS) [19,20,21,22,23,24,25,26,27,28], with this value apparently diminishing with increasing molecular size. In spite of this fact, the Tg value for the shortest 1−alkanols decreases from methanol (C1OH) to ethanol (C2OH), followed by a monotonous increasing trend, starting from C2OH in the DMS data [26], or from 1−propanol (C3OH) in the CAL data [17]. As originally proposed by Faucher and Koleske [26], the DMS results could be described by the power law (PL)-type expression as a function of molecular weight, M:
where A and α are empirical parameters of a given homologous series of compounds. As seen in Figure 1A, they exhibit similar trends depending relatively strongly on the method of generation of the amorphous material and the set up used in DMS or DTA, respectively. The latter values of Tg DTA coinciding with Tg CAL in cases of lower 1-alkanols, such as ethanol, 1−propanol and 1−butanol, as studied by the special CAL technique, i.e., quasi-adiabatic calorimetry (QADC) [17,28], are considered to be more reliable, mainly because of the experimental complexity of DMS.
Tg = AM α
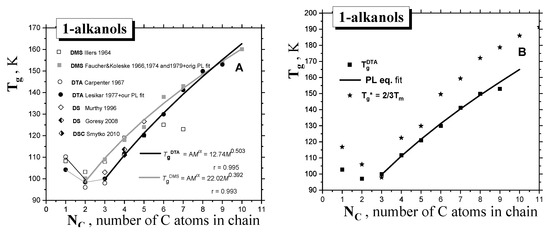
Figure 1.
(A) Glass−to−liquid transition temperature Tg of 1−alkanols as a function of the molecular size expressed by the number of C atoms in the chain, NC. Two fits of the Tg values from the DMS [20,26] and DTA data sets [22] via the PL equation (Tg = AM α) are included, (B) Comparison of the Tg DTA values with the empirical rule: Tg* = (2/3) Tm.
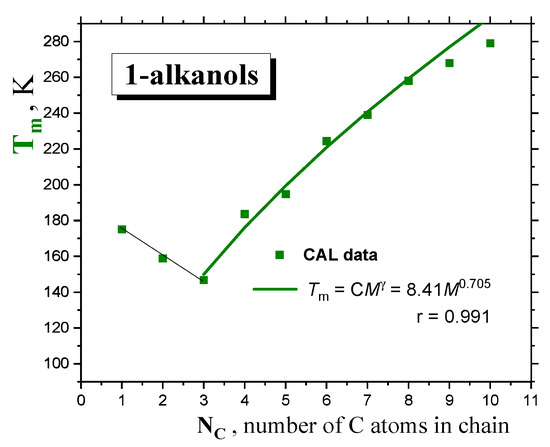
Figure 2.
The melting temperature Tm of 1-alkanols as a function of the molecular size expressed by the number of C atoms in the chain, NC. Fit of the Tm’s from the CAL data set from Ref. [18] via the PL equation Tm = CM γ is included.
As for the melting transition of 1−alkanols, the same expression can be approximately used for the melting points:
where C = 8.41 and γ = 0.705 are empirical parameters of the melting of a given homologous series of compounds, as derived mainly from the calorimetric data [18] in Figure 2. A similar approach has recently been applied for n−alkanes and monoalcohols in spite of the very pronounced zig-zag effect for the former, with a similar γ value of 0.7, as given by Novikov and Rössler [29].
Tm = CM γ
Finally, in Figure 1B, the estimated values of glass transition, Tg*, calculated according to the well-known empirical rule for many organic and inorganic glass-formers (Tg* = (2/3) × Tm—see e.g., Refs. [29,30,31,32]), are also listed. Given their comparison with the measured Tg data from DTA or DMS, it follows that this rule is not valid for our series of the first ten 1-alkanols, with the one exception of C3OH. Alternatively, the measured Tm CAL/Tg DTA ratios fulfill the rather different empirical rule of ~1.70, instead of Tm/Tg* = (3/2) = 1.50, which is valid again for C3OH only—see Figure 3.
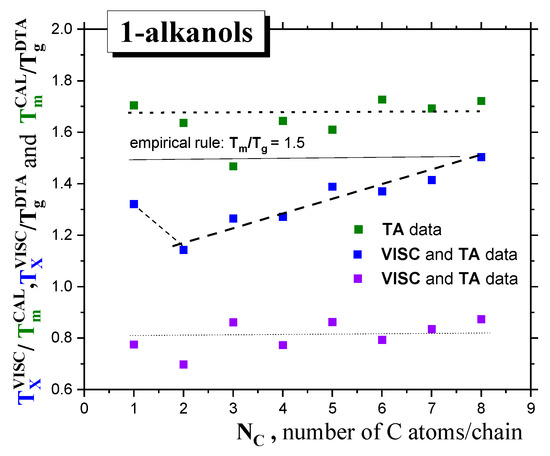
Figure 3.
The Tm/Tg, TX/Tg and Tm/TX ratios of 1-alkanols as a function of the molecular size expressed by the number of C atoms in the chain, NC.
In the case of Tm, the full horizontal lines represent the constant value of Tm/Tg = 1.5 derived from the empirical rule, and the dotted line represents Tm/Tg~1.68 for our series of 1-alkanols, whereas in the case TX, with one exception for C1OH, an increasing linear trend of TX/Tg is found. Finally, TX/Tm~0.81.
2.1.2. Dynamic Crossovers in 1-alkanols
As mentioned in the introduction, the crossover temperature in the supercooled liquid state of many organic compounds can be obtained using the power law (PL) equation, connecting the viscosity of liquids, η, with temperature T in the normal liquid and weakly supercooled liquid states [2,9,16]. Figure 4, Figure 5 and Figure 6, as well as Table 1, give the results of this method of determination of the crossover temperature, TX. Thus, Figure 4 presents compilations of the viscosity data for a series of ten 1-alkanols ranging from methanol (C1OH) up to 1-decanol (C10OH) as a function of temperature, mostly derived from the two large summarizing literature sources [33,34]. Most viscosity data of 1-alkanols fall into the normal liquid state between the melting temperature, Tm, and the boiling temperature, Tb. For the two lower members of this series, namely, ethanol (C2OH) and 1-propa nol (C3OH), the viscosities were also measured in the supercooled liquid state below the corresponding Tm’s; these values were only slightly lower for C2OH [35], but in C3OH they almost reached down to the corresponding Tg, because of its very good glass-forming ability [17,35,36,37]. Moreover, the additional liquid data from ref. [38] are included.
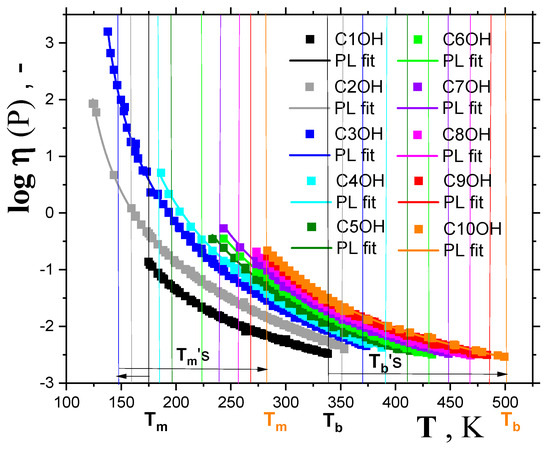
Figure 4.
Viscosities for a series of 1–alkanols as a function of temperature together with the respective power law (PL) fittings given by Equation (3). The vertical lines in the same colors as the corresponding experimental points for each member of a series of 1–alkanols mark the melting temperatures (on the left) and the boiling points (on the right), with two extrema demonstrations for methanol (black points and lines) and 1–decanol (orange points and lines). Trends in Tm and Tb are shown by the two arrows at the bottom of the plot.
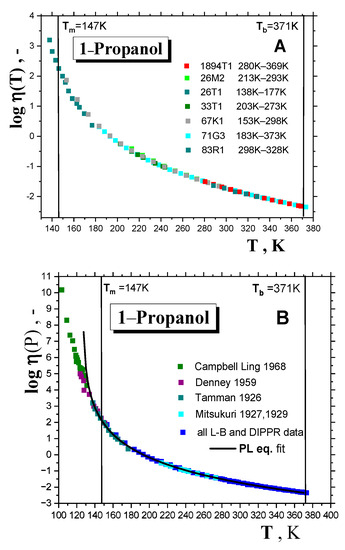
Figure 5.
Viscosity of a good glass–former 1–propanol over (A) a restricted temperature range from Tb down to slightly below Tm using the data sets summarized in Ref. [33] (Landolt–Börnstein Table data) and Ref. [34] (DIPPR data), and (B) an extraordinarily wide temperature range from Tb down almost to Tg with the addition data from Refs. [35,36,37] in the strongly supercooled liquid state, as well as from Ref. [38] in the liquid one, together with the best PL equation fitting. The original references marked, such as 1891T1 and 26M2, can be found in Ref. [33].
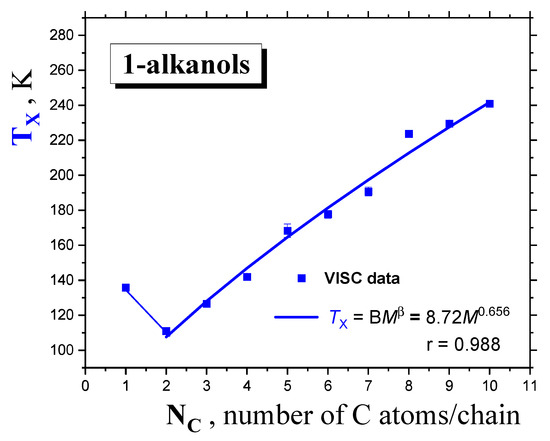
Figure 6.
Dynamic crossover temperature TX of 1−alkanols as a function of the molecular size expressed by the number of C atoms in the chain NC. The fit of the TX values from VISC data via the PL equation of the form TX = BM β is included.
All the viscosity data for the series of the first ten 1-alkanols can be described by the power law (PL) equation:
where η∞ is the pre-exponential factor, TX is the empirical characteristic dynamic PL temperature or the theoretical critical MCT temperature Tc, and μ is a non-universal coefficient. The corresponding fitting curves are plotted in Figure 4 and the obtained crossover temperatures TX are listed in Table 1 and Figure 6. In the above-mentioned case of 1-alkanols for which viscosity data in the supercooled liquid state also exist [35,36,37], the TX values for, e.g., C2OH, extracted from fitting over the usual normal liquid state ranging Tb−Tm = 192 K, and over the whole accessible temperature range of 227 K [35] (Tb−Tmin = 227 K), change by 4 K only. Similarly, for the very good glass-former C3OH, this difference reaches 3 K, which is towards the lower value, as it also includes the strongly supercooled liquid range data from Ref. [35]—see Figure 5A,B. Thus, the PL equation fit over the normal liquid state appears to be a very good approximation only for obtaining the TX values lying in the supercooled liquid state below the corresponding Tm values in strongly crystallizing organics, such as 1-alkanols. These are also listed in Table 1, together with the few determinations based on the first three members, namely, C1OH, C2OH and C3OH, as given by other authors [2,5,9].

Table 1.
Basic physical properties of investigated 1-alkanols.
Table 1.
Basic physical properties of investigated 1-alkanols.
| 1-alkanol | M g/mol | Tg K | TX K | Tma K | Tba K | Azz’ (100K) b G | Aiso (RT) c G | μgd D | μle D | εrd - |
|---|---|---|---|---|---|---|---|---|---|---|
| MeOH | 32.04 | 108.2 f 110.2 g 103 h 104.2 i | 135.8 1352 | 175.2 | 338 | 37.87 | 16.46 | 1.70 | 2.70 | 33 |
| EtOH | 46.07 | 103.2 f 100.2 g 98.4 i 96 j 97 k | 111 1115 | 158.9 | 351 | 36.81 | 16.30 | 1.69 | 2.81 | 25.3 |
| 1-PrOH | 60.10 | 108 f 98 j 109 g 100 i 103 l 110 m | 126.5 1399 | 146.8 | 371 | 36.15 | 16.23 | 1.68 | 2.87 | 20.8 |
| 1-BuOH | 74.12 | 119 f 111.7 i 113.6 n 111 o 119 m | 142 | 183.7 | 391 | 35.95 | 16.16 | 1.66 | 2.88 | 17.8 |
| 1-PentOH | 88.15 | 120.1 f 124 g 120 i 126.1 l 127.4 m | 168 | 194.8 | 411 | 35.9 | 16.05 | 1.70 | 2.97 | 15.1 |
| 1-HxOH | 102.18 | 125 f 138 g 129.9 i 135 m | 178 | 224.4 | 430 | 35.75 | 15.91 | 1.65 | 2.87 | 13.0 |
| 1-HptOH | 116.20 | 123 f 143 g 141.2 i 141.9 m | 199.7 | 239 | 449 | 35.60 | 15.88 | 1.71 | 2.99 | 11.75 |
| 1-OctOH | 130.23 | 149 g 149.9 i 148.3 m | 225.4 | 258.1 | 468 | 35.45 | 15.83 | 1.68 | 2.90 | 10.30 |
| 1-NonOH | 144.25 | 153 i 154.4 m | 236 | 268 | 487 | 35.25 | 15.75 | 1.60 | 2.73 | 8.83 |
| 1-DecOH | 158.28 | (160.1) m | 239 | 279 | 501 | 35.15 | 15.70 | 1.60 | 2.70 | 7.93 |
a Ref. [18]; b uncertainty = 0.05 G; c uncertainty = 0.03 G; d Ref. [33]; e Ref. [39]; f Ref. [19]; g Ref. [20]; h Ref. [21]; i Ref. [22]; j Ref. [23]; k Ref. [24]; l Ref. [25]; m Ref. [26]; n Ref. [27]; o Ref. [28].
It is shown that the PL equation is valid for a large number of organic molecular glass-formers over rather higher temperature range [2,5,9] It is also known that the I-MCT also works very well for the relatively lower viscosity regime [11]. In reality, although the viscosity does not diverge at TX ≈ Tc, several analyses of the slightly supercooled and normal liquid dynamics in various organic glass-formers in terms of the extended mode coupling theory (E-MCT), which removes this singularity, provide the same crossover temperature in the supercooled liquid phase [11,40]. Thus, the TX parameter marks two distinct regimes of the strongly and weakly supercooled liquid dynamics [11]. In particular, it corresponds to the onset of dynamic heterogeneities, i.e., regions with slower dynamics embedded into regions of higher dynamics, when the decoupling of translation from rotation of the molecular tracers and the decoupling of rotation of the molecular tracer from that of the medium constituents occur, and the classic Stokes−Einstein or Debye−Stokes−Einstein laws, respectively, are violated [9,14].
Figure 6 displays the molecular size dependence of the extracted dynamic crossover temperature TX for 1−alkanols, together with its fitting curve, with a similar form to that of Tg and Tm. Similar to the quantities of Tg and Tm, after the initial decrease to the second lowest member the series of nine 1−alkanols, the power law formula below is followed:
with the β exponent, equaling 0.656, lying in between those for the glass temperature, Tg(α = 0.503), and melting point, Tm (γ = 0.705).
TX = BM β
Finally, returning to Figure 3, a comparison of TX/Tg vs. Tm/Tg dependencies as a function of molecular size NC starting from C2OH shows a diametrically different trend for the former quantity with respect to the latter one, i.e., the increasing distance of the particular TX from the respective Tg with the increasing molecular size of 1−alkanol. This finding, together with the almost identical relative distance of TX from Tm (ca. 0.81 × Tm), indicates that the larger the molecule of 1−alkanol, the larger the temperature range of the strongly (or deeply) (and correspondingly, the shorter the weakly (slightly)) supercooled liquid state. On the other hand, upon cooling the smaller 1−alkanols, the weakly supercooled liquid state persists for longer, with a correspondingly shorter deeply supercooled liquid range.
2.2. ESR Data
2.2.1. General Spectral and Dynamic Features
Figure 7 presents the 2Azz’ vs. T dependencies for our series of spin systems: TEMPO/1-alkanols. In all cases, the quasi-sigmoidal courses of these plots are found, with higher 2Azz’ values in a slow motional regime at relatively lower temperatures and the lower 2Azz’ ones in a fast motion regime in relatively higher temperature regions. The most pronounced feature of 2Azz’ vs. T dependencies is a more or less sharp change at the main characteristic ESR temperature, T50G at which the 2Azz’(T50G) value reaches just 50 Gauss, corresponding to the correlation time of the TEMPO in a typical organic medium around a few ns. Note that the detailed spectral simulations of the TEMPO dynamics in several organics, including one of the investigated 1−alkanols, namely, 1−propanol [41], reveal that the spin probe population even at T50G is not completely situated in the fast motion regime, which occurs at a slightly higher temperature, TX1fast. In addition to these main characteristic ESR temperatures, T50G and TX1fast, other effects appear at TX1slow and TX2fast, a discussion of which goes beyond the scope of this work, and will therefore be addressed elsewhere. All the 2Azz’ vs. T plots also include the afore-mentioned thermodynamic and dynamic temperatures: Tg, Tm or TX, respectively. The mutual relationships of these three basic characteristic thermodynamic and dynamic temperatures with T50G and TX1fast in a series of 1−alkanols will be discussed below, in Section 2.2.2.
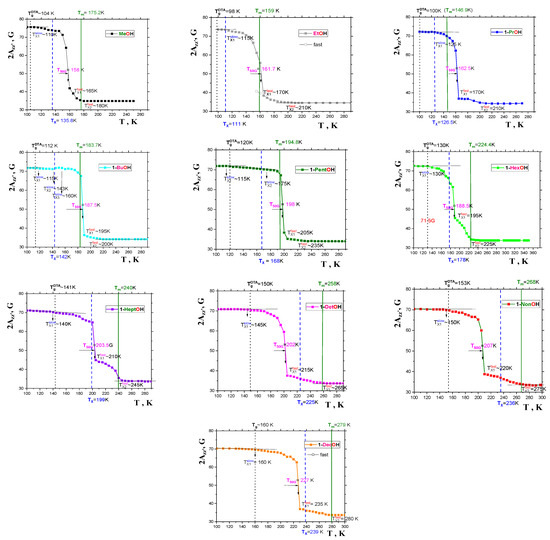
Figure 7.
Spectral parameter 2Azz’ in a series of 1−alkanols as a function of temperature. The colors for the ESR data for the individual 1−alkanols are the same as for their viscosity data in Figure 4. The characteristic ESR temperatures TXi slow, T50G and TXi fast are marked, and the thermodynamic temperatures TgDTA and TmCAL, as well as the dynamic one, TX, are depicted by the black, olive and blue lines, consistently with Figure 1, Figure 2 and Figure 6 and are discussed in the text in detail.
In principle, the main slow-to-fast motion transition of the TEMPO in any organics is related not only to these thermodynamic and dynamic transitions, but it may also be influenced by further factors, such as the potential mutual interaction of a polar spin probe with organic media, especially polar ones. The values of anisotropic hyperfine constants Azz’ (100 K) at the lowest measured temperature of 100 K, and of isotropic ones Aiso (RT) at room temperature, are summarized in Table 1. Their dependencies on NC, as well as on some relevant bulk properties of the media, such as the bulk polarity of media, through their dielectric constant, εr, will be discussed in the Section 2.2.3.
Finally, the mutual connections between the temperature parameters of the slow-to-fast transition, and the thermodynamic as well as dynamic ones, in relation to the polarization interaction of the polar TEMPO probe with a series of polar 1−alkanols, are discussed in Section 2.2.4.
2.2.2. The Mutual Relationships of T50G and TX1fast with Thermodynamic and Dynamic Transitions
In Figure 8, global comparisons of the characteristic ESR temperatures T50G and TX1fast with the aforementioned thermodynamic and dynamic temperatures Tg, TX and Tm are presented. In all the cases, the slow-to-fast transition in all 1−alkanols occurs above the corresponding glass−to−liquid transition, Tg, i.e., in the amorphous phase of liquid sample or in the local amorphous liquid zones of partially crystalline matrices, at least. Figure 9 expresses these comparisons in terms of the corresponding ratios: T50G/Tm, T50G/TX and TX1fast/Tm, TX1fast/TX. We can approximately distinguish two distinct regions of these ratios with a boundary at around C5OH
low M region: C1OH-C5OH with T50G/Tm ≈ 1
high M region: C6OH-C10OH with T50G/TX ≈ 1
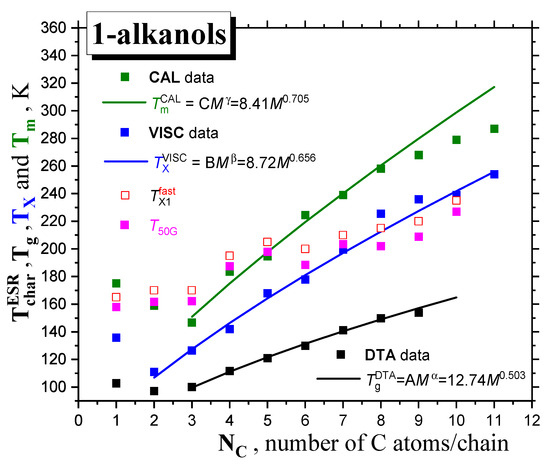
Figure 8.
Comparison of the characteristic ESR temperatures, T50G and TX1fast, with the thermodynamic and dynamic temperatures Tg DTA, TX VISC and Tm CAL, together with their corresponding PL fits.
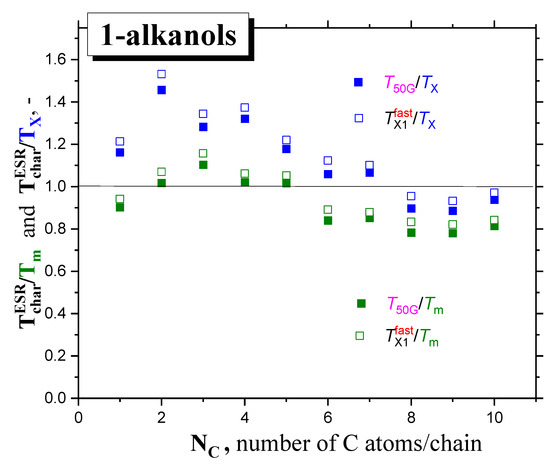
Figure 9.
Comparison of the ratios of the characteristic ESR temperatures, T50G and TX1fast, with the dynamic and thermodynamic temperatures, TX or Tm, respectively, as a function of NC.
So, for higher members starting at C6OH to C10OH, with a relatively longer aliphatic part, we observe a plausible closeness between the characteristic ESR temperatures and the TX’s, indicating that the main ESR transition is related to the dynamic crossover between the deeply and slightly supercooled liquid state. This basic finding is similar to the previous one for a series of n−alkanes [16], with the fact that the TX’s of 1−alkanols are higher than the TX values for the corresponding n-alkanes with the same number of C atoms in the molecule. This difference indicates that the spin probe TEMPO is not fully surrounded by the apolar aliphatic parts of the 1−alkanol molecules, and that the polar -OH groups influence its dynamics, as will be discussed later in Section 2.2.3. This indicates that the immediate environment of the molecular-sized spin probe TEMPO is locally disordered, and subsequently, sensitive to the crossover transition in this local amorphous phase. On the other hand, for low M members from C1OH to C5OH, the T50G and TX1fast values lie significantly above the corresponding TX values, and they are situated in the vicinity of the corresponding melting temperatures, Tm. This indicates that the slow-to−fast transition of TEMPO appears to be related to the global disordering process connected with the solid−to−liquid state phase transition in the otherwise partially crystallized samples.
2.2.3. Isotropic and Anisotropic Hyperfine Constants Aiso (RT) and Azz’ (100 K) as a Function of the NC, Polarity and Proticity of 1−alkanols
Figure 10 displays the anisotropic hyperfine constant, Azz’ (100K), and the isotropic. hyperfine constant, Aiso(RT), of the TEMPO as a function of the chain length in the series of 1-alkanols studied. Our values of Aiso (RT) for TEMPO are quite consistent with the few obtained for lower members of our series, namely, C1OH–C4OH [42,43,44]. Although both the quantities decrease with increasing chain size, a significant difference can be found in the corresponding trends. The former quantity shows two clear regions of distinct behavior: a sharper decreasing trend for low-M members, and a weaker one for higher M members above NC~4. On the other hand, the Aiso (RT) parameter is slightly reduced with a suggestion of a slight change at NC~6 as the number of C atoms in the molecule, NC, increases.
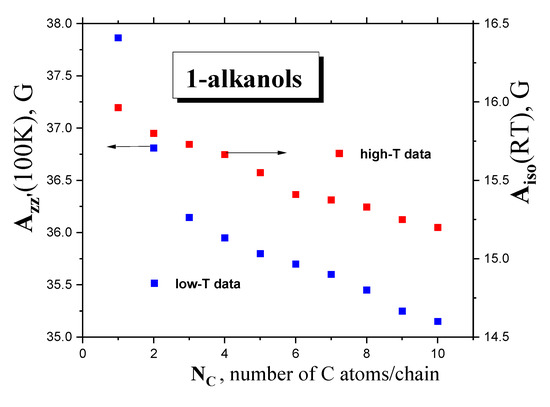
Figure 10.
Hyperfine constants of TEMPO Azz’(100 K) and Aiso (RT) in a series of ten 1−alkanols as a function of NC.
These basic empirical findings can be discussed in relation to the polarity of a set of polar media, with the dissolved polar spin probe TEMPO μTEMPO~3 D [45], from both the phenomenological and theoretical viewpoints. First, the Aiso(RT) values can be related to various measures of the polarity of the medium, e.g., the dipole moment of the medium‘s molecule, μentity,x, as a measure of the polarity of the individual entity in a given phase state x = gaseous or liquid state or the static dielectric constant of the medium, εr (RT), as a measure of the polarity of the bulk liquid medium, as listed in Table 1. In the first case, evidently, no relationship can be found due to the quasi-constant values of the gaseous-phase μg = 1.66 ± 0.05 D [34] or the liquid-phase μl = 2.84 ± 0.15 D dipole moments [39]. On the other hand, Figure 11 displays the mutual relationships between the isotropic hyperfine constant, Aiso (RT), and the dielectric constant, εr (RT), of 1−alkanols [34], together with those of the latter quantity at RT as a function of the number of C atoms in the chain inserted. Both the quantities decrease with NC, resulting in an Aiso (RT) vs. εr (RT) relationship, with approximately two regions showing distinct behavior: (i) for the lower polar 1−alkanols (C10OH-C5OH) with εr < ~17, with a strong sensitivity of Aiso (RT) to polarity and a weak one to proticity, and (ii) for higher polar 1−alkanols (C3OH-C1OH) with εr > ~17, with the weak sensitivity of Aiso (RT) to polarity and a stronger sensitivity to proticity, due to the increased population of HO-groups potentially interacting with the spin probe TEMPO molecule. The apparent boundary between both regions occurs at NC = 4–5, i.e., for 1−butanol or 1−pentanol, where the εr (RT) vs. NC (RT) plot changes rather notably from a sharply decreasing dependence to a slightly decreasing one, and where, at the same time, conformational degrees of freedom and related enhanced alignments of the apolar parts of the molecules start to occur. Interestingly, in spite of the absence of εr (100 K) data facilitating their direct comparison with Azz’ (100K), the boundary for this quantity seems to be consistent with that for Aiso (RT), suggesting a significant role of polarity and proticity in both the mobility states of the spin probe TEMPO. These findings of the solvent dependence of the different ESR parameters are consistent with the previous ones for Aiso (RT) [46,47], as well as for Azz’ (77 K) [48,49].
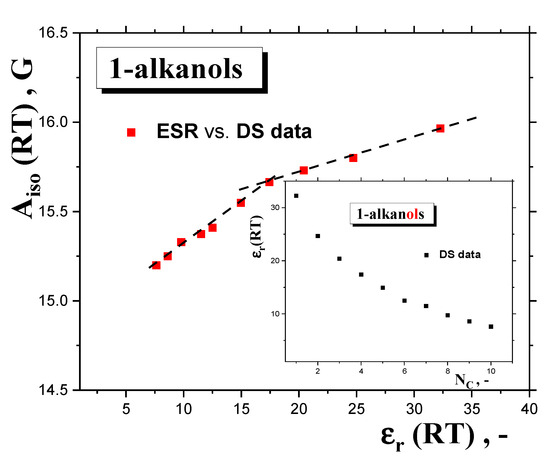
Figure 11.
Empirical relationship between the isotropic hyperfine constant, Aiso (RT), and the relative permitivity, εr (RT), of 1−alkanols. Insert contains the latter quantity at RT from Table 1 as a function of NC.
Our basic finding is similar to that derived for another larger nitroxide spin probe, 1−oxyl−2,2,5,5−tetramethyl pyrroline−3−methyl)methanethiosulfonate (MTSSL), in a series of 17 solvents ranging from apolar methylbenzene (toluene) (εr (RT) = 2.4) to highly polar water (εr (RT) = 80.4) and even more polar formamid (εr (RT) = 109), including most of the members of our 1−alkanol series, with one exception for 1−pentanol [50]. These authors similarly distinguished the following two regions, i.e., an “apolar”region for εr(RT) < 25, where the sensitivity of Aiso(RT) and Azz’(77K) to the polarity expressed by εr (RT) is large, and a “polar” region for εr (RT) > 25, where the sensitivity of Aiso (RT) and Azz’ (77 K) to the polarity is small, and the change is ascribed to the medium proticity.
However, it is evident that this division is rather arbitrary and very rough, because the former “apolar” region also includes many of our polar 1−alkanols. In connection with the aforementioned empirical relation between spectral parameters and bulk polarity, more elaborate theoretical approaches, based on models using the medium as a dielectric continuum with dielectric constant, εr, and the molecular solute, e.g., polar spin probe, as a molecular entity localized in a spherical cavity [47,51], can be discussed. Within the reaction field concept of the polarization of the continuum medium by the polar solute, one obtains for the Onsager’s reaction field [52] and Böttcher’s reaction field [53] the following functional relations: Aiso = f [(εr − 1)/(εr + 1)] [47], or Aiso = f [(2εr + 1)/(2εr + nD2)], where nD is the refraction index of the pure nitroxide [51]. Figure 12 displays a test of the validity of the first functional dependence for two basic groups of organic compounds at RT doped by TEMPO. The first is represented by a series of apolar and aprotic polar solvents, which range from apolar benzene (BZ) with εr (RT) = 2.3, to a highly polar but aprotic dimethyl sulfoxide (DMSO) with εr (RT) = 48.9, as taken from Ref. [50]. The other group, including our series of ten 1−alkanols from methanol with εr (RT) = 33 to 1−decanol with εr(RT) = 7.9, differs significantly from the predicted linear trend due to the specific protic character of the molecules, allowing for H-bond formation between the polar spin probe TEMPO molecule and the alkanol’s one. This is quite consistent with the maximal value of Aiso (RT) = 17 Gauss [44] for highly polar and protic water, where εr (RT) = 80.4 [50]. The relatively large difference between water and the first member of the alkanol family is determined on the basis of theoretical calculations using density functional theory (DFT) interpreted in terms of the complexation of nitroxide with two water or one methanol molecules, respectively [45,50]. Moreover, a closer inspection of this group of protic polar compounds confirms the distinction of a series of 1−alkanols into two subgroups, with distinct slopes of Aiso (RT) as a function of the corresponding dielectric function: (i) weaker for higher members from C10OH to C6OH, and (ii) stronger for shorter ones from C5OH to C1OH, with an approximate boundary between C5OH and C6OH, i.e., for εr (RT)~16.5. A similar situation can be found for the Böttcher reaction field due to the linearity between the respective functional forms. Both these findings appear to be consistent with the empirically determined boundary at C4OH-C5OH, as seen from the Aiso (RT) vs. εr (RT) plot without the inclusion of the polarization interaction between the polar solute and the solvent, as shown in Figure 11.
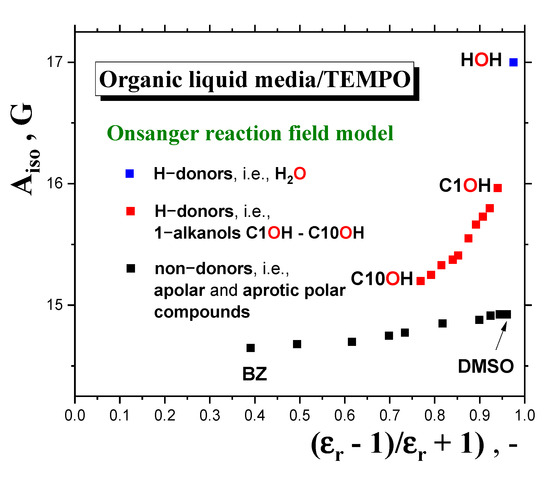
Figure 12.
Test of the Griffith−Onsanger model for isotropic hyperfine constant, Aiso (RT), as a function of the polarization expression εr (RT) – 1)/(εr (RT) + 1) of the Onsanger reaction field model for three types of media: apolar, such as benzene (BZ) [44]; aprotic polar, such as dimethylsulfoxide (DMSO) [44]’ and protic polar compounds, such as water [44] and our series of ten 1−alkanols.
2.2.4. Connection of the Main Slow-to-Fast Motion Transition of the Spin Probe TEMPO with the Polarity, Proticity and Thermodynamic and Dynamic Transition Behaviors of 1−alkanols
In Figure 8 and Figure 9 in Section 2.2.2, we compare the characteristic ESR temperatures T50G and TX1fast of the slow-to-fast transition of TEMPO in a series of 1−alkanols with the dynamic crossover TX and thermodynamic transition temperatures Tm, and also shown their mutual ratios as a function of the molecular size, NC, of the media. In particular, we revealed a step-like change in the main spin probe TEMPO transition from that seen at the dynamic crossovers at around TX for the longer chains, to that related to thermodynamic transitions at around Tm for the shorter molecules, at NC~5.
Next, in Figure 10, Figure 11 and Figure 12 in Section 2.2.3, we present the relations of spectral parameters Aiso (RT) and Azz’ (100 K) to NC, as well as their phenomenological and theoretical relationships, especially for that of Aiso (RT) to the polarity properties of a set of 1−alkanols. Here, we have observed a change in the trend of hyperfine interactions with the polarity and proticity of 1-alkanol media at NC~4. Now, a combination of these findings indicates that the slow−to-fast transition in the mobility of TEMPO in a series of 1-alkanols is relatively strongly dependent on the strength of intermolecular interactions between the polar constituents of the polar media, and between the polar spin probe and the polarity and proticity of the 1−alkanols investigated. In the longer members of the 1-alkanol family, with the relatively higher population of apolar aliphatic methylene groups related to a weakly changing polarity, the slow-to-fast transition is related mainly to the dynamic crossover process around TX, similar to what is seen for the apolar n−alkanes [16]. On the other hand, in the shorter members with relatively higher dielectric constants and proticity due to the relatively higher populations in the polar hydroxyl groups, a larger-scale disorder process connected with the solid-to-liquid phase transition around Tm is needed in order to destroy not only the dense H-bonding network between the medium’s molecules, but also to destroy the clustering of polar TEMPO molecules with them, and subsequently, and the appearance of slow−to−fast transition in the mobility of TEMPO. The critical molecular size of 1−alkanol for this step-like change in the slow-to-fast transition of TEMPO lies at NC~5, below which the polarity and proticity aspects of the media become dominating factors.
3. Experimental
3.1. Materials and Methods
A series of 1−alkanols ranging from methanol to 1−decanol, received from Sigma-Aldrich, Inc., St. Louis, MO, USA, was used as the model protic polar media. Our choice of this series stems from the fact that all of the used 1−alkanols are in the liquid state at room temperature, making spin probe system preparation easier. As an extrinsic particle, the spin probe 2,2,6,6−tetramethyl−1−piperidinyloxy (TEMPO) with a quasi-spherical shape and a relatively high dipole moment of μTEMPO = 3 D [45] was applied in the deoxygenated 1−alkanols at a very low concentration of ~5 × 10−4 M.
3.2. ESR
ESR measurements of the very diluted spin systems 1-alkanol/TEMPO were performed on the X-band Bruker–ER 200 SRL spectrometer operating at 9.4 GHz with a Bruker BVT 100 temperature variation controller unit. ESR spectra were recorded after cooling at a rate ~− 4 K/min in a heating mode over a wide temperature range from 100 K up to 300 K, with steps of 5–10 K. To reach the thermal equilibrium, the sample was kept at a given temperature for 10 min before starting three spectra collections. The temperature stability was ± 0.5 K. The microwave power and the amplitude of field modulation were optimized to avoid signal distortion. The ESR spectra were evaluated in terms of the spectral parameter of mobility, 2Azz’ (T), i.e., the z-component of the anisotropic tensor of the hyperfine interaction A(T), corresponding to the outermost peak separation of the triplet spectra of the spin probe of nitroxide type in a given medium, as a function of temperature and the subsequent determination of the spectral T50G parameter [50,54]. This is the characteristic ESR temperature, at which 2Azz’ reaches the conventional value of 50 Gauss (G). Additional characteristic ESR temperatures can be obtained, and used to describe in detail the slow-to-fast regime transition zones over more or less wide temperature intervals around T50G, as well as in both slow and fast motion regimes: TXi slow, TXi fast [55,56,57]. In addition to the anisotropic hyperfine splitting parameter, Azz’ (100 K), the isotropic hyperfine constant values, Aiso (RT), of the spin probe TEMPO, as a measure of its interaction with a given medium, were determined at room temperature under the fast motion condition in a low-viscosity media [50].
4. Conclusions
The spectral and dynamic behaviors of the spin probe TEMPO in a series of 1−alkanols ranging from methanol to 1−decanol, over a wide temperature range from 100 K up to 300 K, using electron spin resonance (ESR) are reported. For all of the alkanols, the main characteristic ESR temperatures connected with slow-to-fast motion regime transition, namely, T50 and TX1fast, are situated above the corresponding glass temperatures, Tg, and for the first five shorter members, the T50G values lie in the vicinity of the melting point, Tm, while for the longer ones, the T50G < Tm relationship indicates that the TEMPO molecules are in the local disordered regions of the crystalline media. The TX1fast values are compared with the dynamic crossover temperatures, parametrized as TXVISC = 8.72M0.66, which were obtained by fitting the viscosity data for the liquid 1−alkanols with the empirical power law. In particular, for NC = 6–10, the TX1fast values lie relatively close to the TXVISC seen for apolar n-alkanes, while for NC = 1–5, they are situated above the respective TXVISC values in the vicinity of Tm. The absence of such a coincidence for lower 1−alkanols indicates that the slow-to-fast motion transition is significantly influenced by the mutual interaction between the polar TEMPO and the protic polar medium, due to the increased polarity and proticity, which are destroyed at higher temperatures associated with the larger-scale solid-to-liquid transition.
Author Contributions
Conceptualization, J.B.; methodology, J.B.; investigation, J.B. and H.Š.; analyses, J.B. and H.Š.; data curation, J.B.; writing—original draft preparation, J.B.; writing—review and editing, J.B. and H.Š.; visualization, J.B.; project administration, J.B. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the VEGA, grant number 2/0005/20.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors thank C. Corsaro for the fruitful discussion.
Conflicts of Interest
The authors declare no conflict of interests.
References
- Barlow, A.J.; Lamb, J.; Matheson, A.J. Viscous behavior of supercooled liquids. Proc. Roy. Soc. London Ser. A 1966, 292, 322–342. [Google Scholar]
- Taborek, P.; Kleinman, R.N.; Bishop, D.J. Power law behavior in the viscosity of supercooled liquids. Phys. Rev. B 1986, 34, 1835–1840. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Fischer, E.W.; Richert, R. Dynamics of glass-forming liquids. I. Temperature-derivative analysis of dielectric relaxation data. J. Chem. Phys. 1995, 102, 6251–6257. [Google Scholar] [CrossRef]
- Stickel, F.; Fischer, E.W.; Richert, R. Dynamics of glass-forming liquids. II. Detailed comparison of dielectric relaxation, dc-conductivity and viscosity data. J. Chem. Phys. 1996, 104, 2043–2055. [Google Scholar] [CrossRef]
- Martinez-Garcia, J.C.; Martinez-Garcia, J.; Rzoska, S.; Huellinger, J. The new insight into dynamic crossover in glass forming liquids from the apparent enthalpy analysis. J. Chem. Phys. 2012, 137, 064501-8. [Google Scholar] [CrossRef] [PubMed]
- Leon, L.; Ngai, K.L. Rapidity of the Change of the Kohlrausch Exponent of the α-Relaxation of Glass-Forming Liquids at TB or Tβ and Consequence. J. Phys. Chem. B 1999, 103, 4045–4051. [Google Scholar] [CrossRef]
- Schönhals, A. Evidence for a universal crossover behavior of the dynamic glass transition. Europhys. Lett. 2001, 56, 815. [Google Scholar] [CrossRef]
- Johari, G.P.; Goldstein, M. Viscous Liquids and the Glass Transition. II. Secondary Relaxations in Glasses of Rigid Molecules. J. Chem. Phys. 1970, 53, 2372–2388. [Google Scholar] [CrossRef]
- Mallamace, F.; Branca, C.; Corsaro, C.; Leone, N.; Spooren, J.; Chen, S.H.; Stanley, H.E. Transport properties of glass-forming liquids suggest that dynamic crossover temperature is as important as the glass transition temperature. Proc. Natl. Acad. Sci. USA 2010, 107, 22457–22462. [Google Scholar] [CrossRef]
- Roland, C.M. Characteristic relaxation times and their invariance to thermodynamic conditions. Soft Matter 2008, 4, 2316–2322. [Google Scholar] [CrossRef]
- Götze, W.; Sjögren, L. Relaxation processes in supercooled liquids. Rep. Progr. Phys. 1992, 55, 241–376. [Google Scholar] [CrossRef]
- Novikov, V.N.; Sokolov, A.P. Universality of the dynamic crossover in glass-forming liquids: A “magic” relaxation time. Phys. Rev. E 2003, 67, 031507. [Google Scholar] [CrossRef] [PubMed]
- Hyde, P.D.; Evert, T.E.; Cicerone, M.T.; Ediger, M.D. Rotational motion of molecular probes in orto-terphenyl and cis-poly-isoprene. J. Non-Cryst. Solids 1991, 131–133, 42–47. [Google Scholar] [CrossRef]
- Andreozzi, L.; Schinoy, A.D.; Giordano, M.; Leporini, D. A study of the Debye–Stokes–Einstein law in supercooled fluids. J. Phys.-Cond. Matter 1996, 8, 9605–9608. [Google Scholar] [CrossRef]
- Bartoš, J.; Šauša, O.; Bandžuch, P.; Zrubcová, J.; Krištiak, J. Free volume factor in supercooled liquid dynamics. J. Non-Cryst. Solids 2002, 307–310, 417–425. [Google Scholar] [CrossRef]
- Bartoš, J.; Corsaro, C.; Mallamace, D.; Švajdlenková, H.; Lukešová, M. ESR evidence of the dynamic crossover in the supercooled liquid states of a series of solid n-alkanes. Phys. Chem. Chem. Phys. 2018, 20, 11145–11151. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.A.; Talon, C.; Jimenez-Riobóo, R.J.; Vieira, S. Low-temperature specific heat of structural and orientational glasses of simple alcohols. J. Phys.-Cond. Matter 2003, 15, S1007. [Google Scholar] [CrossRef]
- NIST Chemistry WebBook, NIST Standard Reference3 Database Number 69. Last Update to Data: 2023. Available online: http://webbooknistgov/chemistry/ (accessed on 7 September 2023).
- Illers, K.H. Innere Rotationen in Festkörpern aus hoch- und niedrigmolekularen organischen Molekülen. Rheol. Acta 1964, 3, 183–193. [Google Scholar] [CrossRef]
- Faucher, J.A.; Koleske, J.V. Glass Transitions of Organic Compounds. 1. Lower Aliphatic Alcohols. Phys. Chem. Glasses 1966, 7, 202–208. [Google Scholar]
- Sugisaki, H.; Suga, H.; Seki, S. Calorimetric Study of the Glassy state. III. Novel Type Calorimeter for Study of Glassy State and heat Capacity of Glassy Methanol. Bull. Chem. Soc. Jpn. 1968, 41, 2586–2591. [Google Scholar] [CrossRef]
- Lesikar, A.V. On the self-association of the normal alcohols and the glass transition in alcohol-alcohol solutions. J. Solut. Chem. 1977, 6, 81–93. [Google Scholar] [CrossRef]
- Carpenter, M.R.; Davis, D.B.; Matheson, A.J. Measurement of the Glass Transition Temperature of Simple Liquids. J. Chem. Phys. 1967, 48, 2451–2454. [Google Scholar] [CrossRef]
- Haida, O.; Suga, H.; Seki, S. Calorimetric study of the glassy state XII. Plural glass transition phenomena of ethanol. J. Chem. Thermodyn. 1977, 9, 1133–1148. [Google Scholar] [CrossRef]
- Murthy, S.S.N. Experimental study of dielectric relaxation in supercooled alcohols and polyols. Mol. Phys. 1996, 97, 691–709. [Google Scholar] [CrossRef]
- Koleske, J.V.; Faucher, J.A. Glass Transitions of Organic Compounds. III. Cellulose Substrate Technique and Aliphatic Alcohols. Polym. Eng. Sci. 1979, 19, 716–721. [Google Scholar]
- El Goresy, T.; Böhmer, B. Diluting the hydrogen bonds in viscous solutions of n-butanol with n-bromobutane: A dielectric study. J. Chem. Phys. 2008, 28, 154520. [Google Scholar] [CrossRef] [PubMed]
- Hassaine, M.; Jimenez-Rioboo, R.J.; Ramos, M.A.; Sharapova, I.V.; Koroyluk, O.A.; Krivchikov, A.I. Thermal properties and Brillouin scattering study of glass, crystal and “glacial” state in n-butanol. J. Chem. Phys. 2009, 131, 174508. [Google Scholar] [CrossRef] [PubMed]
- Novikov, V.N.; Rössler, E.A. Correlation between glass transition temperature and molecular mass in non-polymeric and polymer glass formers. Polymer 2013, 54, 6987–6991. [Google Scholar] [CrossRef]
- Kauzmann, W. The nature of the glassy state and the behavior of liquids at low temperature. Chem. Rev. 1948, 43, 219–256. [Google Scholar] [CrossRef]
- Boyer, R. The relation of transition temperatures to chemical structure in high polymers. Rubber Chem. Technol. 1963, 36, 1303–1421. [Google Scholar] [CrossRef]
- Sakka, S.; McKenzie, J.D. Relation between apparent glass transition temperature and liquidus temperature for inorganic glasses. J. Non-Cryst. Solids 1971, 6, 145–162. [Google Scholar] [CrossRef]
- Landolt-Börnstein—Group IV Physical Chemistry. Pure Organic Liquids, Subvolume B ‘Pure Organic Liquids’ of Volume 18. In Viscosity of Pure Organic Liquids and Binary Liquid Mixtures; Landolt-Börnstein—Group IV Physical Chemistry, Springer: Berlin-Heidelberg, Germany, 2002. [Google Scholar]
- Rowley, R. DIPPR Data Compilation of Pure Chemical Properties; Ref Type: Electronic Citation; Design Institute for Physical Properties: AIChE: New York, NY, USA, 2010. [Google Scholar]
- Tamman, G.; Hesse, W.Z. Die Abhängigkeit der Viskosität von der Temperatur die unterkühlten Flüssigkeiten. Zeitsch. Anorg. Allgem. Chem. 1926, 156, 245–257. [Google Scholar] [CrossRef]
- Denney, D.J. Viscosities of some undercooled liquid alkylhalides. J. Chem. Phys. 1959, 30, 159–162. [Google Scholar] [CrossRef]
- Campbell Ling, A.A.C.; Willard, J.E. Viscosities of some organic glasses used as trapping matrixes. J. Phys. Chem. 1968, 72, 1918–1923. [Google Scholar] [CrossRef]
- Mitsukuri, S.; Tonomura, T. Viscosities of liquids at constant volume. Proc. Imper. Acad. Jpn. 1927, 3, 155, and 1929, 5, 23. [Google Scholar] [CrossRef][Green Version]
- Barrera, M.C.; Jorge, M. A Polarization-Consistent Model for Alcohols to Predict Solvation Free Energies. J. Chem. Info Model. 2020, 60, 1352–1367. [Google Scholar] [CrossRef] [PubMed]
- Krakoviack, V.; Alba-Simionesco, C.; Krauzman, M. Study of the depolarized light scattering spectra of supercooled liquids by a simple mode-coupling model. J. Chem. Phys. 1997, 107, 3417–3427. [Google Scholar] [CrossRef]
- Bartoš, J.; Švajdlenková, H.; Šauša, O.; Lukešová, M.; Ehlers, D.; Michl, M.; Lunkenheimer, P.; Loidl, A. Molecular probe dy-namics and free volume in organic glass-formers and their relationships to structural relaxation: 1-propanol. J. Phys.-Cond. Matt. 2016, 28, 015101. [Google Scholar] [CrossRef]
- Dodd, G.H.; Barratt, M.D.; Rayner, L. Spin probes for binding site polarity. FEBS 1970, 8, 286–288. [Google Scholar] [CrossRef]
- Jolicoeur, C.; Friedman, H.L. Hydrophobic nitroxide radicals as probes to investigate the hydrophobic interaction. J. Sol. Chem. 1974, 3, 15–43. [Google Scholar] [CrossRef]
- Al-Bala, R.D.; Bates, R.D., Jr. Medium Effects on ESR Spectra in Studies of Hydrogen-Bonded Transient Solvent-Solute Complexes. J. Magn. Reson. 1987, 73, 78–89. [Google Scholar] [CrossRef]
- Laleveé, J.; Allonas, X.; Jacques, P. Electronic distribution and solvatochromism of investigation a model radical 2,2,6,6-tetramethyl piperidine N—Oxyl: TEMPO through TD-DFT calculation including PCM solvation. J. Mol. Struct. Theochem. 2006, 767, 143–147. [Google Scholar] [CrossRef]
- Kawamura, T.; Matsunami, S.; Yonezawa, T. Solvent Effects on the g-value of Di-t-butyl Nitric oxide. Bull. Chem. Soc. Jpn. 1967, 40, 1111–1115. [Google Scholar] [CrossRef]
- Griffith, O.H.; Dehlinger, P.J.; Van, S.P. Shape of the Hydrophobic Barrier of Phospholipid Bilayers (Evidence for Water Penetration in Biological Membranes). J. Membrane Biol. 1974, 15, 159–192. [Google Scholar] [CrossRef] [PubMed]
- Krinichnyi, V.I.; Grinberg, O.Y.; Bogatyrenko, V.R.; Likhtenshtein, G.I.; Lebedev, Y.S. Study of the influence of the microsurroundings on magneto-resonance parameters of spin labelled human serum albumin in the 2 mm e.s.r. range. Biophysics 1985, 30, 233–237. [Google Scholar]
- Ondar, M.A.; Grinberg, O.Y.; Dubinskii, A.A.; Lebedev, Y.S. Study of the effect of the medium on the magnetic-resonance parameters of nitroxyl radicals by high-resolution EPR spectroscopy. Sov. J. Chem. Phys. 1985, 3, 781–792. [Google Scholar]
- Owenius, R.; Engström, M.R.; Lindgren, M. Influence of Solvent Polarity and Hydrogen Bonding on the EPR parameters of a Nitroxide Spin label Studied by 9-GHZ and 95-GHz EPR Spectroscopy and DFT calculations. J. Phys. Chem. 2001, 105, 10967–10977. [Google Scholar] [CrossRef]
- Seelig, J.; Limacher, H.; Bader, P. Molecular Architecture of Liquid Crystalline Bilayer. J. Amer. Chem. Soc. 1972, 94, 6364–6371. [Google Scholar] [CrossRef]
- Onsanger, L. Electric Moments of Molecules in Liquids. J. Amer. Chem. Soc. 1936, 58, 1486–1493. [Google Scholar] [CrossRef]
- Böttcher, C.J.F. Theory of Electric Polarisation; Elsevier: New York, NY, USA; Amsterdam, The Netherlands, 1952; p. 70. [Google Scholar]
- Rabold, G.P. Spin-Probe Studies. II. Applications to Polymer Characterization. J. Polym. Sci. A 1969, 7, 1203–1223. [Google Scholar] [CrossRef]
- Bartoš, J.; Švajdlenková, H.; Zaleski, R.; Edelmann, M.; Lukešová, M. Spin probe dynamics in relation to free volume in crystalline organics by means of ESR and PALS: N-Hexadecane. Phys. B Cond. Mat. 2013, 430, 99–105. [Google Scholar] [CrossRef]
- Švajdlenková, H.; Iskrová, M.; Šauša, O.; Dlubek, G.; Krištiak, J.; Bartoš, J. The Spin probe Dynamics and the Free Volume in a series of Amorphous Polymer Glass-Formers. Macromol. Symp. 2011, 305, 108–115. [Google Scholar] [CrossRef]
- Švajdlenková, H.; Arrese-Igor, S.; Nógellová, Z.; Alegría, A.; Bartoš, J. Molecular dynamic heterogeneity in relation to free volume and relaxation dynamics in organic glass-formers: Oligomeric cis-1,4-poly(isoprene). Phys. Chem. Chem. Phys. 2017, 19, 15215–15226. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).