Abstract
The most prevalent and aggressive type of brain cancer, namely, glioblastoma (GBM), is characterized by intra- and inter-tumor heterogeneity and strong spreading capacity, which makes treatment ineffective. A true therapeutic answer is still in its infancy despite various studies that have made significant progress toward understanding the mechanisms behind GBM recurrence and its resistance. The primary causes of GBM recurrence are attributed to the heterogeneity and diffusive nature; therefore, monitoring the tumor’s heterogeneity and spreading may offer a set of therapeutic targets that could improve the clinical management of GBM and prevent tumor relapse. Additionally, the blood–brain barrier (BBB)-related poor drug delivery that prevents effective drug concentrations within the tumor is discussed. With a primary emphasis on signaling heterogeneity, tumor infiltration, and computational modeling of GBM, this review covers typical therapeutic difficulties and factors contributing to drug resistance development and discusses potential therapeutic approaches.
1. Introduction
Glioma is a term used to describe a neuroepithelial tumor originating from glial cells, which are the most common type of supporting cells in the CNS. One of the deadliest types of human cancer, namely, glioblastoma multiforme (GBM), is classified as a WHO grade IV brain tumor. A combination of surgical resection, radiation therapy, and chemotherapy are the current accepted treatments for GBM [1].
Surgery to remove a GBM tumor carries a significant danger for the patient since it frequently invades vital brain regions. Following surgery, patients receive concurrent Temozolomide (TMZ) and radiation therapy. Unfortunately, these methods only slightly improve the prognosis for GBM patients, with a median survival of 14–15 months and a 5-year survival rate of about 10% [2,3].
In this review, we highlight three major problems in GBM treatment: tumor heterogeneity, GBM infiltration, and the blood–brain barrier (BBB) (Figure 1).
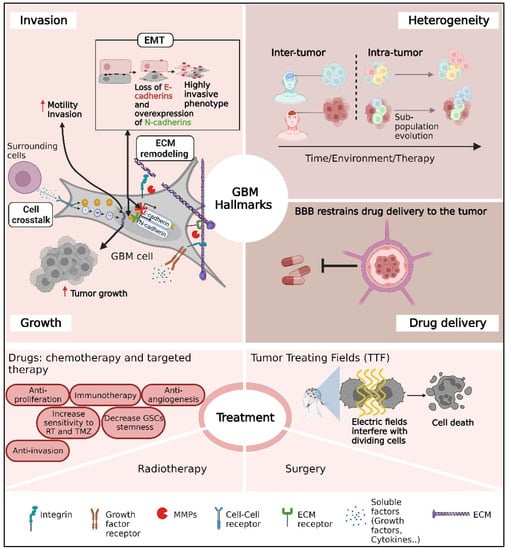
Figure 1.
A schematic overview of the main challenges for glioblastoma (GBM) clinical care mentioned in this review. The challenges include the infiltrative nature of GBM tumors, substantial heterogeneity between patients or cells inside the tumor, drug delivery through the BBB, and therapeutic limitations.
Significant efforts have been made to subtype GBM in order to address the problem of inter-tumor heterogeneity, including methylation (six subtypes), transcriptomic subtyping (proneural, neural, classical, and mesenchymal), and the World Health Organization definition (IDH wild type, IDH mutated, NOS) [4,5]. However, when protein–protein expression patterns are estimated over a large population of GBM tumors in proteome variability studies, they reveal even more variations between GBM patients, resulting in dozens of subgroups [6]. Furthermore, tumor cell variability (intra-tumor), which is often dynamic and develops over time, adds another layer of complexity to tumor classification [7,8,9].
Another clinical difficulty is the highly diffuse nature of GBM. Even when a tumor is successfully removed, followed by a combination of radiotherapy and chemotherapy, GBM recurs. Tumor relapse is frequently caused by highly infiltrative cells that penetrate the brain and are not recognized during surgery [10]. Enhanced cellular infiltration is caused by several processes, including signaling pathways involved in EMT (epithelial–mesenchymal transition), ECM remodeling, and increased spreading due to cell–cell communication inside GBM [11].
The anatomical position of GBM provides a further significant barrier to treatment, as GBM cells are inaccessible to most systemically administered medicines [1].
Taken as a whole, the review expands on these issues (Figure 1) and recent initiatives to solve them, whether through novel treatment options, recent research, or technological improvements. Other important topics, such as immune checkpoint treatment, CAR T-based immunotherapy, oncolytic viral medicines, gene and thermo-therapies, and tailored neoantigen-based vaccinations, were recently reviewed elsewhere [12,13,14].
2. Current Glioblastoma Treatments
The degree of GBM resection is a crucial element in patients’ survival. The gross total resection (GTR) was found to be favorably connected to survival time [15,16]. However, achieving GTR and removing the entire tumor is rare. An exact high-level determination of the tumor margins is required for a successful GTR. Surgical removal becomes extremely difficult when a tumor is located in an unresectable brain area or next to an area responsible for neurological function. Furthermore, due to the invasive nature of GBM, tumor cells infiltrate alone or in small groups into the healthy area, making identifying tumor margins and, ultimately, resecting the entire tumor without harming vital parts impossible [1]. As a result, post- or pre-surgical treatments and non-surgical treatment modalities are required to prevent tumor recurrence.
The following sections contain a list of available anti-GBM treatments and a discussion of recent developments in GBM research that aim to increase the field’s current understanding of the disease.
2.1. Temozolomide (TMZ) Therapy
Temozolomide (TMZ) is a first-line chemotherapeutic drug used to treat post-surgical GBM [17]. It is an oral alkylating drug that produces DNA adducts by methylating purine bases in DNA [18]. Its chemical nature makes it a preferred chemotherapeutic agent for GBM because of its stability at stomach acidic pH and ability to pass the blood–brain barrier (BBB) [19].
The treatment regimen has mostly stayed the same since a clinical trial in 2005 that confirmed the efficacy of adding TMZ to RT in a group of patients with newly diagnosed GBM [20]. Unfortunately, despite its contribution to patient survival (a 37% increase in survival at a median follow-up of 28 months) [20], the development of TMZ resistance and thus, the incidence of tumor recurrence, remains very high [21].
TMZ resistance can be developed through a variety of mechanisms [19,22], including MGMT (O6 Methylguanine DNA Methyltransferase) overexpression [23]. MGMT is a DNA repair protein that removes the methyl group from O6-methylguanine to prevent DNA alkylation [24].
Another mechanism shows that repeated TMZ treatment of GBM cells may transform non-GSCs (GBM stem cells) into new GSCs [25], enhancing their potential to self-renew, proliferate indefinitely, and differentiate into many lineages [26].
TMZ can also cause thrombocytopenia in patients, which is caused by TMZ-dependent DNA damage in healthy cells [27,28].
Furthermore, recent research has shown that TMZ treatment may contribute to tumor progression by promoting tumor invasion and EMT (epithelial–mesenchymal transition). Kubelt et al., for example, demonstrated that TMZ therapy enhances mRNA expression of many EMT markers in T98G glioma cells in vitro, including Vimentin, TGF-β, and Fibronectin [29]. Kochanowski et al. revealed that Cx43 signaling promotes GBM cell invasiveness in both MGMThigh (T98G) and MGMTlow (U87) populations [30]. The “GO OR GROW” phenomenon, which was reported in slow-dividing but spreading cells, was also detected in IDH-mutant astrocytoma and IDH-WT GBM cells after TMZ treatment. These cells reduced their proliferation rate significantly in response to TMZ but moved 2–3 times faster [31].
2.2. Radiotherapy (RT)
Radiotherapy is recommended either alone or in conjunction with TMZ after a surgical GBM resection. Despite the long history of radiation therapy in GBM therapy, ongoing debates exist about its usefulness as a standard treatment technique and its involvement in GBM recurrence.
The major mechanism through which RT causes cell death is DNA damage. The manner in which cells respond to this injury, however, is determined by cell-intrinsic and microenvironmental factors [32]. Current clinical practice and research show that radiotherapy is less effective in some brain cancers than others [33,34]. Glioblastomas are among the most radio-resistant aggressive forms of cancer [35]. Several efforts were made to identify biomarkers to determine and select patients with radio-sensitive tumors [36].
For example, in a study that ranked the radiosensitivity of 40 human cell lines based on survival components, three glioblastoma cell lines, namely, U87, U251, and T98G, were found to be the most radio-resistant cell line models [34]. The radiosensitivity was found to be mainely associated with p53 status and the expression of the ATM gene, which plays a critical role in regulating the DNA damage response [33]. A similar pattern was reported in several in vivo models [37,38,39]. For instance, the combined effects of radiation and the ATM kinase inhibitor (KU-60019) significantly increased mice survival by 2-3-fold compared with controls [38]. Furthermore, the mutant p53 group was substantially more radiosensitive to KU-60019 than p53 WT [39,40].
MDM2-mediated p53 suppression is another axis that was found to be disturbed in at least 25% of primary and 60% of secondary GBM. The MDM2/p53 axis was reported to reduce the efficacy of RT in the treatment of cancer [41]. Thus, inhibiting the MDM2/X-p53 interaction is recognized as a potential anti-cancer strategy, including in the treatment of glioblastoma [42].
Radio-resistance may also arise from the cancer stem cell (CSC) subpopulation, hypoxia, and the increased expression of DNA repair pathways [43]. Moreover, irradiation, similarly to TMZ, can promote cancer progression. For instance, it was observed that primary malignancies treated with brain radiation had a seven-fold greater chance of developing secondary CNS tumors [2].
Besides the radio-resistance mechanisms, RT was found to induce enhanced invasion and cell “escaping” from the primary tumor [44], which is one of the main reasons for tumor relapse [45], especially at the post-surgical margins, which occurs at rates of up to 90% [46].
Cells that survive the lethal effects of RT are frequently aggressive, multiply more quickly, and have improved migratory and invasion capabilities [47]. It was found that modest dosages of 5-8 Gy, which cannot be increased due to safety reasons [48], enhance tumor cell invasion [49,50]. It is yet not fully clear why low radiation exposures increase cell migration. Several studies found that RT changes the expression and functional activities of adhesion molecules [51], such as the upregulation of αv β3 integrin expression following RT [52]. Another study found that RT can activate Src-dependent EGFR, which activates the p38/Akt and PI3K/Akt signaling pathways, resulting in enhanced MMP-2 secretion and invasiveness of PTEN mutant glioma cells [53].
2.3. Tumor-Treating Fields (TTFs)
A TTF is a relatively recent non-invasive therapeutic approach that involves administering alternating, low-intensity, intermediate-frequency electric fields (100–300 kHz) to tumor cells [54]. The TTF devices comprise nine insulated electrodes placed on the patient’s scalp to deliver electric fields that aim to disrupt cell division and reduce tumor growth [55]. TTFs, in particular, interrupt the normal polymerization–depolymerization process of microtubules during mitosis. In terms of cell morphological changes in response to therapy, their effect is comparable with that of Taxol [55].
The US Food and Drug Administration (FDA) approved a TTF device in 2011 to treat recurrent or resistant GBM. Together with the National Comprehensive Cancer Network (NCCN), they recently approved the TTF device as an adjuvant treatment for newly diagnosed GBM patients who have completed standard-of-care surgery and chemoradiation [56]. However, a TTF is not regarded as a “standard of treatment” for GBM patients, owing to incomplete clinical trials that lacked a placebo-control “sham” device and demonstrated poor safety, in addition to the device’s high cost [56].
The poor response of GBM tumors to existing treatments spurred researchers worldwide to define the mechanisms and GBM phenotypes that cause therapeutic resistance. The primary mechanisms are listed below.
3. Infiltration and Invasion of GBM Cells
The invasiveness of GBM is a major reason for therapeutic failure. Cancer cells that remain after surgery or treatment frequently create a new mass within 2–3 cm of the original lesion [57]. While other aggressive cancers spread to organs via the circulatory or lymphatic systems, high-grade glioma cells migrate actively through two types of extracellular space in the brain: (1) perivascular space near blood vessels and (2) space between neurons and glial cells that make up the brain parenchyma and white matter fiber tracts [58].
3.1. Epithelial–Mesenchymal Transition (EMT)
The EMT is a critical mechanism in physiological and pathological processes, such as embryogenesis, wound healing, and cancer development, enabling cells to transit from an epithelial to a mesenchymal state [59]. The literature defines three distinct EMT subtypes, each of which occurs in different biological circumstances and has a range of functional outcomes [60,61].
Normal tissue homeostasis, including embryonic development, is influenced by type 1 EMT, while type 2 EMT occurs in wound healing, organ fibrosis, and tissue regeneration [61]. Type 3 EMT, on the other hand, is associated with neoplastic cells that have undergone genetic and epigenetic changes, boosting tumor-initiating and metastatic potential, as well as resistance to different treatment regimens [62].
EMT causes epithelial cells to separate when cells lose their apical–basal polarity and connections through tight junctions. In other words, epithelial cells lose their ability to adhere and instead develop a fibroblast-like shape and enhanced mobility during EMT. EMT simultaneously promotes the expression of mesenchymal marker proteins and aids in developing mesenchymal characteristics and attachment to the extracellular matrix (ECM) [63].
Type 3 EMT plays a dominant role in GBM tumors [64], where it is associated with poor prognosis, tumors’ immune escape [65], and invasive behavior of GBMs, coupled with autophagy, which is a cellular process involved in the degradation of cytosolic protein aggregates, supporting cellular and organismal homeostasis [66]. GBM cells become more capable of migrating and invading through blood vessels and basement membranes as they approach the mesenchymal state. Since GBM cells do not originate from epithelial cells, the EMT process in GBM is referred to as EMT-like.
N-cadherin overexpression and E-cadherin loss are two of the most noticeable characteristics of EMT [57,67]. The loss of E-cadherin causes changes in cell adhesion and increases cell spreading since it is an epithelial marker that prevents epithelial cells from separating from the parent tissue [68]. On the other hand, it was demonstrated that brain tumor invasion is increased by overexpressing N-cadherin, which is a transmembrane adhesion protein [68,69].
Eukaryotic Translation Elongation Factor 1 Delta (EEF1D) and Calponin and LIM Domain Containing 2 (MICAL2) are two further examples of EMT-related markers. It was demonstrated that EEF1D increases glioma proliferation and invasion via modifying the EMT process and that blocking EEF1D might reverse the EMT properties of glioma cells, decreasing cell growth and tumor progression [70]. In addition, a TGF-B/p-Smad2/EMT-like signaling pathway was discovered to increase GBM growth and invasion [71].
Recently, it was discovered that the radioresistant characteristics of some gliomas and GBMs are related to the overexpression of EMT genes, such as ACTN1, CCND1, HCLS1, ITGB5, PFN2, PTPRC, RAB13, and WAS [72]. Irradiation was also shown to increase the expression of Vimentin and other EMT-associated proteins, which may play a role in cell migration [46,73]. Furthermore, it was proposed that Vimentin can cause EMT movement by upregulating N-cadherin and downregulating E-cadherin [74].
3.2. The Extracellular Matrix (ECM) Has a Role in Cell Invasion
GBM cells invade by undergoing many biological changes that ultimately result in remodeling the cellular cytoskeleton and the surrounding extracellular matrix (ECM). Even though the ECM is a physical barrier that GBM cells must overcome, it also provides ligands, such as integrins [58], to which the tumor cells can anchor to propel themselves forward and carry out the mesenchymal migration [75]. It was shown, for example, that RT can alter the ECM by upregulating integrins, facilitating GBM infiltration [76].
Cell adhesion, migration, and cell fate decisions are significantly influenced by the molecular and physical properties of the ECM. Normal brain tissue’s ECM comprises hyaluronan, proteoglycans, and tenascin-C, and it lacks fibrillar collagens’ capacity to create stiff ECM structures [77]. In contrast, the ECM in GBM is associated with a significant increase in basement membrane constituents like collagens and laminin [10,78].
Laminin expression and peri-tumor collagen production were both found to be upregulated at the invasive edge of GBMs [79], favoring cancer cell invasion and metastasis in different cancer models [80,81].
Collagen type I, which acts as a structural framework for cells and other scaffold proteins [82], is one of the most abundant connective tissue components. Collagen I may be prevalent in some tumor types, like lung carcinoma [83]; however, in GBM [84], collagen types IV and VI were shown to be more essential for GBM tumor growth, vascularization, and migration [85,86,87]. Thus, ECM elements, including collagens, were proposed as potential therapeutic targets for GBM [47,78,88].
4. Signal Transduction Pathways and Targeted Therapies
The primary regulatory pathways that support GBM tumor invasion, survival, drug resistance, and anti-apoptotic characteristics are listed below.
4.1. Epidermal Growth Factor Receptor (EGFR)
The epidermal growth factor receptor (EGFR) has been designated as a “signature molecule” for glioblastoma [89,90]. EGFR is a tyrosine kinase receptor that regulates multiple signaling pathways involved in cell proliferation, differentiation, and migration [91], including PI3K/AKT, RAS/RAF/MEK/MAPK, and STAT cascades [92,93,94] (Figure 2). The EGFR gene amplification is common in all gliomas, accounting for 40 to 50 percent of primary GBMs [95].
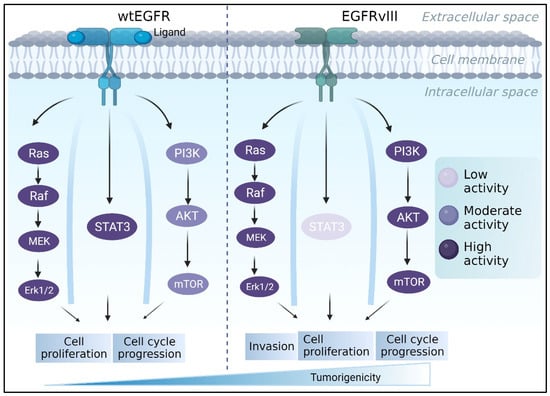
Figure 2.
The EGFR/EGFRvIII pathway contributes to the progression of GBM. While PI3K and RAS/MAPK activation is stronger in EGFRvIII-expressing cells, EGFRwt commonly stimulates MAPK and STAT3, encouraging tumor growth. Meanwhile, EGFRvIII is constitutively activated and primarily initiates the PI3K pathway, which is involved in tumor invasion and survival.
EGFRvIII is a mutant EGF receptor that lacks the extracellular ligand-binding domain (exons 2–7 deletion) and is constitutively active in 50-60% of EGFR-amplified GBMs [96]. GBM tumors that express wtEGFR frequently express the EGFR lesion [97,98,99]. EGFRvIII expression, on the other hand, is uncommon in the absence of wtEGFR amplification [98,100]. The difference between the two receptors also appears in their activated downstream pathways. For instance, it was reported that EGFRvIII cells had substantially higher PI3K activity than did cells with wtEGFR [101]. EGFRvIII, unlike wtEGFR, does not appear to activate the STAT3 pathway via direct phosphorylation [102]. Furthermore, a study found that U87MG GBM cells engineered to express EGFRvIII increased RAS activity twice as much as parental cells [103] (Figure 2). EGFRvIII was regularly found to be more tumorigenic than wtEGFR [104,105,106]. Nude mice injected with U87MG GBM cell lines harboring EGFRvIII, for example, form tumors faster than parental U87MG cells or cells expressing wtEGFR [107]. Furthermore, NR6 murine fibroblasts harboring EGFRvIII demonstrated increased motility, while U87 MG cells transfected with EGFRvIII demonstrated increased migration and invasion [108,109]. This phenomenon was compatible with clinical observations [110].
Recently, it was demonstrated that murine astrocytes expressing EGFRvIII were significantly less adhesive by reducing their focal adhesion size and number and displayed enhanced migration compared with cells bearing mutations in Ink4a or PTEN [111].
4.2. Vascular Endothelial Growth Factor Receptor (VEGFR)
Three Receptor Tyrosine Kinases (RTKs) are members of the Vascular Endothelial Growth Factor Receptors family (VEGFR1-3). It was shown to influence vasculogenesis and angiogenesis [112,113]. Because of unregulated angiogenesis and vascularization in GBM tumors, VEGFR2 signaling is disrupted, resulting in uncontrolled survival, migration, and vessel permeability [114]. Furthermore, the surrounding hypoxic microenvironment of glioblastoma induces VEGFR signaling, allowing the tumors to compensate for the hypoxia [115]. In addition to its angiogenic involvement in glioblastoma, a few studies revealed that VEGFR may be involved in the irradiation-dependent motility and proliferation of GBM cells [116].
4.3. Fibroblast Growth Factor Receptor (FGFR)
The Fibroblast Growth Factor Receptor family consists of four receptors, namely, FGFR 1-4. They control essential biological processes involved in development, including differentiation and proliferation, as well as CNS growth and tissue repair [117,118].
Although FGFR genetic mutations are regarded as infrequent in glioblastoma, FGFR overexpression in astrocytes may contribute to malignant transformation and GBM progression [119]. FGFR3 and FGFR4 were shown to be upregulated in invasive GBM cells [120]. Furthermore, FGRF1 and FGFR3 gene fusions with transforming acidic coiled-coil genes (FGFR-TACC) were discovered in GBM, leading to constitutive receptor activation and aneuploidy [121].
4.4. Platelet-Derived Growth Factor Receptor (PDGFR)
The platelet-derived growth factor receptor family includes two receptors, namely, PDGFRα and PDGFRβ, which are required for tissue development during embryogenesis. PDGF mutations were implicated in GBM development and metastasis [122]. The two receptors contribute to the progression of GBM in separate ways. While PDGFRα, which is the second most amplified receptor in glioblastoma after EGFR, is primarily upregulated in the proneural subtype, PDGFRβ is preferentially expressed in glioma stem cells (GSCs), where it regulates the level of stem cell markers, like SOX2 [4,123]. Furthermore, PDGFRα mutations are strongly linked to the occurrence and poor prognosis of GBMs [124].
4.5. Hepatocyte Growth Factor Receptor (HGFR)
The hepatocyte growth factor receptor, also known as c-MET, is recognized to play an essential role in the interaction of mesenchymal and epithelial cells throughout embryogenesis and tissue homeostasis. HGFR-activating ligands are abundantly produced by GBM cells, resulting in significant activation of PI3K, STAT3, and RAS pathways [125,126]. HGFR amplification was found in 1.6-4 percent of GBM samples, and its presence was linked to a poor prognosis [98,127].
4.6. Insulin-like Growth Factor Receptor 1 (IGF1R)
The insulin-like growth factor receptor 1 is a member of the IGF receptor family, which is known to play important functions in prenatal and postnatal development [128]. While IGF1R expression in normal tissue regulates cell proliferation and differentiation during brain development, its upregulation in GBMs is associated with enhanced activation of PI3K/Akt and MAPK pathways, leading to neoplastic transformation, TMZ resistance, and poor patient survival [129,130].
4.7. Discoidin Domain Receptors (DDRs)
Triple-helical collagen activates the discoidin domain receptors, namely, DDR1 and DDR2, in a delayed and sustained manner [131,132]. DDRs play a vital role in embryonic development by regulating a variety of processes, including proliferation, migration, adhesion, and ECM remodeling [133]. GBMs are distinguished by a high amount of collagens, which alters DDR signaling and ECM stiffness, thereby influencing tumor progression [86]. DDR1 overexpression in GBM cells is associated with increased migration and invasion [134] and poor clinical outcomes [135]. In DDR2-mutated GBM, increased cell–ECM interactions were connected to tumor invasion [136].
4.8. Tyrosine Kinase with Immunoglobulin-like and EGF-like Domains (Tie) Receptors
Tie1 and Tie2 are members of the Tie receptor family. They are required for angiogenic vascular remodeling during embryogenesis and to control lymphangiogenic responses [137]. Tie2 expression in nonvascular glioma compartments correlates with glioma development and grade [138,139]. Following this finding, it was suggested that Tie2 signaling may facilitate cross-talk in the tumor microenvironment (TME) between glioma cells and vascular endothelial cells [139]. Ang-2, which is its agonist, was shown to be overexpressed in GBMs, where it is related to Tie2 activation, increased invasion, and reduced VEGF inhibition [140,141,142].
4.9. RTK Downstream Signaling
Ras/MAPK, PI3K/AKT/PTEN, FAK/SRC, and DNA repair signaling cascades are commonly disturbed in GBM [139,143].
Despite lacking RAS mutations, the protein is significantly activated in glioblastoma due to Receptor Tyrosine Kinase (RTK) activation, such as EGFR [144]. The upregulation of H-Ras or K-Ras was found to induce astrocyte transformation into malignant and infiltrating gliomas [145,146]. In addition to contributing to gliomagenesis, the RAS/MAPK signaling pathway is also involved in tumor maintenance [147].
The PI3K/AKT/PTEN and FAK/SRC pathways, which control cell proliferation, invasion, metastasis, and metabolism, are additional critical signaling pathways in GBM [148,149,150]. PI3K/AKT/PTEN is often active in GBMs due to PTEN and PI3K mutations or Akt amplifications [151], and it plays an important role in the development and progression of gliomas [152]. The upregulation of FAK/SRC induces the invasion and metastasis of GBM malignancies. Furthermore, SRC was linked to GBM maintenance via TME inflammation and metabolic rewiring [153].
DNA damage response [154,155,156] is one of the fundamental mechanisms driving radiation or chemotherapy resistance. It was shown that EGFR may activate ATM, which is one of the main regulators and effectors of the DNA-damage-activated checkpoint system, which causes radiation resistance in EGFRvIII tumor cells [157,158].
4.10. Targeted Therapy
Although significant progress has been made recently in understanding the molecular mechanisms underlying the malignancy of glioblastomas [159,160,161], which also resulted in the identification and validation of prognostic and predictive biomarkers [162,163], targeted therapies have so far demonstrated only modest clinical trial efficacy [160] (Figure 3).
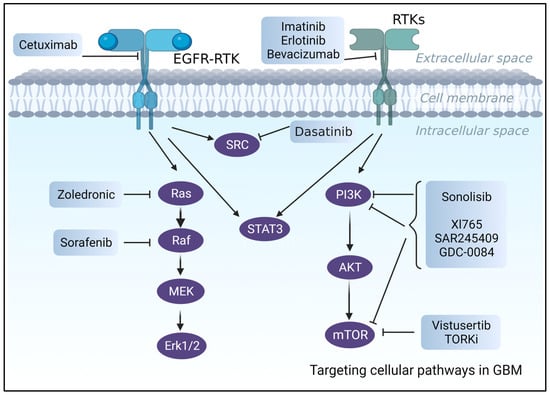
Figure 3.
An illustration of the suggested pharmacological treatments and biological targets for the treatment of GBM.
Even though more than >70% of GBM patients have overexpressed RTK [164], such as EGFR and PDGFRA with active downstream pathways (e.g., PI3K/AKT/mTOR) [165], clinical trials including RTK inhibitors, such as Cetuximab, an FDA-approved anti-EGFR [166] antibody, or Imatinib (anti-PDGFRA inhibitor), have failed. Poor BBB permeability, intertumor heterogeneity, unfavorable side effects, and toxicities were the leading causes of these failures [160,167]. Despite promising results in preclinical investigations [168], erlotinib, which is another EGFR tyrosine kinase inhibitor (TKI), exhibited poor efficacy and unacceptable side effects [169] in phase 2 clinical trials of newly diagnosed or recurrent GBM patients.
Other examples are VEGF inhibitors [167,170], such as Bevacizumab, which is an FDA-approved, humanized monoclonal antibody that failed clinical trials due to low overall patient survival [170].
Several studies were undertaken to evaluate the idea of using inhibitors of downstream pathways (PTEN/PI3K/AKT/mTOR) rather than targeting upstream receptors [160,171]. However, despite promising results in in vitro and in vivo preclinical studies, PI3K and RAS inhibitors failed in clinical trials. PI3K inhibitors, for example, were ineffective at low acceptable doses, but large dosages or long-term therapy resulted in significant side effects and toxicities, as seen with LY294002 [17,172,173,174].
Significant efforts have been made to provide novel therapeutic technologies to dissect the complexity of GBM biology, as well as crossing the BBB [175,176]. Nevertheless, poor BBB permeability and intertumor heterogeneity remain the primary therapeutic challenges for GBM [13,160,167].
5. Tumor Heterogeneity
Inter-tumor heterogeneity is the term used to describe the heterogeneity among patients harboring tumors of various histological or molecular types. The existence of phenotypically and molecularly distinct cell populations within a tumor that exhibit varying degrees of resistance to existing treatments is known as intra-tumor heterogeneity (Figure 4). Using transcriptional profiling data from bulk tumor tissues, four subgroups of GBM tumors were identified: mesenchymal, classical, proneural, and neural. A recent study identified 18 driver genes, including MGMT, ATRX, H3F3A, TP53, EGFR, NES, VIM, MIK67, and OLIG2, with differential expression profiles in different molecular subtypes [177]. For instance, Herrera-Oropeza et al. found that the classical subtype showed overexpression of EGFR, NES, VIM, and TP53, while the proneural subtype was characterized by the overexpression of MKi67 and OLIG2. The mesenchymal subtype showed the overexpression of MGMT and VIM, and the repression of EGFR, H3F3Q, OLIG2, S100, and TP53. In fact, it was discovered that NES, OLIG2, VIM, and EGFR were sufficient to subtype GBM into four subgroups, as confirmed by another investigation [178].
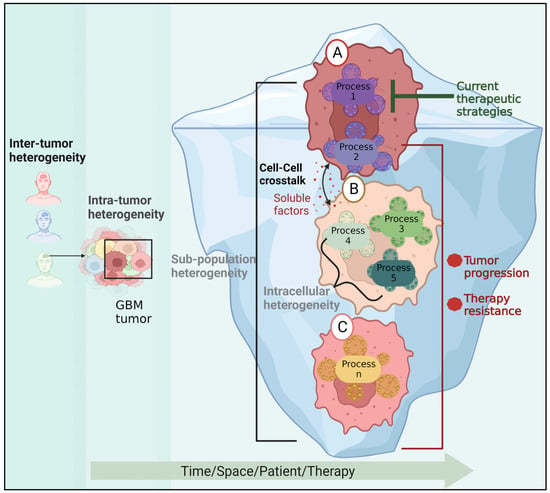
Figure 4.
The role of GBM heterogeneity in GBM recurrence. In addition to patient variability, the cells within the tumor may belong to different subpopulations and carry a variety of active molecular processes. An untargeted process in cell (A) may trigger survival molecular mechanisms in cell (B) via soluble factors, resulting in the emergence of a new resistant subpopulation and tumor progression. Cell (C) represents another innately resistant subpopulation.
High-throughput proteomics studies, which quantify protein–protein expression patterns across a large sample of GBM tumors, show a great diversity among GBM patients, resulting in dozens of subgroups. Some patients do not fit into any groupings because they have specific changes in protein–protein expression networks [6,179].
Recent investigations showed that the expression of GBM biomarkers is not homogenous, indicating that there is not only variation across patients but also within the tumor cells [1]. Sottoriva et al. demonstrated that fragments from the same tumor can be categorized into several GBM subgroups by genomic analysis of samples from various locations of a single GBM tumor. In this regard, they discovered that one tumor clone exhibited EGFR, CDK6, and MET amplification, whereas another subclone received a copy of chromosome 3 with PIK3CA, resulting in PIK3CA amplification [7].
The existence of diverse subpopulations (Figure 4) underlies tumor plasticity, resulting in resistance [8] to RTK inhibitors [180] or radiotherapy [181]. Moreover, different regions within the tumor tissue can have varying degrees of radiosensitivity, as was demonstrated using patient-derived neurosphere cultures [182].
Furthermore, microenvironmental pressures like hypoxia, acidosis, and reactive oxygen species can arbitrarily cause genetic instability, resulting in the formation of de-novo therapy-resistant subpopulations [9]. Resolving and targeting the expanding cellular subpopulations in response to therapy, as has recently been demonstrated in other cancer types, may be an effective method for reducing drug resistance development [183].
Cell–Cell Communication within GBM
Cell–cell communication between GBM subpopulations or between GBM cells and the cells in the tumor microenvironment is crucial for maintaining GBM development [96]. Factors released by microenvironment cells, like Chi3l1, or ligands, like IL-6 or HGF, that mediate the interaction of GBM subpopulations facilitate cell–cell contact [96,184], resulting in the generation and maintenance of a diversity of transcriptome and phenotypic states inside the tumor [185]. They contribute to the tumor’s aggressiveness by creating a cellular network through which cancer cells communicate with one another or other tumoral microenvironment components to stimulate their growth, invasion, neo-angiogenesis, oncogenic transformation, and immune suppression [186].
For instance, it was found that astrocytes interact closely with tumor cells, forming a network of communication crucial to tumor development [186]. Astrocytes secrete a large number of soluble factors that encourage GBM invasiveness and growth by activating several intracellular signaling pathways, such as NF-Kb and STAT1, in GBM cells [187,188,189]. As a result, GBM tumors inhibit the expression of p53 in astrocytes, promoting the survival of GBM cells through ECM remodeling [190]. Another study revealed that the communication between GBM cells and non-cancerous astrocytes drives tumor growth through mitochondrial transfer from astrocytes to GBM cells [191].
In the research on the GBM microenvironment, macrophages have also received a lot of interest since they comprise the majority and up to 30% of the tumoral mass [192]. Several signaling pathways involved in glioma invasion, including the TGF-B, EGF, and PDGF signaling pathways, were found to be stimulated by macrophages [193,194]. Another illustration of macrophage–tumor interaction is macrophage-dependent angiogenesis. IL-6 secretion, JAK-STAT activation, and increased Src-PI3K-YAP signaling all contribute to the process [195,196]. Furthermore, recent research showed that the crosstalk between GBM cells and macrophages promotes tumor growth and progression by evolving heterogeneous mechanisms that permit malignant glioma cells to enfeeble microglia and brain macrophage defense systems [197]. These mechanisms include various pathways, such as IL-6, IL-33, m-TOR, CCN4, miR-155-3p, and miR-1246 [197].
Additionally, communication between the tumor subpopulations is essential for the progression of GBM. It was discovered, using U87 model cell lines and GBM patient-derived cells, that paracrine interactions between the subpopulations expressing activating mutations (EGFRvIII) and subpopulations harboring epidermal growth factor receptor amplification (EGFRwt) play a significant role in the diffuse architectures of GBM tumors. It was shown that aggressive EGFRvIII cells alter the ability of EGFRwt cells to migrate and invade. HGF and IL6 released by EGFRvIII cells activate Src protein in EGFRwt cells, enhancing the EGFRwt cell-spreading ability and velocity [184]. Additionally, communication between these cell subtypes was found to support the tumor growth and heterogeneity of GBM [96].
6. Blood–Brain Barrier (BBB) in GBM
Although there is a growing list of the mechanisms underlying GBM development and resistance, and there is a high potential for using this knowledge to provide new therapeutic strategies, brain cancer medicine still needs to overcome an additional barrier to be able to implement potential therapeutic strategies. The blood–brain barrier (BBB) is a significant hurdle to the delivery of therapeutic drugs.
The BBB serves as a protection barrier between the circulatory system and the central nervous system’s extracellular space. The endothelial cells that make up the majority of the BBB, form a tight barrier along the blood vessel wall and regulate which substances can enter the parenchyma [198]. Thus, more than 98% of small molecules cannot penetrate tight junctions, which are smaller than 1 nm.
Although the BBB in GBM may have increased permeability as a result of poorly developed, leaky blood vessels, upregulated transporter proteins, and downregulated tight junction proteins [199,200], the disruption of the tumor’s BBB is not uniform, and almost all GBM patients have large tumor regions with an intact BBB [201].
In order to effectively treat GBM patients with drugs, several technologies are being developed [202,203]. For instance, chemical delivery systems (CDSs), which link an active drug molecule to a lipophilic carrier to boost a drug’s solubility and cell permeability or lipidation of the therapeutic molecule, are examples of such efforts [167].
Using polymeric nanoparticles (NPs) is another promising therapeutic method [204,205]. NPs are carriers with diameters ranging from 10 to 1000 nm that can be engineered from various materials, including metal-, lipid-, and polymer-based materials. They can be conjugated into multiple chemotherapeutic and targeted drugs. For instance, Alessandro Sacchetti et al. designed a gel formulation that enhances the release of TMZ locally beyond the BBB in orthotopic human xenograft models [206]. Another study suggested a novel design of flexibility-tunable polymer-drug conjugates to deliver drug combinations with ratiometric dosing and reported that focused ultrasound (FUS) improved the penetration of the drug conjugates into murine brain GBM models [207]. Furthermore, a clinical study was conducted on the use of an implantable ultrasound device, for delivering albumin-bound paclitaxel in patients with recurrent GBM [208]. The device could transiently open the BBB, allowing a safe and repeated penetration of cytotoxic drugs into the brain. Following these results, a phase 2 clinical trial is taking place to further evaluate the safety and efficacy of the approach and is registered with ClinicalTrials.gov (NCT04528680).
7. Computational Modeling of GBM: New Insights toward Understanding and Treating GBM
Computational GBM research is a rapidly growing multidisciplinary subject frequently utilized to investigate and define tumor heterogeneity; the effect of BBB constraints on treatment effectiveness; and GBM behavior, such as aggressiveness and recurrence.
For example, Randles et al. described the development of a spatially explicit stochastic process modeling to investigate the impact of the perivascular niche spatiotemporal dynamics in GBM, which can be used to optimize standard treatment (chemotherapy and radiation) schedules for this disease [209]. Another study proposed a model informed by in silico signaling pathways and kinetics characteristics to predict outcomes and prescribe customized therapy in GBM patients treated with radiation and TMZ [210].
Partial least-squares regression (PLSR) data-driven models that were constructed based on the paired signaling and phenotype data to predict the efficacy of phosphatase inhibition in GBM treatment [211] or to identify new targetable GBM markers are additional examples [212].
Information-theoretical approaches were used [6,179] to identify patient-specific signaling signatures in each GBM patient. Based on these signatures, patient-specific targeted drug combinations can be designed. This method was verified for other types of cancer [183,213].
Multiple machine learning (ML)/deep learning approaches have been extensively used to identify new potential therapeutic targets (reviewed in [214]). For example, based on progression-free survival (PFS), ML-based models (random forest classifier (RFD), extreme gradient boosting (XGBoost), naïve Bayes, and support vector machine (SVM)), algorithms were created to stratify newly diagnosed GBM patients into prognostic subclasses, identifying those at increased risk of early recurrence [215,216]. Wang et al. used machine learning algorithms (least absolute shrinkage and selection operator (LASSO) regression, SVM, RFB, and XGBoost) to identify individuals who may react better to immunotherapy and have higher overall survival [217].
Further examples include mathematical and bioinformatics models that attempt to predict BBB permeability and drug delivery efficacy [218,219]. For example, based on experimental information from preclinical subjects treated with anti-EGFR targeted therapy, an ordinary differential equation (ODE) model was developed to characterize the heterogeneous sensitivity of drug response and blood–brain barrier penetration [220].
Another study created a geometrical model that employed computational fluid dynamics to predict blood flow behavior with injected magnetic nanoparticles under various conditions, such as blood flow fluctuations. This provided information on the permeability of the BBB [221]. One more study used three-compartment cellular modeling (apical, cell monolayer, and basolateral) and statistical approaches to successfully simulate the time course of drug cellular uptake and accumulation, based on its BBB passive permeability, in order to demonstrate the functional relevance of uptake and efflux transporters to BBB penetration of drugs [222].
Overall, computational and theoretical models provide important insights into GBM heterogeneity, drug permeability across the BBB, tumor growth, and treatment responses, hence improving the efficacy of GBM dissection and individualized therapies.
8. Conclusions
Despite several advances in developing effective treatment regimens, GBM is incurable. The standard course of treatment includes radiotherapy, chemotherapy, and surgery. However, most patients experience tumor relapse and recurrence. From a therapeutic standpoint, invasion, intra- and inter-tumor heterogeneity, and BBB pose significant barriers to curative treatment.
The current primary challenge is to develop computational and experimental approaches for designing individualized multimodal treatments. The therapy should target ongoing patient-specific processes responsible for GBM infiltration and drug resistance. The proposed therapy should also consider basal or evolving states in response to treatment, as well as the interaction between the microenvironment and GBM, and should be designed to effectively transport a drug cocktail via the BBB.
Author Contributions
Conceptualization, N.R., F.-E.A.M. and N.K.-B.; writing—original draft preparation, N.R., F.-E.A.M. and N.K.-B.; writing—review and editing, N.R., F.-E.A.M. and N.K.-B.; supervision, N.K.-B. All authors have read and agreed to the published version of the manuscript.
Funding
The funding sources for this work were from Nataly Kravchenko-Balasha’s research endowments from the Hebrew University of Jerusalem.
Acknowledgments
All figures were made in BioRender.com, accessed on 13 September 2023.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| GBM | Glioblastoma |
| CNS | Central nervous system |
| TMZ | Temozolomide |
| BBB | Blood–brain barrier |
| IDH | Isocitrate Dehydrogenase 1 |
| NOS | Nitric Oxide Synthase |
| EMT | Epithelial–mesenchymal transition |
| ECM | Extracellular matrix |
| MMPs | Matrix Metalloproteinases |
| GTR | Gross total resection |
| MGMT | O6 Methylguanine DNA Methyltransferase |
| GSCs | GBM stem cells |
| EMT | Epithelial–mesenchymal transition |
| CAR T | Chimeric antigen receptor T-cells |
| TGF-β | Transforming Growth Factor Beta |
| Cx43 | Connexin43 |
| RT | Radiotherapy |
| P53 | Tumor Protein 53 |
| ATM gene | Ataxia-Telangiesctasia Mutated |
| MDM2 | Mouse Double Minute 2 |
| CSC | Cancer Stem Cell |
| PTEN | Phosphatase and Tensin Homolog |
| Akt | Protein Kinase B |
| PI3K | Phosphoinositide 3-Kinases |
| TTFs | Tumor-treating fields |
| EEF1D | Elongation Factor 1-Delta |
| MICAL2 | Calponin and LIM Domain Containing 2 |
| p-Smad2 | Mothers against decapentaplegic homolog 2 |
| ACTN1 | Alpha-Actinin-3 |
| CCND1 | Cyclin D1 |
| HCLS1 | Hematopoietic Cell-Specific Lyn Substrate 1 |
| ITGB5 | Integrin Subunit Beta 5 |
| PFN2 | Profilin 2 |
| PTPRC | Protein Tyrosine Phosphatase Receptor Type C |
| Gy | Gray (unit) |
| MAPK | Mitogen-Activated Protein Kinases |
| EGFR | Epidermal Growth Factor Receptor |
| EGF | Epidermal Growth Factor |
| VEGFR | Vascular Endothelial Growth Factor Receptor |
| FGFR | Fibroblast Growth Factor Receptor |
| PDGFR | Platelet-derived Growth Factor Receptor |
| SOX2 | SRY-Box Transcription Factor 2 |
| HGFR | Hepatocyte Growth Factor Receptor |
| IGFR | 1R-Insulin-like growth factor receptor 1 |
| DDR | Discoidin Domain Receptor |
| TME | Tumor microenvironment |
| RTK | Receptor tyrosine kinase |
| DDR | DNA damage response |
| TKI | Tyrosine Kinase Inhibitor |
| RTK | Receptor tyrosine kinases |
| NES | Nestin |
| OLIG2 | Oligodendrocyte Transcription Factor |
| VIM | Vimentin |
| Il-6 | Interleukin-6 |
| Chi3l1 | Chitinase 3 Like 1 |
| NF-Kb | Nuclear Factor Kappa Light Chain Enhancer of Activated B Cells |
| STAT1 | Signal Transducer and Activator of Transcription 1 |
| JAK-STAT | Janus kinase/signal transducers and activators of transcription |
| ATRX | Alpha Thalassemia X-linked |
| H3F3A | Histone H3.3A |
| H3F3Q | Histone H3.3Q |
| MIK67 | Marker of Proliferation Ki-67 |
| mTOR | Mammalian Target of Rapamycin |
| IL-33 | Interleukin 33 |
| CCN4 | Cellular Communication Network Factor 4 |
| MiR | MicroRNA |
| YAP | Yes-Associated Protein 1 |
| CDS | Chemical delivery system |
| NPs | Polymeric nanoparticles |
| FUS | Focused ultrasound |
| PLSR | Partial least-squares regression |
| ML | Machine learning |
| XGBoost | eXtreme gradient boosting |
| SVM | Support vector machine |
| LASSO | Least absolute shrinkage and selection operator |
| RFB | Receptive field block |
References
- Wu, W.; Klockow, J.L.; Zhang, M.; Lafortune, F.; Chang, E.; Jin, L.; Wu, Y.; Daldrup-Link, H.E. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol. Res. 2021, 171, 105780. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.E. Epidemiology and Overview of Gliomas. Semin. Oncol. Nurs. 2018, 34, 420–429. [Google Scholar] [CrossRef] [PubMed]
- Gallego, O. Nonsurgical Treatment of Recurrent Glioblastoma. Curr. Oncol. 2015, 22, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Jill, P.; Alexe, G.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef]
- Perez-Garcia, A.; Carrion-Navarro, J.; Bosch-Fortea, M.; Lazaro-Ibanez, E.; Prat-Acin, R.; Ayuso-Sacido, A. Genomic instability of surgical sample and cancer-initiating cell lines from human glioblastoma. Front. Biosci. 2012, 171, 1469–1479. [Google Scholar] [CrossRef]
- Flashner-Abramson, E.; Vasudevan, S.; Adejumobi, I.A.; Sonnenblick, A.; Kravchenko-Balasha, N. Decoding cancer heterogeneity: Studying patient-specific signaling signatures towards personalized cancer therapy. Theranostics 2019, 9, 5149. [Google Scholar] [CrossRef]
- Sottoriva, A.; Spiteri, I.; Piccirillo, S.G.M.; Touloumis, A.; Collins, V.P.; Marioni, J.C.; Curtis, C.; Watts, C.; Tavaré, S. Intratumor heterogeneity in human glioblastoma reflects cancer evolutionary dynamics. Proc. Natl. Acad. Sci. USA 2013, 110, 4009–4014. [Google Scholar] [CrossRef]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Sidhu, S.S.; Moffat, J.; Swanton, C.; Singh, S.K. Intratumoral heterogeneity: Pathways to treatment resistance and relapse in human glioblastoma. Ann. Oncol. 2017, 28, 1448–1456. [Google Scholar] [CrossRef]
- Gillies, R.J.; Verduzco, D.; Gatenby, R.A. Evolutionary dynamics of carcinogenesis and why targeted therapy does not work. Nat. Rev. Cancer 2012, 12, 487–493. [Google Scholar] [CrossRef]
- Seker-Polat, F.; Degirmenci, N.P.; Solaroglu, I.; Bagci-Onder, T. Tumor Cell Infiltration into the Brain in Glioblastoma: From Mechanisms to Clinical Perspectives. Cancers 2022, 14, 443. [Google Scholar] [CrossRef]
- Crivii, C.B.; Boșca, A.B.; Melincovici, C.S.; Constantin, A.M.; Mărginean, M.; Dronca, E.; Suflețel, R.; Gonciar, D.; Bungărdean, M.; Șovrea, A. Glioblastoma Microenvironment and Cellular Interactions. Cancers 2022, 14, 1092. [Google Scholar] [CrossRef] [PubMed]
- Aldoghachi, A.F.; Aldoghachi, A.F.; Breyne, K.; Ling, K.H.; Cheah, P.S. Recent Advances in the Therapeutic Strategies of Glioblastoma Multiforme. Neuroscience 2022, 491, 240–270. [Google Scholar] [PubMed]
- Bikfalvi, A.; da Costa, C.A.; Avril, T.; Barnier, J.V.; Bauchet, L.; Brisson, L.; Cartron, P.F.; Castel, H.; Chevet, E.; Chneiweiss, H.; et al. Challenges in glioblastoma research: Focus on the tumor microenvironment. Trends Cancer 2023, 9, 9–27. [Google Scholar] [CrossRef]
- Rong, L.; Li, N.; Zhang, Z. Emerging therapies for glioblastoma: Current state and future directions. J. Exp. Clin. Cancer Res. 2022, 41, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Abi-Said, D.; Fourney, D.R.; Gokaslan, Z.L.; Shi, W.; DeMonte, F.; Lang, F.F.; McCutcheon, I.E.; Hassenbusch, S.J.; Holland, E.; et al. A multivariate analysis of 416 patients with glioblastoma multiforme: Prognosis, extent of resection, and survival. J. Neurosurg. 2001, 95, 190–198. [Google Scholar] [CrossRef]
- Stummer, W.; Reulen, H.J.; Meinel, T.; Pichlmeier, U.; Schumacher, W.; Tonn, J.C.; Rohde, V.; Oppel, F.; Turowski, B.; Woiciechowsky, C.; et al. Extent of resection and survival in glioblastoma multiforme: Identification of and adjustment for bias. Neurosurgery 2008, 62, 564–576. [Google Scholar] [CrossRef]
- Ohka, F.; Natsume, A.; Wakabayashi, T. Current Trends in Targeted Therapies for Glioblastoma Multiforme. Neurol. Res. Int. 2012, 13, 2012. [Google Scholar] [CrossRef]
- Wang, T.; Pickard, A.J.; Gallo, J.M. Histone Methylation by Temozolomide; A Classic DNA Methylating Anticancer Drug. Anticancer Res. 2016, 36, 3289. [Google Scholar]
- Newlands, E.S.; Stevens, M.F.G.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A review of glioblastoma immunotherapy. J. Neuro-Oncol. 2020, 1511, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Tomar, M.S.; Kumar, A.; Srivastava, C.; Shrivastava, A. Elucidating the mechanisms of Temozolomide resistance in gliomas and the strategies to overcome the resistance. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188616. [Google Scholar] [CrossRef] [PubMed]
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol. Med. Chir. 2018, 58, 405. [Google Scholar] [CrossRef] [PubMed]
- Alifieris, C.; Trafalis, D.T. Glioblastoma multiforme: Pathogenesis and treatment. Pharmacol. Ther. 2015, 152, 63–82. [Google Scholar] [PubMed]
- Auffinger, B.; Tobias, A.L.; Han, Y.; Lee, G.; Guo, D.; Dey, M.; Lesniak, M.S.; Ahmed, A.U. Conversion of differentiated cancer cells into cancer stem-like cells in a glioblastoma model after primary chemotherapy. Cell Death Differ. 2014, 21, 1119–1131. [Google Scholar] [CrossRef]
- Ahmed, A.U.; Auffinger, B.; Lesniak, M.S. Understanding glioma stem cells: Rationale, clinical relevance and therapeutic strategies. Expert Rev. Neurother. 2014, 13, 545–555. [Google Scholar] [CrossRef]
- Carlsson, S.K.; Brothers, S.P.; Wahlestedt, C. Emerging treatment strategies for glioblastoma multiforme. EMBO Mol. Med. 2014, 6, 1359. [Google Scholar] [CrossRef]
- Gerber, D.E.; Grossman, S.A.; Zeltzman, M.; Parisi, M.A.; Kleinberg, L. The impact of thrombocytopenia from temozolomide and radiation in newly diagnosed adults with high-grade gliomas. Neuro Oncol. 2007, 9, 47. [Google Scholar] [CrossRef]
- Kubelt, C.; Hattermann, K.; Sebens, S.; Mehdorn, H.M.; Held-Feindt, J. Epithelial-to-mesenchymal transition in paired human primary and recurrent glioblastomas. Int. J. Oncol. 2015, 46, 2515–2525. [Google Scholar] [CrossRef]
- Kochanowski, P.; Catapano, J.; Pudełek, M.; Wróbel, T.; Madeja, Z.; Ryszawy, D.; Czyż, J. Temozolomide induces the acquisition of invasive phenotype by o6-methylguanine-dna methyltransferase (Mgmt)+ glioblastoma cells in a snail-1/cx43-dependent manner. Int. J. Mol. Sci. 2021, 22, 4150. [Google Scholar] [CrossRef]
- Feldheim, J.; Kessler, A.F.; Feldheim, J.J.; Schulz, E.; Wend, D.; Lazaridis, L.; Kleinschnitz, C.; Glas, M.; Ernestus, R.I.; Brandner, S.; et al. Effects of Long-Term Temozolomide Treatment on Glioblastoma and Astrocytoma WHO Grade 4 Stem-like Cells. Int. J. Mol. Sci. 2022, 23, 5238. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Zhang, Y.; Liu, C.; Zhang, M.; Han, S. Application of Radiosensitizers in Cancer Radiotherapy. Int. J. Nanomed. 2021, 16, 1083. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.R.; Zhang, Y.; Zhou, H.; Gridley, D.S.; Koch, C.J.; Russell, J.; Slater, J.S.; Little, J.B. A quantitative overview of radiosensitivity of human tumor cells across histological type and TP53 status. Int. J. Radiat. Biol. 2009, 84, 253–264. [Google Scholar] [CrossRef] [PubMed]
- Merrick, M.; Mimlitz, M.J.; Weeder, C.; Akhter, H.; Bray, A.; Walther, A.; Nwakama, C.; Bamesberger, J.; Djam, H.; Abid, K.; et al. In vitro radiotherapy and chemotherapy alter migration of brain cancer cells before cell death. Biochem. Biophys. Rep. 2021, 27, 101071. [Google Scholar] [CrossRef]
- Caragher, S.; Chalmers, A.J.; Gomez-Roman, N. Glioblastoma’s Next Top Model: Novel Culture Systems for Brain Cancer Radiotherapy Research. Cancers 2019, 11, 44. [Google Scholar] [CrossRef]
- Aiyappa-Maudsley, R.; Chalmers, A.J.; Parsons, J.L. Factors affecting the radiation response in glioblastoma. Neuro-Oncol. Adv. 2022, 4, vdac156. [Google Scholar] [CrossRef]
- Halliday, J.; Helmy, K.; Pattwell, S.S.; Pitter, K.L.; LaPlant, Q.; Ozawa, T.; Holland, E.C. In vivo radiation response of proneural glioma characterized by protective p53 transcriptional program and proneural-mesenchymal shift. Proc. Natl. Acad. Sci. USA 2014, 111, 5248–5253. [Google Scholar] [CrossRef]
- Beckta, J.M.; Ahmad, S.F.; Yang, H.; Valerie, K. Revisiting p53 for cancer-specific chemo- and radiotherapy: Ten years after. Cell Cycle 2014, 13, 710–713. [Google Scholar] [CrossRef]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef]
- Frosina, G.; Marubbi, D.; Marcello, D.; Vecchio, D.; Daga, A. The efficacy and toxicity of ATM inhibition in glioblastoma initiating cells-driven tumor models. Crit. Rev. Oncol. Hematol. 2019, 138, 214–222. [Google Scholar] [CrossRef]
- Okazaki, R. Role of p53 in Regulating Radiation Responses. Life 2022, 12, 1099. [Google Scholar] [CrossRef] [PubMed]
- Miles, X.; Vandevoorde, C.; Hunter, A.; Bolcaen, J. MDM2/X Inhibitors as Radiosensitizers for Glioblastoma Targeted Therapy. Front. Oncol. 2021, 11, 703442. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.Y.; Oliva, C.R.; Noman, A.S.M.; Allen, B.G.; Goswami, P.C.; Zakharia, Y.; Monga, V.; Spitz, D.R.; Buatti, J.M.; Griguer, C.E. Radioresistance in Glioblastoma and the Development of Radiosensitizers. Cancers 2020, 12, 2511. [Google Scholar] [CrossRef] [PubMed]
- Kargiotis, O.; Geka, A.; Rao, J.S.; Kyritsis, A.P. Effects of irradiation on tumor cell survival, invasion and angiogenesis. J. Neuro-Oncol. 2010, 100, 323–338. [Google Scholar] [CrossRef]
- Fiveash, J.B.; Spencer, S.A. Role of radiation therapy and radiosurgery in glioblastoma multiforme. Cancer J. 2003, 9, 222–229. [Google Scholar] [CrossRef]
- Kouam, P.N.; Rezniczek, G.A.; Kochanneck, A.; Priesch-Grzeszkowiak, B.; Hero, T.; Adamietz, I.A.; Bühler, H. Robo1 and vimentin regulate radiation-induced motility of human glioblastoma cells. PLoS ONE 2018, 13, e0198508. [Google Scholar]
- Gupta, K.; Burns, T.C. Radiation-induced alterations in the recurrent glioblastoma microenvironment: Therapeutic implications. Front. Oncol. 2018, 8, 503. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, J.; Xu, J.; Zhao, K.; Liu, X.; Wang, G.; Zhang, Z.; Ge, Y.; Zong, Y.; Xu, D.; et al. Effects of combined radiosurgery and temozolomide therapy on epidermal growth factor receptor and variant III in glioblastoma multiforme. Oncol. Lett. 2018, 15, 5751–5759. [Google Scholar] [CrossRef]
- Li, J.; Wu, D.M.; Han, R.; Yu, Y.; Deng, S.H.; Liu, T.; Zhang, T.; Xu, Y. Low-Dose Radiation Promotes Invasion and Migration of A549 Cells by Activating the CXCL1/NF-κB Signaling Pathway. OncoTargets Ther. 2020, 13, 3619–3629. [Google Scholar]
- Vala, I.S.; Martins, L.R.; Imaizumi, N.; Nunes, R.J.; Rino, J.; Kuonen, F.; Carvalho, L.M.; Rüegg, C.; Grillo, I.M.; Barata, J.T.; et al. Low Doses of Ionizing Radiation Promote Tumor Growth and Metastasis by Enhancing Angiogenesis. PLoS ONE 2010, 5, e1122. [Google Scholar]
- Baluna, R.G.; Eng, T.Y.; Thomas, C.R. Adhesion molecules in radiotherapy. Radiat. Res. 2006, 166, 819–831. [Google Scholar] [CrossRef]
- Cao, Q.; Cai, W.; Li, T.; Yang, Y.; Chen, K.; Xing, L.; Chen, X. Combination of integrin siRNA and irradiation for breast cancer therapy. Biochem. Biophys. Res. Commun. 2006, 351, 726–732. [Google Scholar] [CrossRef] [PubMed]
- Park, C.M.; Park, M.J.; Kwak, H.J.; Lee, H.C.; Kim, M.S.; Lee, S.H.; Park, I.C.; Rhee, C.H.; Hong, S.I. Ionizing Radiation Enhances Matrix Metalloproteinase-2 Secretion and Invasion of Glioma Cells through Src/Epidermal Growth Factor Receptor–Mediated p38/Akt and Phosphatidylinositol 3-Kinase/Akt Signaling Pathways. Cancer Res. 2006, 66, 8511–8519. [Google Scholar] [CrossRef] [PubMed]
- Rominiyi, O.; Vanderlinden, A.; Clenton, S.J.; Bridgewater, C.; Al-Tamimi, Y.; Collis, S.J. Tumour treating fields therapy for glioblastoma: Current advances and future directions. Br. J. Cancer 2020, 1244, 697–709. [Google Scholar]
- Kirson, E.D.; Gurvich, Z.; Schneiderman, R.; Dekel, E.; Itzhaki, A.; Wasserman, Y.; Schatzberger, R.; Palti, Y. Disruption of Cancer Cell Replication by Alternating Electric Fields. Cancer Res. 2004, 64, 3288–3295. [Google Scholar] [CrossRef] [PubMed]
- Fabian, D.; Eibl, M.d.P.G.P.; Alnahhas, I.; Sebastian, N.; Giglio, P.; Puduvalli, V.; Gonzalez, J.; Palmer, J.D. Treatment of Glioblastoma (GBM) with the Addition of Tumor-Treating Fields (TTF): A Review. Cancers 2019, 11, 174. [Google Scholar] [CrossRef]
- Gaspar, L.E.; Fisher, B.J.; Macdonald, D.R.; Leber, D.V.; Halperin, E.C.; Schold, S.C.; Cairncross, J.G. Supratentorial malignant glioma: Patterns of recurrence and implications for external beam local treatment. Int. J. Radiat. Oncol. Biol. Phys. 1992, 24, 55–57. [Google Scholar] [CrossRef]
- Paw, I.; Carpenter, R.C.; Watabe, K.; Debinski, W.; Lo, H.W. Mechanisms regulating glioma invasion. Cancer Lett. 2015, 362, 1–7. [Google Scholar] [CrossRef]
- Jayachandran, J.; Srinivasan, H.; Mani, K.P. Molecular mechanism involved in epithelial to mesenchymal transition. Arch. Biochem. Biophys. 2021, 710, 108984. [Google Scholar] [CrossRef]
- Pasupulati, A.K.; Nishad, R.; Nakuluri, K.; Motrapu, M. Epithelial–mesenchymal Transition of Glomerular Podocytes: Implications in Proteinuria. MGM J. Med. Sci. 2017, 4, 1–9. [Google Scholar] [CrossRef]
- Marconi, G.D.; Fonticoli, L.; Rajan, T.S.; Pierdomenico, S.D.; Trubiani, O.; Pizzicannella, J.; Diomede, F. Epithelial-mesenchymal transition (Emt): The type-2 emt in wound healing, tissue regeneration and organ fibrosis. Cells 2021, 10, 1587. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial–mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Involvement of partial EMT in cancer progression. J. Biochem. 2018, 164, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Brabletz, T.; Kalluri, R.; Nieto, M.A.; Weinberg, R.A. EMT in cancer. Nat. Rev. Cancer 2018, 182, 128–134. [Google Scholar] [CrossRef]
- Ning, W.; Qiu, Z.; Ji, X.; Wang, X.; An, Y.; Wang, S.; Zhang, H. The Prognostic Value of EMT in Glioma and its Role in the Glioma Immune Microenvironment. J. Mol. Neurosci. 2020, 70, 1501–1511. [Google Scholar] [CrossRef]
- Colella, B.; Faienza, F.; Di Bartolomeo, S. EMT regulation by autophagy: A new perspective in glioblastoma biology. Cancers 2019, 11, 312. [Google Scholar] [CrossRef]
- Loh, C.Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Na, T.Y.; Schecterson, L.; Mendonsa, A.M.; Gumbiner, B.M. The functional activity of E-cadherin controls tumor cell metastasis at multiple steps. Proc. Natl. Acad. Sci. USA 2020, 117, 5931–5937. [Google Scholar] [CrossRef]
- Noronha, C.; Ribeiro, A.S.; Taipa, R.; Castro, D.S.; Reis, J.; Faria, C.; Paredes, J. Cadherin expression and emt: A focus on gliomas. Biomedicines 2021, 9, 1328. [Google Scholar] [CrossRef]
- Xie, C.; Zhou, M.; Lin, J.; Wu, Z.; Ding, S.; Luo, J.; Zhan, Z.; Cai, Y.; Xue, S.; Song, Y. EEF1D Promotes Glioma Proliferation, Migration, and Invasion through EMT and PI3K/Akt Pathway. BioMed Res. Int. 2020. [Google Scholar] [CrossRef]
- Pu, B.; Zhang, X.; Yan, T.; Li, Y.; Liu, B.; Jian, Z.; Mahgoub, O.K.; Gu, L.; Xiong, X.; Zou, N. MICAL2 Promotes Proliferation and Migration of Glioblastoma Cells Through TGF-β/p-Smad2/EMT-Like Signaling Pathway. Front. Oncol. 2021, 11, 735180. [Google Scholar] [CrossRef] [PubMed]
- Meng, J.; Li, P.; Zhang, Q.; Yang, Z.; Fu, S. A radiosensitivity gene signature in predicting glioma prognostic via EMT pathway. Oncotarget 2014, 5, 4683. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Jang, S.J.; Kang, S.W.; Park, S.; Hwang, S.-G.; Kim, W.-J.; Kang, J.H.; Um, H.-D. Establishment of animal model for the analysis of cancer cell metastasis during radiotherapy. Radiat. Oncol. 2012, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Osuka, S.; Zhu, D.; Zhang, Z.; Li, C.; Stackhouse, C.T.; Sampetrean, O.; Olson, J.J.; Gillespie, G.Y.; Saya, H.; Willey, C.D.; et al. N-cadherin upregulation mediates adaptive radioresistance in glioblastoma. J. Clin. Investig. 2021, 131, e136098. [Google Scholar] [CrossRef] [PubMed]
- Lintz, M.; Muñoz, A.; Reinhart-King, C.A. The Mechanics of Single Cell and Collective Migration of Tumor Cells. J. Biomech. Eng. 2017, 139, 021005. [Google Scholar] [CrossRef]
- Barker, H.E.; Paget, J.T.E.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef]
- Bellail, A.C.; Hunter, S.B.; Brat, D.J.; Tan, C.; Van Meir, E.G. Microregional extracellular matrix heterogeneity in brain modulates glioma cell invasion. Int. J. Biochem. Cell Biol. 2004, 36, 1046–1069. [Google Scholar] [CrossRef]
- Mohiuddin, E.; Wakimoto, H. Extracellular matrix in glioblastoma: Opportunities for emerging therapeutic approaches. Am. J. Cancer Res. 2021, 11, 3742. [Google Scholar]
- Tysnes, B.B.; Mahesparan, R.; Thorsen, F.; Haugland, H.K.; Porwol, T.; Enger, P.Ø.; Lund-Johansen, M.; Bjerkvig, R. Laminin expression by glial fibrillary acidic protein positive cells in human gliomas. Int. J. Dev. Neurosci. 1999, 17, 531–539. [Google Scholar] [CrossRef]
- Paszek, M.J.; Zahir, N.; Johnson, K.R.; Lakins, J.N.; Rozenberg, G.I.; Gefen, A.; Reinhart-King, C.A.; Margulies, S.S.; Dembo, M.; Boettiger, D.; et al. Tensional homeostasis and the malignant phenotype. Cancer Cell 2005, 8, 241–254. [Google Scholar] [CrossRef]
- Goetz, J.G.; Minguet, S.; Navarro-Lérida, I.; Lazcano, J.J.; Samaniego, R.; Calvo, E.; Tello, M.; Osteso-Ibáñez, T.; Pellinen, T.; Echarri, A.; et al. Biomechanical Remodeling of the Microenvironment by Stromal Caveolin-1 Favors Tumor Invasion and Metastasis. Cell 2011, 146, 148–163. [Google Scholar] [CrossRef] [PubMed]
- Friedl, P.; Alexander, S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell 2011, 147, 992–1009. [Google Scholar] [CrossRef] [PubMed]
- Sarazin, T.; Collin, G.; Buache, E.; Van Gulick, L.; Charpentier, C.; Terryn, C.; Morjani, H.; Saby, C. Type I Collagen Aging Increases Expression and Activation of EGFR and Induces Resistance to Erlotinib in Lung Carcinoma in 3D Matrix Model. Front. Oncol. 2020, 10, 1593. [Google Scholar] [CrossRef] [PubMed]
- Zamecnik, J. The extracellular space and matrix of gliomas. Acta Neuropathol. 2005, 110, 435–442. [Google Scholar] [CrossRef] [PubMed]
- Mammoto, T.; Jiang, A.; Jiang, E.; Panigrahy, D.; Kieran, M.W.; Mammoto, A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am. J. Pathol. 2013, 183, 1293–1305. [Google Scholar] [CrossRef] [PubMed]
- Payne, L.S.; Huang, P.H. The pathobiology of collagens in glioma. Mol. Cancer Res. 2013, 11, 1129–1140. [Google Scholar] [CrossRef]
- Rape, A.; Ananthanarayanan, B.; Kumar, S. Engineering strategies to mimic the glioblastoma microenvironment. Adv. Drug Deliv. Rev. 2014, 79, 172–183. [Google Scholar] [CrossRef]
- Gupta, A.; Kheur, S.; Palaskar, S.; Narang, B. Deciphering the “Collagen code” in tumor progression. J. Cancer Res. Ther. 2021, 17, 29–32. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a Target for Glioblastoma Treatment: An Unfulfilled Promise. CNS Drugs 2017, 31, 723. [Google Scholar] [CrossRef]
- Dhawan, A.; Manem, V.S.K.; Yeaney, G.; Lathia, J.D.; Ahluwalia, M.S. EGFR Pathway Expression Persists in Recurrent Glioblastoma Independent of Amplification Status. Cancers 2023, 15, 670. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Paplomata, E.; O’regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets, trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef]
- Ou, A.; Ott, M.; Fang, D.; Heimberger, A.B. The role and therapeutic targeting of JAK/STAT signaling in glioblastoma. Cancers 2021, 13, 437. [Google Scholar] [CrossRef] [PubMed]
- Ramaswamy, P.; Nanjaiah, N.D.; Borkotokey, M. Role of MEK-ERK signaling mediated adhesion of glioma cells to extracellular matrix: Possible implication on migration and proliferation. Ann. Neurosci. 2019, 26, 52–56. [Google Scholar] [CrossRef] [PubMed]
- Roth, P.; Weller, M. Challenges to targeting epidermal growth factor receptor in glioblastoma: Escape mechanisms and combinatorial treatment strategies. Neuro Oncol. 2014, 16, viii14–viii19. [Google Scholar] [CrossRef]
- Inda, M.-D.; Bonavia, R.; Mukasa, A.; Narita, Y.; Sah, D.W.; Vandenberg, S.; Brennan, C.; Johns, T.G.; Bachoo, R.; Hadwiger, P.; et al. Tumor heterogeneity is an active process maintained by a mutant EGFR-induced cytokine circuit in glioblastoma. Genes Dev. 2010, 24, 1731–1745. [Google Scholar] [CrossRef]
- An, Z.; Aksoy, O.; Zheng, T.; Fan, Q.-W.W.; Weiss, W.A. Epidermal growth factor receptor and EGFRvIII in glioblastoma: Signaling pathways and targeted therapies. Oncogene 2018, 37, 1561–1575. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Lassman, A.B.; Aldape, K.D.; Ansell, P.J.; Bain, E.; Curran, W.J.; Eoli, M.; French, P.J.; Kinoshita, M.; Looman, J.; Mehta, M.; et al. Epidermal growth factor receptor (EGFR) amplification rates observed in screening patients for randomized trials in glioblastoma. J. Neuro-Oncol. 2019, 144, 205–210. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Hlatky, R.; Suki, D.; Yang, D.; Weinberg, J.; Gilbert, M.; Sawaya, R.; Aldape, K. Prognostic effect of epidermal growth factor receptor and EGFRvIII in glioblastoma multiforme patients. Clin. Cancer Res. 2005, 11, 1462–1466. [Google Scholar] [CrossRef]
- Moscatello, D.K.; Holgado-Madruga, M.; Emlet, D.R.; Montgomery, R.B.; Wong, A.J. Constitutive activation of phosphatidylinositol 3-kinase by a naturally occurring mutant epidermal growth factor receptor. J. Biol. Chem. 1998, 273, 200–206. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia, N.; Konopka, G.; Puram, S.V.; Chan, J.A.; Bachoo, R.M.; You, M.J.; Levy, D.E.; DePinho, R.A.; Bonni, A. Identification of a PTEN-regulated STAT3 brain tumor suppressor pathway. Genes Dev. 2008, 22, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Prigent, S.A.; Nagane, M.; Lin, H.; Huvar, I.; Boss, G.R.; Feramisco, J.R.; Cavenee, W.K.; Huang, H.-J.S. Enhanced tumorigenic behavior of glioblastoma cells expressing a truncated epidermal growth factor receptor is mediated through the Ras-Shc-Grb2 pathway. J. Biol. Chem. 1996, 271, 25639–25645. [Google Scholar] [CrossRef] [PubMed]
- Feldkamp, M.M.; Lala, P.; Lau, N.; Roncari, L.; Guha, A. Expression of activated epidermal growth factor receptors, Ras-guanosine triphosphate, and mitogen-activated protein kinase in human glioblastoma multiforme specimens. Neurosurgery 1999, 45, 1442. [Google Scholar] [CrossRef]
- Frederick, L.; Eley, G.; Wang, X.Y.; James, C.D. Analysis of genomic rearrangements associated with EGFRvIII expression suggests involvement of Alu repeat elements. Neuro Oncol. 2000, 2, 159–163. [Google Scholar] [CrossRef]
- Huang, H.-J.S.; Nagane, M.; Klingbeil, C.K.; Lin, H.; Nishikawa, R.; Ji, X.-D.; Huang, C.-M.; Gill, G.N.; Wiley, H.S.; Cavenee, W.K. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J. Biol. Chem. 1997, 272, 2927–2935. [Google Scholar] [CrossRef]
- Nishikawa, R.; Ji, X.D.; Harmon, R.C.; Lazar, C.S.; Gill, G.N.; Cavenee, W.K.; Huang, H.J.S. A mutant epidermal growth factor receptor common in human glioma confers enhanced tumorigenicity. Proc. Natl. Acad. Sci. USA 1994, 91, 7727–7731. [Google Scholar] [CrossRef]
- Cai, X.-M.; Tao, B.-B.; Wang, L.-Y.; Liang, Y.-L.; Jin, J.-W.; Yang, Y.; Hu, Y.-L.; Zha, X.-L. Protein phosphatase activity of PTEN inhibited the invasion of glioma cells with epidermal growth factor receptor mutation type III expression. Int. J. Cancer 2005, 117, 905–912. [Google Scholar] [CrossRef]
- Pedersen, M.W.; Tkach, V.; Pedersen, N.; Berezin, V.; Poulsen, H.S. Expression of a naturally occurring constitutively active variant of the epidermal growth factor receptor in mouse fibroblasts increases motility. Int. J. Cancer 2004, 108, 643–653. [Google Scholar] [CrossRef]
- Bakas, S.; Akbari, H.; Pisapia, J.; Martinez-Lage, M.; Rozycki, M.; Rathore, S.; Dahmane, N.; O’Rourke, D.M.; Davatzikos, C. In vivo detection of EGFRvIII in glioblastoma via perfusion magnetic resonance imaging signature consistent with deep peritumoral infiltration: The φ-index. Clin. Cancer Res. 2017, 23, 4724–4734. [Google Scholar] [CrossRef]
- Banisadr, A.; Eick, M.; Beri, P.; Parisian, A.D.; Yeoman, B.; Placone, J.K.; Engler, A.J.; Furnari, F. EGFRvIII uses intrinsic and extrinsic mechanisms to reduce glioma adhesion and increase migration. J. Cell Sci. 2020, 133, jcs247189. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Wang, N.; Jain, R.K.; Batchelor, T.T. New Directions in Anti-Angiogenic Therapy for Glioblastoma. Neurotherapeutics 2017, 14, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct. Funct. 2001, 26, 25–35. [Google Scholar] [CrossRef]
- Krcek, R.; Matschke, V.; Theis, V.; Adamietz, I.A.; Bühler, H.; Theiss, C. Vascular endothelial growth factor, irradiation, and axitinib have diverse effects on motility and proliferation of glioblastoma multiforme cells. Front. Oncol. 2017, 7, 182. [Google Scholar] [CrossRef]
- Ornitz, D.M.; Itoh, N. Protein family review: Fibroblast growth factors. Genome Biol. 2001, 2, 3005.1–3005.12. [Google Scholar] [CrossRef]
- Guillemot, F.; Zimmer, C. From cradle to grave: The multiple roles of fibroblast growth factors in neural development. Neuron 2011, 71, 574–588. [Google Scholar] [CrossRef]
- Morrison, R.S.; Yamaguchi, F.; Saya, H.; Bruner, J.M.; Yahanda, A.M.; Donehower, L.A.; Berger, M. Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J. Neurooncol. 1994, 18, 207–216. [Google Scholar] [CrossRef]
- Katoh, M.; Nakagama, H. FGF Receptors: Cancer Biology and Therapeutics. Med. Res. Rev. 2014, 34, 280–300. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y. Multifarious functions of PDGFs and PDGFRs in tumor growth and metastasis. Trends Mol. Med. 2013, 19, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. J. Bone Jt. Surg. 2012, 26, 1247–1262. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Gao, X.; Chi, Y.; Zhang, M.; Lin, H.; Chen, H.; Sun, C.; Ma, X. Molecular Alterations and Their Correlation with the Survival of Glioblastoma Patients with Corpus Callosum Involvement. Front. Neurosci. 2021, 15, 701426. [Google Scholar] [CrossRef]
- Abounader, R.; Laterra, J. Scatter factor/hepatocyte growth factor in brain tumor growth and angiogenesis. Neuro-Oncol. 2005, 7, 436–451. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 270. [Google Scholar] [CrossRef]
- Kong, D.-S.; Song, S.-Y.; Kim, D.-H.; Joo, K.M.; Yoo, J.-S.; Koh, J.S.; Dong, S.M.; Suh, Y.-L.; Lee, J.-I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2008, 115, 140–148. [Google Scholar] [CrossRef]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin receptor isoforms in physiology and disease: An updated view. Endocr. Rev. 2017, 38, 379–431. [Google Scholar] [CrossRef]
- Trojan, J.; Cloix, J.F.; Ardourel, M.Y.; Chatel, M.; Anthony, D.D. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience 2007, 145, 795–811. [Google Scholar] [CrossRef]
- Maris, C.; D'Haene, N.; Trépant, A.-L.; Le Mercier, M.; Sauvage, S.; Allard, J.; Rorive, S.; Demetter, P.; Decaestecker, C.; Salmon, I. IGF-IR: A new prognostic biomarker for human glioblastoma. Br. J. Cancer 2015, 113, 729–737. [Google Scholar] [CrossRef]
- Carafoli, F.; Hohenester, E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The Discoidin Domain Receptor Tyrosine KinasesAre Activated by Collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Lorente, G.; Nikolich, K.; Urfer, R.; Foehr, E.; Nagavarapu, U. Discoidin domain receptor-1a (DDR1a) promotes glioma cell invasion and adhesion in association with matrix metalloproteinase-2. J. Neurooncol. 2006, 76, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Arao, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Tanaka, R.; Sano, M.; Oide, A.; Sekijima, M.; Nishio, K. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006, 25, 5994–6002. [Google Scholar] [CrossRef]
- El Husseini, K.; Marguet, F.; Lamy, A.; Magne, N.; Fontanilles, M. Major response to temozolomide as first-line treatment for newly-diagnosed DDR2-mutated glioblastoma: A case report. Rev. Neurol. 2020, 176, 402–404. [Google Scholar] [CrossRef]
- Jones, N.; Iljin, K.; Dumont, D.J.; Alitalo, K. Tie receptors: New modulators of angiogenic and lymphangiogenic responses. Nat. Rev. Mol. Cell Biol. 2001, 2, 257–267. [Google Scholar] [CrossRef]
- Lee, O.-H.; Xu, J.; Fueyo, J.; Fuller, G.N.; Aldape, K.D.; Alonso, M.M.; Piao, Y.; Liu, T.-J.; Lang, F.F.; Bekele, B.N.; et al. Expression of the receptor tyrosine kinase Tie2 in neoplastic glial cells is associated with integrin β1-dependent adhesion to the extracellular matrix. Mol. Cancer Res. 2006, 4, 915–926. [Google Scholar] [CrossRef]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor tyrosine kinase signaling and targeting in glioblastoma multiforme. Int. J. Mol. Sci. 2021, 22, 1831. [Google Scholar] [CrossRef]
- Teichert-Kuliszewska, K.; Maisonpierre, P.C.; Jones, N.; Campbell, A.I.; Master, Z.; Bendeck, M.P.; Alitalo, K.; Dumont, D.J.; Yancopoulos, G.D.; Stewart, D.J. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc. Res. 2001, 49, 659–670. [Google Scholar] [CrossRef]
- Hu, B.; Guo, P.; Fang, Q.; Tao, H.-Q.; Wang, D.; Nagane, M.; Huang, H.-J.S.; Gunji, Y.; Nishikawa, R.; Alitalo, K.; et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc. Natl. Acad. Sci. USA 2003, 100, 8904–8909. [Google Scholar] [CrossRef]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target Ther. 2017, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rajasekhar, V.K.; Viale, A.; Socci, N.D.; Wiedmann, M.; Hu, X.; Holland, E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 2003, 12, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Abel, T.W.; Clark, C.; Bierie, B.; Chytil, A.; Aakre, M.; Gorska, A.; Moses, H.L. GFAP-Cre-mediated activation of oncogenic K-ras results in expansion of the subventricular zone and infiltrating glioma. Mol. Cancer Res. 2009, 7, 645–653. [Google Scholar] [CrossRef]
- Ding, H.; Roncari, L.; Shannon, P.; Wu, X.; Lau, N.; Karaskova, J.; Gutmann, D.; Squire, J.; Nagy, A.; Guha, A. Astrocyte-specific expression of activated p21-ras results in malignant astrocytoma formation in a transgenic mouse model of human gliomas. Cancer Res. 2001, 61, 3826–3836. [Google Scholar]
- Holmen, S.L.; Williams, B.O. Essential role for Ras signaling in glioblastoma maintenance. Cancer Res. 2005, 65, 8250–8255. [Google Scholar] [CrossRef]
- Cheng, C.K.; Fan, Q.W.; Weiss, W.A. PI3K signaling in glioma—Animal models and therapeutic challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef]
- Hauck, C.R.; Hsia, D.A.; Schlaepfer, D.D. The focal adhesion kinase—A regulator of cell migration and invasion. IUBMB Life 2002, 53, 115–119. [Google Scholar] [CrossRef]
- Hu, J.; Mukhopadhyay, A.; Truesdell, P.; Chander, H.; Mukhopadhyay, U.K.; Mak, A.S.; Craig, A.W.B. Cdc42-interacting protein 4 is a Src substrate that regulates invadopodia and invasiveness of breast tumors by promoting MT1-MMP endocytosis. J. Cell Sci. 2011, 124, 1739–1751. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; MMastrogianakis, G.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.G.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Cirotti, C.; Contadini, C.; Barilà, D. SRC kinase in Glioblastoma: News from an Old Acquaintance. Cancers 2020, 12, 1558. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.K.; Hung, M.C. Regulation of therapeutic resistance in cancers by receptor tyrosine kinases. Am. J. Cancer Res. 2016, 6, 827. [Google Scholar] [PubMed]
- Grabocka, E.; Pylayeva-Gupta, Y.; Jones, M.J.; Lubkov, V.; Yemanaberhan, E.; Taylor, L.; Jeng, H.H.; Bar-Sagi, D. Wild-Type H- and N-Ras Promote Mutant K-Ras-Driven Tumorigenesis by Modulating the DNA Damage Response. Cancer Cell 2014, 25, 243–256. [Google Scholar] [CrossRef]
- Hähnel, P.S.; Enders, B.; Sasca, D.; Roos, W.P.; Kaina, B.; Bullinger, L.; Theobald, M.; Kindler, T. Targeting components of the alternative NHEJ pathway sensitizes KRAS mutant leukemic cells to chemotherapy. Blood 2014, 123, 2355–2366. [Google Scholar] [CrossRef]
- Bensimon, A.; Aebersold, R.; Shiloh, Y. Beyond ATM: The protein kinase landscape of the DNA damage response. FEBS Lett. 2011, 585, 1625–1639. [Google Scholar] [CrossRef]
- Golding, S.E.; Rosenberg, E.; Khalil, A.; McEwen, A.; Holmes, M.; Neill, S.; Povirk, L.F.; Valerie, K. Double Strand Break Repair by Homologous Recombination Is Regulated by Cell Cycle-independent Signaling via ATM in Human Glioma Cells. J. Biol. Chem. 2004, 279, 15402–15410. [Google Scholar] [CrossRef]
- Sasmita, A.O.; Wong, Y.P.; Ling, A.P.K. Biomarkers and therapeutic advances in glioblastoma multiforme. Asia Pac. J. Clin. Oncol. 2018, 14, 40–51. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Qin, A.; Musket, A.; Musich, P.R.; Schweitzer, J.B.; Xie, Q. Receptor tyrosine kinases as druggable targets in glioblastoma: Do signaling pathways matter? Neuro-Oncol. Adv. 2021, 3, vdab133. [Google Scholar] [CrossRef]
- Cruz Da Silva, E.; Mercier, M.C.; Etienne-selloum, N.; Dontenwill, M.; Choulier, L. A Systematic Review of Glioblastoma-Targeted Therapies in Phases II, III, IV Clinical Trials. Cancers 2021, 13, 1795. [Google Scholar] [CrossRef]
- Le Rhun, E.; Preusser, M.; Roth, P.; Reardon, D.A.; van den Bent, M.V.; Wen, P.; Reifenberger, G.; Weller, M. Molecular targeted therapy of glioblastoma. Cancer Treat. Rev. 2019, 80, 101896. [Google Scholar] [CrossRef]
- Liu, H.; Qiu, W.; Sun, T.; Wang, L.; Du, C.; Hu, Y.; Liu, W.; Feng, F.; Chen, Y.; Sun, H. Therapeutic strategies of glioblastoma (GBM): The current advances in the molecular targets and bioactive small molecule compounds. Acta Pharm. Sin. B 2022, 12, 1781–1804. [Google Scholar] [CrossRef] [PubMed]
- Ohgaki, H.; Kleihues, P. Genetic alterations and signaling pathways in the evolution of gliomas. Cancer Sci. 2009, 100, 2235–2241. [Google Scholar] [CrossRef] [PubMed]
- Dreier, A.; Barth, S.; Goswami, A.; Weis, J. Cetuximab induces mitochondrial translocalization of EGFRvIII, but not EGFR: Involvement of mitochondria in tumor drug resistance? Tumour Biol. 2012, 33, 85–94. [Google Scholar] [CrossRef]
- Shergalis, A.; Bankhead, A.; Luesakul, U.; Muangsin, N.; Neamati, N. Current challenges and opportunities in treating glioblastomas. Pharmacol. Rev. 2018, 70, 412–445. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Desjardins, A.; Vredenburgh, J.J.; Gururangan, S.; Friedman, A.H.; Herndon, J.E.; Marcello, J.; Norfleet, J.A.; McLendon, R.E.; Sampson, J.H.; et al. Phase 2 trial of erlotinib plus sirolimus in adults with recurrent glioblastoma. J. Neuro-Oncol. 2009, 96, 219–230. [Google Scholar] [CrossRef]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.J.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neuro-Oncol. 2009, 98, 93–99. [Google Scholar] [CrossRef]
- Kim, M.M.; Umemura, Y.; Leung, D. Bevacizumab and Glioblastoma: Past, Present, and Future Directions. Cancer J. 2018, 24, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wu, W.; Bi, H.; Yang, D.; Zhang, C. Glioblastoma precision therapy: From the bench to the clinic. Cancer Lett. 2020, 475, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.F.; Wang, J.; Shao, W.; Wu, C.P.; Chen, Z.P.; To, S.S.T.; Li, W.P. Recent advances in the use of PI3K inhibitors for glioblastoma multiforme: Current preclinical and clinical development. Mol. Cancer 2017, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.Y.; Omuro, A.; Ahluwalia, M.S.; Fathallah-Shaykh, H.M.; Mohile, N.; Lager, J.; Laird, A.D.; Tang, J.; Jiang, J.; Egile, C.; et al. Phase I dose-escalation study of the PI3K/mTOR inhibitor voxtalisib (SAR245409, XL765) plus temozolomide with or without radiotherapy in patients with high-grade glioma. Neuro-Oncol. 2015, 17, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Pitz, M.W.; Eisenhauer, E.A.; MacNeil, M.V.; Thiessen, B.; Easaw, J.C.; Macdonald, D.R.; Eisenstat, D.D.; Kakumanu, A.S.; Salim, M.; Chalchal, H.; et al. Phase II study of PX-866 in recurrent glioblastoma. Neuro-Oncol. 2015, 17, 1270–1274. [Google Scholar] [CrossRef]
- Oprita, A.; Baloi, S.C.; Staicu, G.A.; Alexandru, O.; Tache, D.E.; Danoiu, S.; Micu, E.S.; Sevastre, A.S. Updated insights on EGFR signaling pathways in glioma. Int. J. Mol. Sci. 2021, 22, 587. [Google Scholar] [CrossRef]
- Yang, K.; Wu, Z.; Zhang, H.; Zhang, N.; Wu, W.; Wang, Z.; Dai, Z.; Zhang, X.; Zhang, L.; Peng, Y.; et al. Glioma targeted therapy: Insight into future of molecular approaches. Mol. Cancer 2022, 21, 1–32. [Google Scholar] [CrossRef]
- Herrera-Oropeza, G.E.; Angulo-Rojo, C.; Gástelum-López, S.A.; Varela-Echavarría, A.; Hernández-Rosales, M.; Aviña-Padilla, K. Glioblastoma multiforme: A multi-omics analysis of driver genes and tumour heterogeneity. Interface Focus 2021, 11, 20200072. [Google Scholar] [CrossRef]
- Teo, W.-Y.; Sekar, K.; Seshachalam, P.; Shen, J.; Chow, W.-Y.; Lau, C.C.; Yang, H.; Park, J.; Kang, S.-G.; Li, X.; et al. Relevance of a TCGA-derived Glioblastoma Subtype Gene-Classifier among Patient Populations. Sci. Rep. 2019, 9, 7442. [Google Scholar] [CrossRef]
- Kravchenko-Balasha, N.; Johnson, H.; White, F.M.; Heath, J.R.; Levine, R.D. A thermodynamic-based interpretation of protein expression heterogeneity in different glioblastoma multiforme tumors identifies tumor-specific unbalanced processes. J. Phys. Chem. B 2016, 120, 5990–5997. [Google Scholar] [CrossRef]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- McAbee, J.H.; Degorre-Kerbaul, C.; Valdez, K.; Wendler, A.; Shankavaram, U.T.; Watts, C.; Camphausen, K.; Tofilon, P.J. Detection of glioblastoma intratumor heterogeneity in radiosensitivity using patient-derived neurosphere cultures. J. Neuro-Oncol. 2020, 149, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Alkhatib, H.; Rubinstein, A.M.; Vasudevan, S.; Flashner-Abramson, E.; Stefansky, S.; Chowdhury, S.R.; Oguche, S.; Peretz-Yablonsky, T.; Granit, A.; Granot, Z.; et al. Computational quantification and characterization of independently evolving cellular subpopulations within tumors is critical to inhibit anti-cancer therapy resistance. Genome Med. 2022, 14, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Jubran, M.R.; Rubinstein, A.M.; Cojocari, I.; Adejumobi, I.A.; Mogilevsky, M.; Tibi, S.; Sionov, R.V.; Verreault, M.; Idbaih, A.; Karni, R.; et al. Dissecting the role of crosstalk between glioblastoma subpopulations in tumor cell spreading. Oncogenesis 2020, 9, 11. [Google Scholar] [CrossRef]
- Guetta-Terrier, C.; Karambizi, D.; Akosman, B.; Zepecki, J.P.; Chen, J.-S.; Kamle, S.; Fajardo, J.E.; Fiser, A.; Singh, R.; Toms, S.A.; et al. Chi3l1 is a modulator of glioma stem cell states and a therapeutic target in glioblastoma. Cancer Res. 2023, 83, 1984–1999. [Google Scholar] [CrossRef]
- Nieland, L.; Morsett, L.M.; Broekman, M.L.D.; Breakefield, X.O.; Abels, E.R. Extracellular Vesicle-Mediated Bilateral Communication between Glioblastoma and Astrocytes. Trends Neurosci. 2021, 44, 215–226. [Google Scholar] [CrossRef]
- Zhang, H.; Zhou, Y.; Cui, B.; Liu, Z.; Shen, H. Novel insights into astrocyte-mediated signaling of proliferation, invasion and tumor immune microenvironment in glioblastoma. Biomed. Pharmacother. 2020, 126, 110086. [Google Scholar] [CrossRef]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, S.; Spray, D.C. Glioblastoma–Astrocyte Connexin 43 Gap Junctions Promote Tumor Invasion. Mol. Cancer Res. 2022, 20, 319–331. [Google Scholar] [CrossRef]
- Biasoli, D.; Sobrinho, M.F.; Da Fonseca, A.C.C.; De Matos, D.G.; Romao, L.; de Moraes Maciel, R.; Rehen, S.K.; Moura-Neto, V.; Borges, H.L.; Lima, F.R.S. Glioblastoma cells inhibit astrocytic p53-expression favoring cancer malignancy. Oncogenesis 2014, 3, e123. [Google Scholar] [CrossRef]
- Watson, D.C.; Bayik, D.; Storevik, S.; Moreino, S.S.; Sprowls, S.A.; Han, J.; Augustsson, M.T.; Lauko, A.; Sravya, P.; Røsland, G.V.; et al. GAP43-dependent mitochondria transfer from astrocytes enhances glioblastoma tumorigenicity. Nat. Cancer 2023, 4, 648–664. [Google Scholar] [CrossRef] [PubMed]
- Pires-Afonso, Y.; Niclou, S.P.; Michelucci, A. Revealing and harnessing tumour-associated microglia/macrophage heterogeneity in glioblastoma. Int. J. Mol. Sci. 2020, 21, 689. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Xu, S.; Xin, Y.; Yu, S.-C.; Ping, Y.-F.; Chen, L.; Xiao, H.-L.; Wang, B.; Yi, L.; Wang, Q.-L.; et al. Tumor-Associated Microglia/Macrophages Enhance the Invasion of Glioma Stem-like Cells via TGF-β1 Signaling Pathway. J. Immunol. 2012, 189, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, S.; Blank, A.; Bungert, A.D.; Vajkoczy, P. Distinction of microglia and macrophages in glioblastoma: Close relatives, different tasks? Int. J. Mol. Sci. 2021, 22, 194. [Google Scholar] [CrossRef]
- Zhu, C.; Kros, J.M.; Cheng, C.; Mustafa, D. The contribution of tumor-Associated macrophages in glioma neo-Angiogenesis and implications for anti-Angiogenic strategies. Neuro Oncol. 2017, 19, 1435–1446. [Google Scholar] [CrossRef]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking macrophage-associated immunosuppression for regulating glioblastoma angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef]
- Zheng, Y.; Graeber, M.B. Microglia and Brain Macrophages as Drivers of Glioma Progression. Int. J. Mol. Sci. 2022, 23, 15612. [Google Scholar] [CrossRef]
- Ou, A.; Alfred Yung, W.K.; Majd, N. Molecular Mechanisms of Treatment Resistance in Glioblastoma. Int. J. Mol. Sci. 2021, 22, 351. [Google Scholar] [CrossRef]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef]
- Huang, B.; Li, X.; Li, Y.; Zhang, J.; Zong, Z.; Zhang, H. Current Immunotherapies for Glioblastoma Multiforme. Front. Immunol. 2020, 11, 603911. [Google Scholar] [CrossRef]
- Sarkaria, J.N.; Hu, L.S.; Parney, I.F.; Pafundi, D.H.; Brinkmann, D.H.; Laack, N.N.; Giannini, C.; Burns, T.C.; Kizilbash, S.H.; Laramy, J.K.; et al. Is the blood–brain barrier really disrupted in all glioblastomas? A critical assessment of existing clinical data. Neuro-Oncol. 2017, 20, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Rocka, A.; Psiuk, D.; Nowak, E.; Madras, D.; Szumna, K. Passing across the blood-brain barrier in glioblastoma multiforme (GBM). J. Educ. Health Sport 2020, 10, 627–634. [Google Scholar] [CrossRef]
- Di Filippo, L.D.; de Carvalho, S.G.; Duarte, J.L.; Luiz, M.T.; Dutra, J.A.P.; de Paula, G.A.; Chorilli, M.; Conde, J. A receptor-mediated landscape of druggable and targeted nanomaterials for gliomas. Mater. Today Bio 2023, 20, 100671. [Google Scholar] [CrossRef] [PubMed]
- Jena, L.; Dunne, N.J.; McCarthy, H.O. Nanoparticles beyond the blood-brain barrier for glioblastoma. In Glioblastoma Resistance to Chemotherapy: Molecular Mechanisms and Innovative Reversal Strategies; Academic Press: Cambridge, MA, USA, 2021. [Google Scholar]
- Tang, L.; Feng, Y.; Gao, S.; Mu, Q.; Liu, C. Nanotherapeutics Overcoming the Blood-Brain Barrier for Glioblastoma Treatment. Front. Pharmacol. 2021, 12, 786700. [Google Scholar] [CrossRef]
- Gherardini, L.; Buratti, V.V.; Maturi, M.; Inzalaco, G.; Locatelli, E.; Sambri, L.; Gargiulo, S.; Barone, V.; Bonente, D.; Bertelli, E.; et al. Loco-regional treatment with temozolomide-loaded thermogels prevents glioblastoma recurrences in orthotopic human xenograft models. Sci. Rep. 2023, 13, 4630. [Google Scholar] [CrossRef]
- Sun, T.; Krishnan, V.; Pan, D.C.; Filippov, S.K.; Ravid, S.; Sarode, A.; Kim, J.; Zhang, Y.; Power, C.; Aday, S.; et al. Ultrasound-mediated delivery of flexibility-tunable polymer drug conjugates for treating glioblastoma. Bioeng. Transl. Med. 2022, 8, e10408. [Google Scholar] [CrossRef]
- Sonabend, A.M.; Gould, A.; Amidei, C.; Ward, R.; Schmidt, K.A.; Zhang, D.Y.; Gomez, C.; Bebawy, J.F.; Liu, B.P.; Bouchoux, G.; et al. Repeated blood–brain barrier opening with an implantable ultrasound device for delivery of albumin-bound paclitaxel in patients with recurrent glioblastoma: A phase 1 trial. Lancet Oncol. 2023, 24, 509–522. [Google Scholar] [CrossRef]
- Randles, A.; Wirsching, H.-G.; Dean, J.A.; Cheng, Y.-K.; Emerson, S.; Pattwell, S.S.; Holland, E.C.; Michor, F. Computational modelling of perivascular-niche dynamics for the optimization of treatment schedules for glioblastoma. Nat. Biomed. Eng. 2021, 5, 346–359. [Google Scholar] [CrossRef]
- Rahman, R.; Trippa, L.; Alden, S.; Fell, G.; Abbasi, T.; Mundkur, Y.; Singh, N.K.; Talawdekar, A.; Husain, Z.; Vali, S.; et al. Prediction of Outcomes with a Computational Biology Model in Newly Diagnosed Glioblastoma Patients Treated with Radiation Therapy and Temozolomide. Int. J. Radiat. Oncol. 2020, 108, 716–724. [Google Scholar] [CrossRef]
- Day, E.K.; Zhong, Q.; Purow, B.; Lazzara, M.J. Data-driven computational modeling identifies determinants of glioblastoma response to shp2 inhibition. Cancer Res. 2021, 81, 2056–2070. [Google Scholar] [CrossRef]
- Xue, L.; Liu, H.; Chen, Y.; Wei, L.; Hong, J. Computational analysis and verification of molecular genetic targets for glioblastoma. Biosci. Rep. 2020, 40, BSR20201401. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Adejumobi, I.A.; Alkhatib, H.; Chowdhury, S.R.; Stefansky, S.; Rubinstein, A.M.; Kravchenko-balasha, N. Drug-induced resistance and phenotypic switch in triple-negative breast cancer can be controlled via resolution and targeting of individualized signaling signatures. Cancers 2021, 13, 5009. [Google Scholar] [CrossRef] [PubMed]
- Valdebenito, J.; Medina, F. Machine learning approaches to study glioblastoma: A review of the last decade of applications. Cancer Rep. 2019, 2, e1226. [Google Scholar] [CrossRef]
- Della Pepa, G.M.; Caccavella, V.M.; Menna, G.; Ius, T.; Auricchio, A.M.; Sabatino, G.; La Rocca, G.; Chiesa, S.; Gaudino, S.; Marchese, E.; et al. Machine Learning-Based Prediction of Early Recurrence in Glioblastoma Patients: A Glance Towards Precision Medicine. Neurosurgery 2021, 89, 873–883. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K.; Hung, T.N.K.; Do, D.T.; Lam, L.H.T.; Dang, L.H.; Huynh, T.T. Radiomics-based machine learning model for efficiently classifying transcriptome subtypes in glioblastoma patients from MRI. Comput. Biol. Med. 2021, 132, 104320. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, Y.; Yang, T.; Xing, H.; Wang, Y.; Gao, L.; Guo, X.; Xing, B.; Wang, Y.; Ma, W. Machine learning revealed stemness features and a novel stemness-based classification with appealing implications in discriminating the prognosis, immunotherapy and temozolomide responses of 906 glioblastoma patients. Brief. Bioinform. 2021, 22, bbab032. [Google Scholar] [CrossRef] [PubMed]
- Stanković, T.; Ranđelović, T.; Dragoj, M.; Burić, S.S.; Fernández, L.; Ochoa, I.; Pérez-García, V.M.; Pešić, M. In vitro biomimetic models for glioblastoma-a promising tool for drug response studies. Drug Resist. Update 2021, 55, 100753. [Google Scholar] [CrossRef]
- Falco, J.; Agosti, A.; Vetrano, I.G.; Bizzi, A.; Restelli, F.; Broggi, M.; Schiariti, M.; DiMeco, F.; Ferroli, P.; Ciarletta, P.; et al. In Silico Mathematical Modelling for Glioblastoma: A Critical Review and a Patient-Specific Case. J. Clin. Med. 2021, 10, 2169. [Google Scholar] [CrossRef]
- Massey, S.C.; Urcuyo, J.C.; Marin, B.M.; Sarkaria, J.N.; Swanson, K.R. Quantifying Glioblastoma Drug Response Dynamics Incorporating Treatment Sensitivity and Blood Brain Barrier Penetrance from Experimental Data. Front. Physiol. 2020, 11, 830. [Google Scholar] [CrossRef]
- Gkountas, A.A.; Polychronopoulos, N.D.; Sofiadis, G.N.; Karvelas, E.G.; Spyrou, L.A.; Sarris, I.E. Simulation of magnetic nanoparticles crossing through a simplified blood-brain barrier model for Glioblastoma multiforme treatment. Comput. Methods Programs Biomed. 2021, 212, 106477. [Google Scholar] [CrossRef]
- Bao, X.; Wu, J.; Xie, Y.; Kim, S.; Michelhaugh, S.; Jiang, J.; Mittal, S.; Sanai, N.; Li, J. Protein Expression and Functional Relevance of Efflux and Uptake Drug Transporters at the Blood–Brain Barrier of Human Brain and Glioblastoma. Clin. Pharmacol. Ther. 2020, 107, 1116–1127. [Google Scholar] [CrossRef] [PubMed]
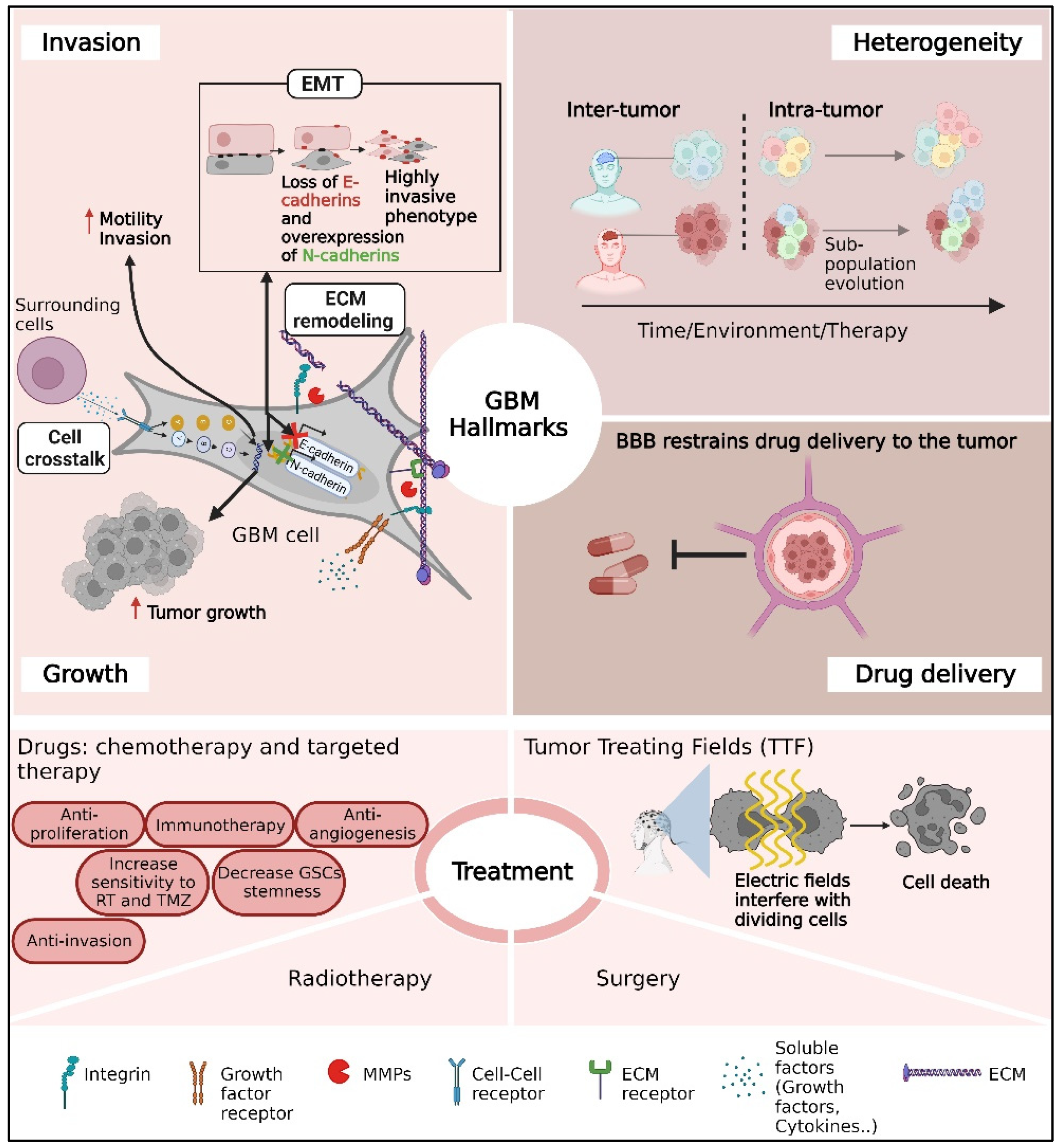
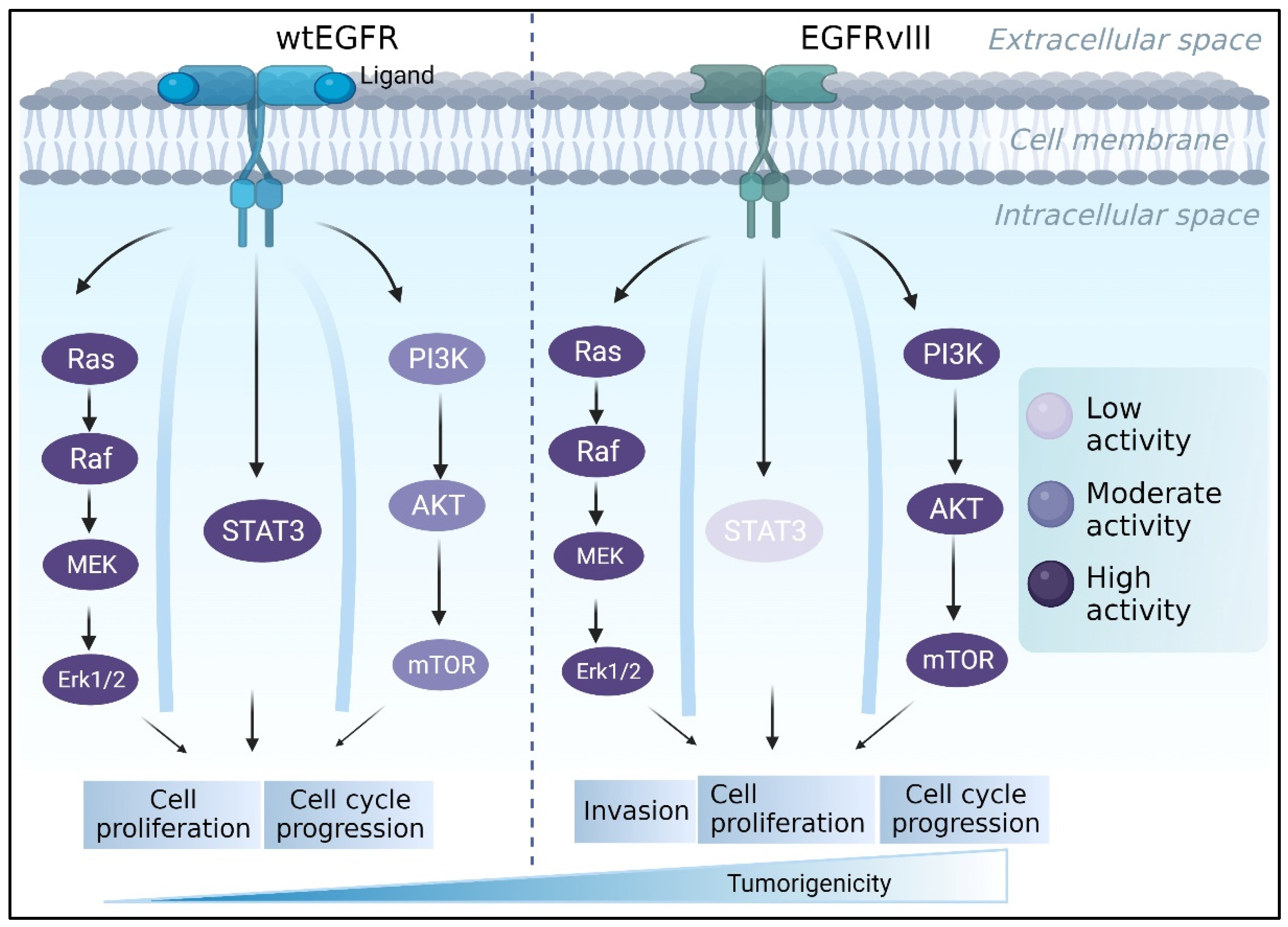
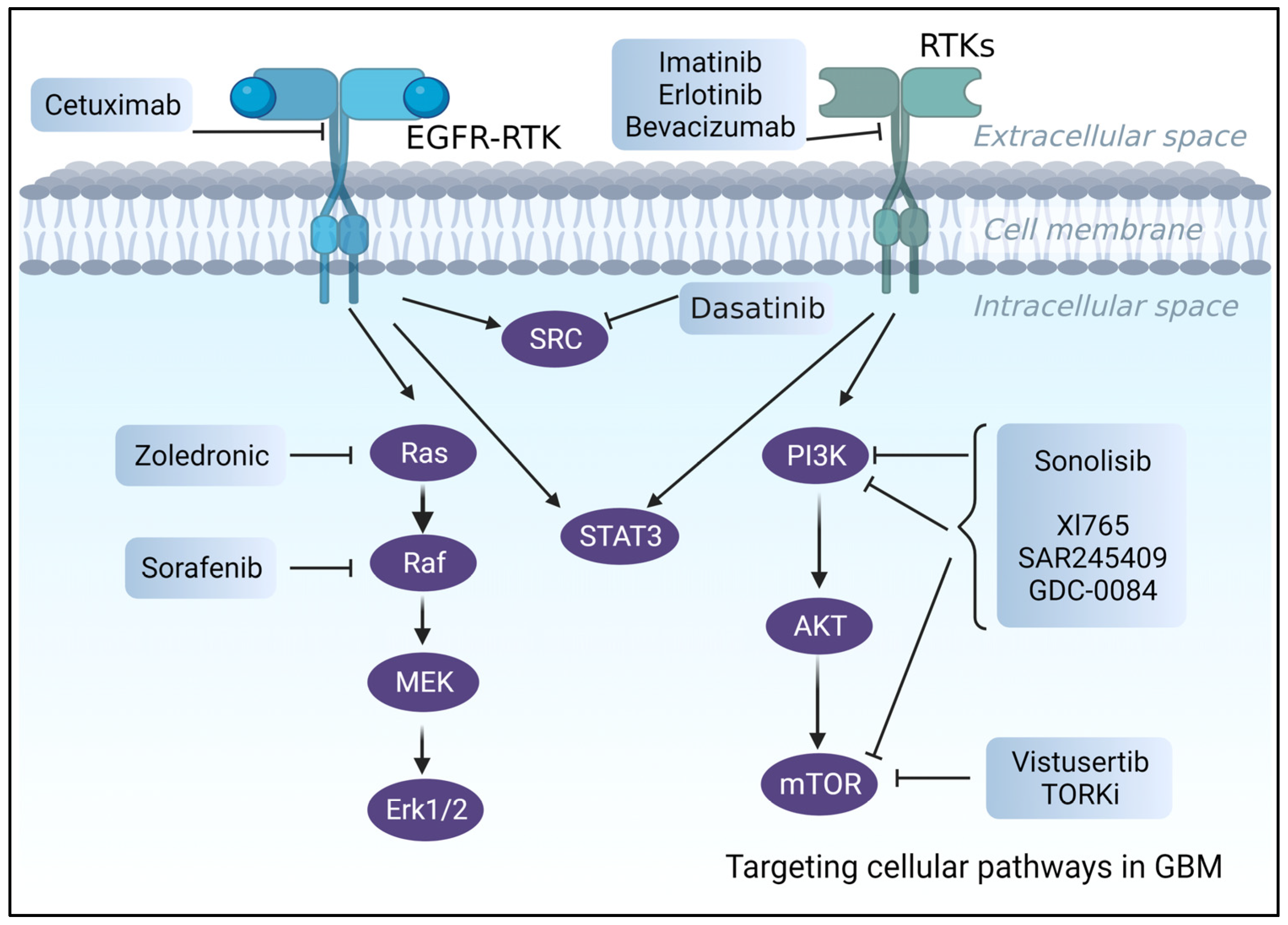
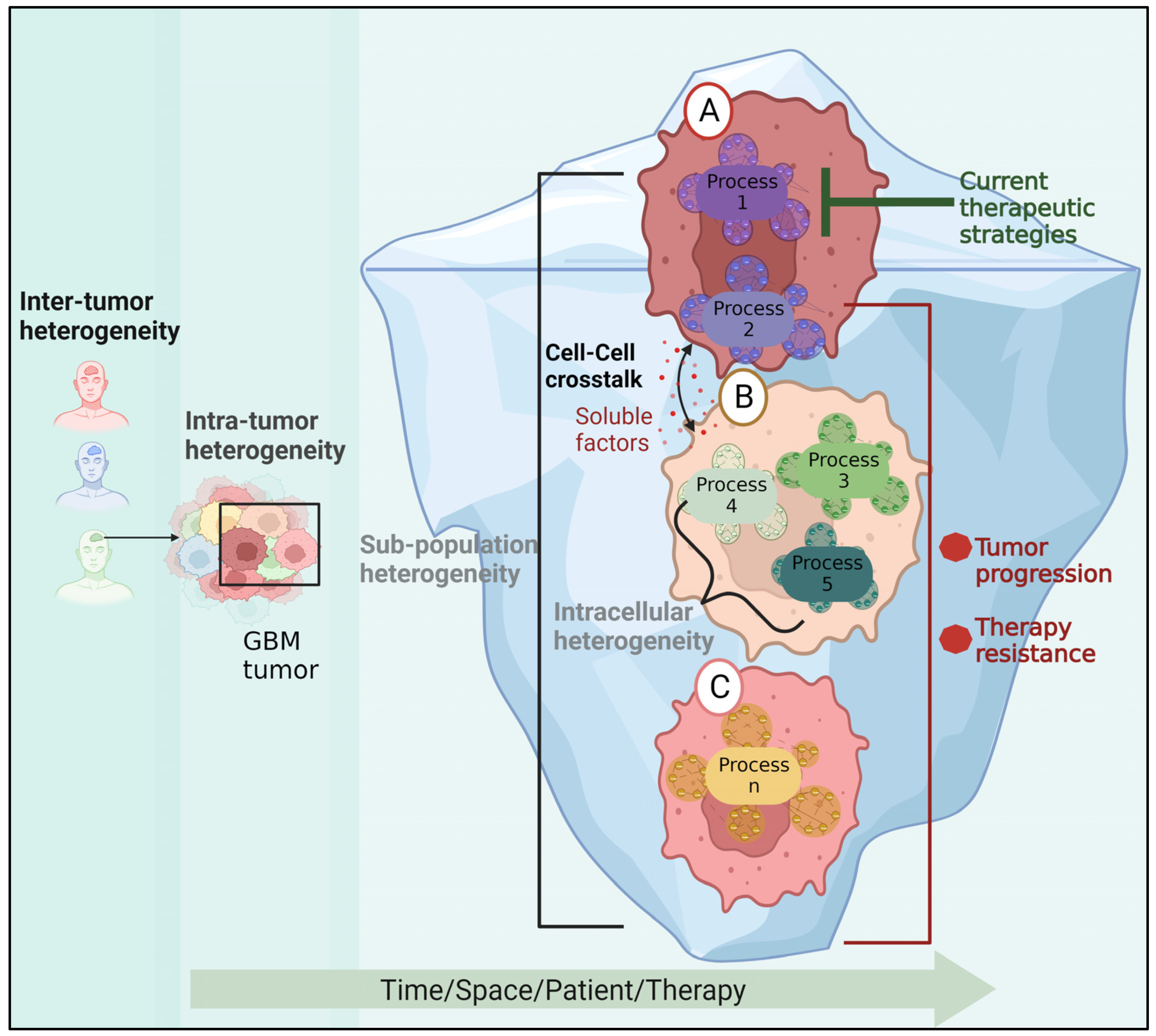
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).