Selective ROCK Inhibitor Enhances Blood Flow Recovery after Hindlimb Ischemia

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
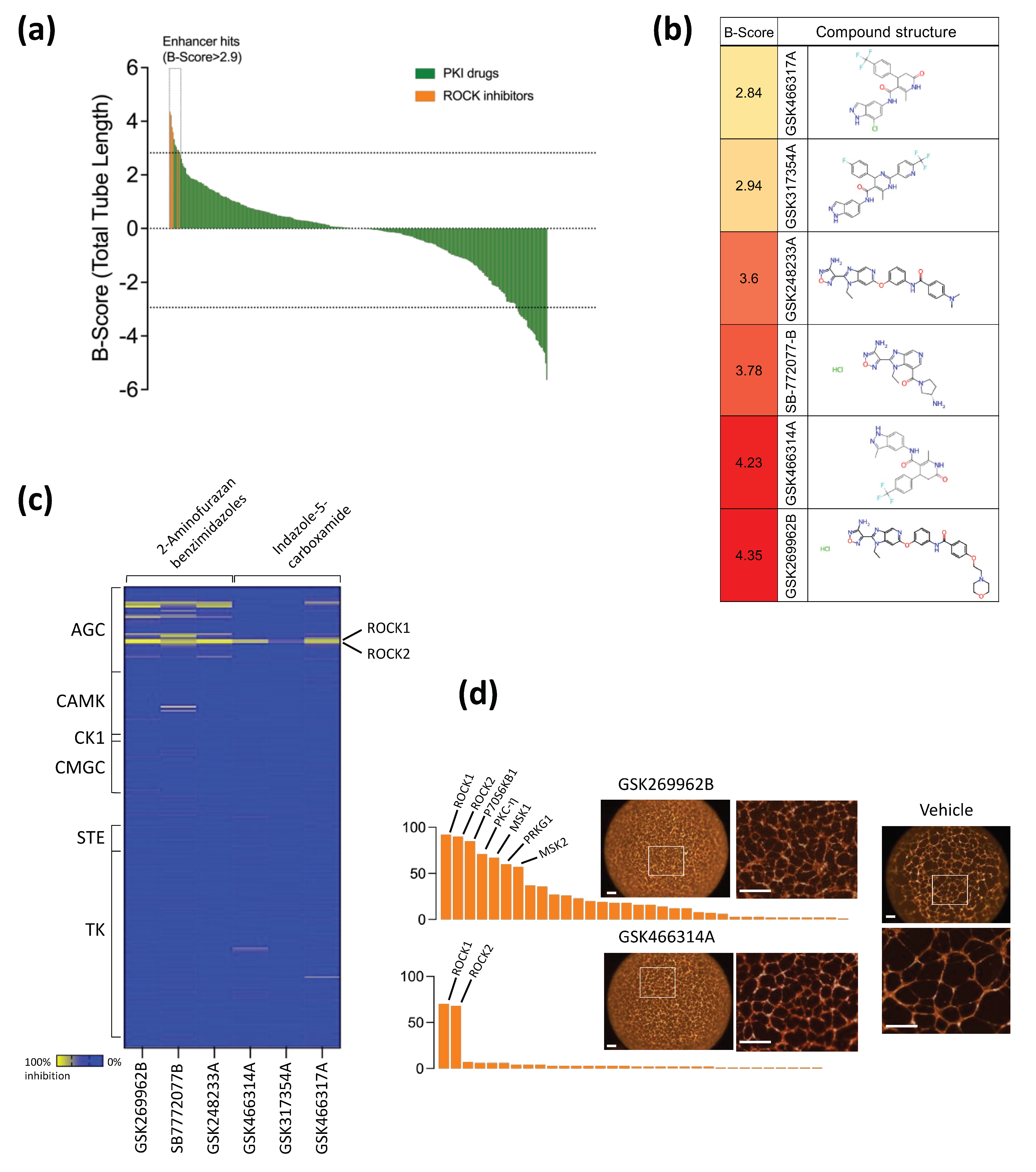
2.1. Phenotypic Drug Screen of Endothelial Tube Formation and Enhancer Hits Identification
2.2. GSK429286 Promotes Endothelial Tube Formation
2.3. GSK429286 Compound Enhanced Blood Flow Recovery after Ischemia
3. Discussion
4. Materials and Methods
4.1. High Content Screening of PKI GSK Library
4.2. siRNA Transfection
4.3. Endothelial Tube Formation Assay
4.4. Western Blotting
4.5. Endothelial Cell Adhesion Assay
4.6. Endothelial Cell Motility Assay
4.7. Co-Culture Organotypic Angiogenesis Assay
4.8. Hindlimb Ischemia Model and Blood Flow Measurement
4.9. Whole-Mount Immunohistochemistry of Ischemic Muscles
4.10. Nitric Oxide Detection Assay
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Potente, M.; Gerhardt, H.; Carmeliet, P. Basic and therapeutic aspects of angiogenesis. Cell 2011, 146, 873–887. [Google Scholar] [CrossRef]
- Ware, J.A.; Simons, M. Angiogenesis in ischemic heart disease. Nat. Med. 1997, 3, 158–164. [Google Scholar] [CrossRef]
- Semenza, G.L. Vascular responses to hypoxia and ischemia. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 648–652. [Google Scholar] [CrossRef]
- Gjedde, A.; Kuwabara, H.; Hakim, A.M. Reduction of functional capillary density in human brain after stroke. J. Cereb. Blood Flow Metab. 1990, 10, 317–326. [Google Scholar] [CrossRef]
- White, S.H.; McDermott, M.M.; Sufit, R.L.; Kosmac, K.; Bugg, A.W.; Gonzalez-Freire, M.; Ferrucci, L.; Tian, L.; Zhao, L.; Gao, Y.; et al. Walking performance is positively correlated to calf muscle fiber size in peripheral artery disease subjects, but fibers show aberrant mitophagy: An observational study. J. Transl. Med. 2016, 14, 284. [Google Scholar] [CrossRef]
- Sano, M.; Minamino, T.; Toko, H.; Miyauchi, H.; Orimo, M.; Qin, Y.; Akazawa, H.; Tateno, K.; Kayama, Y.; Harada, M.; et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature 2007, 446, 444–448. [Google Scholar] [CrossRef]
- Duscha, B.D.; Kraus, W.E.; Jones, W.S.; Robbins, J.L.; Piner, L.W.; Huffman, K.M.; Allen, J.D.; Annex, B.H. Skeletal muscle capillary density is related to anaerobic threshold and claudication in peripheral artery disease. Vasc. Med. 2020, 25, 411–418. [Google Scholar] [CrossRef]
- Banfi, A.; von Degenfeld, G.; Gianni-Barrera, R.; Reginato, S.; Merchant, M.J.; McDonald, D.M.; Blau, H.M. Therapeutic angiogenesis due to balanced single-vector delivery of VEGF and PDGF-BB. FASEB J. 2012, 26, 2486–2497. [Google Scholar] [CrossRef]
- Annex, B.H.; Cooke, J.P. New Directions in Therapeutic Angiogenesis and Arteriogenesis in Peripheral Arterial Disease. Circ. Res. 2021, 128, 1944–1957. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef]
- Adams, R.H.; Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 2007, 8, 464–478. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, M.; Leppanen, V.M.; Saharinen, P.; Alitalo, K. Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a009183. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y.; Thurston, G.; Alitalo, K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 2009, 10, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Nasarre, P.; Thomas, M.; Kruse, K.; Helfrich, I.; Wolter, V.; Deppermann, C.; Schadendorf, D.; Thurston, G.; Fiedler, U.; Augustin, H.G. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res. 2009, 69, 1324–1333. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.H.; Wilkinson, G.A.; Weiss, C.; Diella, F.; Gale, N.W.; Deutsch, U.; Risau, W.; Klein, R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: Demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999, 13, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Claesson-Welsh, L.; Welsh, M. VEGFA and tumour angiogenesis. J. Intern. Med. 2013, 273, 114–127. [Google Scholar] [CrossRef]
- Gotink, K.J.; Verheul, H.M. Anti-angiogenic tyrosine kinase inhibitors: What is their mechanism of action? Angiogenesis 2010, 13, 1–14. [Google Scholar] [CrossRef]
- Qin, S.; Li, A.; Yi, M.; Yu, S.; Zhang, M.; Wu, K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J. Hematol. Oncol. 2019, 12, 27. [Google Scholar] [CrossRef]
- Rust, R.; Gantner, C.; Schwab, M.E. Pro- and antiangiogenic therapies: Current status and clinical implications. FASEB J. 2019, 33, 34–48. [Google Scholar] [CrossRef]
- Isner, J.M.; Pieczek, A.; Schainfeld, R.; Blair, R.; Haley, L.; Asahara, T.; Rosenfield, K.; Razvi, S.; Walsh, K.; Symes, J.F. Clinical evidence of angiogenesis after arterial gene transfer of phVEGF165 in patient with ischaemic limb. Lancet 1996, 348, 370–374. [Google Scholar] [CrossRef]
- Lederman, R.J.; Mendelsohn, F.O.; Anderson, R.D.; Saucedo, J.F.; Tenaglia, A.N.; Hermiller, J.B.; Hillegass, W.B.; Rocha-Singh, K.; Moon, T.E.; Whitehouse, M.J.; et al. Therapeutic angiogenesis with recombinant fibroblast growth factor-2 for intermittent claudication (the TRAFFIC study): A randomised trial. Lancet 2002, 359, 2053–2058. [Google Scholar] [CrossRef] [PubMed]
- Morishita, R.; Makino, H.; Aoki, M.; Hashiya, N.; Yamasaki, K.; Azuma, J.; Taniyama, Y.; Sawa, Y.; Kaneda, Y.; Ogihara, T. Phase I/IIa clinical trial of therapeutic angiogenesis using hepatocyte growth factor gene transfer to treat critical limb ischemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Deev, R.; Plaksa, I.; Bozo, I.; Mzhavanadze, N.; Suchkov, I.; Chervyakov, Y.; Staroverov, I.; Kalinin, R.; Isaev, A. Results of 5-year follow-up study in patients with peripheral artery disease treated with PL-VEGF165 for intermittent claudication. Ther. Adv. Cardiovasc. Dis. 2018, 12, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Deev, R.V.; Bozo, I.Y.; Mzhavanadze, N.D.; Voronov, D.A.; Gavrilenko, A.V.; Chervyakov, Y.V.; Staroverov, I.N.; Kalinin, R.E.; Shvalb, P.G.; Isaev, A.A. pCMV-vegf165 Intramuscular Gene Transfer is an Effective Method of Treatment for Patients with Chronic Lower Limb Ischemia. J. Cardiovasc. Pharmacol. Ther. 2015, 20, 473–482. [Google Scholar] [CrossRef] [PubMed]
- Beltran-Camacho, L.; Rojas-Torres, M.; Duran-Ruiz, M.C. Current Status of Angiogenic Cell Therapy and Related Strategies Applied in Critical Limb Ischemia. Int. J. Mol. Sci. 2021, 22, 2335. [Google Scholar] [CrossRef] [PubMed]
- Yla-Herttuala, S.; Markkanen, J.E.; Rissanen, T.T. Gene therapy for ischemic cardiovascular diseases: Some lessons learned from the first clinical trials. Trends Cardiovasc. Med. 2004, 14, 295–300. [Google Scholar] [CrossRef]
- Tongers, J.; Roncalli, J.G.; Losordo, D.W. Therapeutic angiogenesis for critical limb ischemia: Microvascular therapies coming of age. Circulation 2008, 118, 9–16. [Google Scholar] [CrossRef]
- Makinen, K.; Manninen, H.; Hedman, M.; Matsi, P.; Mussalo, H.; Alhava, E.; Yla-Herttuala, S. Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: A randomized, placebo-controlled, double-blinded phase II study. Mol. Ther. 2002, 6, 127–133. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Mohler, E.R., 3rd; Lederman, R.J.; Mendelsohn, F.O.; Saucedo, J.F.; Goldman, C.K.; Blebea, J.; Macko, J.; Kessler, P.D.; Rasmussen, H.S.; et al. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: A phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 2003, 108, 1933–1938. [Google Scholar] [CrossRef]
- Simons, M.; Ware, J.A. Therapeutic angiogenesis in cardiovascular disease. Nat. Rev. Drug Discov. 2003, 2, 863–871. [Google Scholar] [CrossRef]
- Elkins, J.M.; Fedele, V.; Szklarz, M.; Abdul Azeez, K.R.; Salah, E.; Mikolajczyk, J.; Romanov, S.; Sepetov, N.; Huang, X.P.; Roth, B.L.; et al. Comprehensive characterization of the Published Kinase Inhibitor Set. Nat. Biotechnol. 2016, 34, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Perrin, D.; Fremaux, C.; Shutes, A. Capillary microfluidic electrophoretic mobility shift assays: Application to enzymatic assays in drug discovery. Expert. Opin. Drug Discov. 2010, 5, 51–63. [Google Scholar] [CrossRef]
- Rudolf, A.F.; Skovgaard, T.; Knapp, S.; Jensen, L.J.; Berthelsen, J. A comparison of protein kinases inhibitor screening methods using both enzymatic activity and binding affinity determination. PLoS ONE 2014, 9, e98800. [Google Scholar] [CrossRef] [PubMed]
- Goodman, K.B.; Cui, H.; Dowdell, S.E.; Gaitanopoulos, D.E.; Ivy, R.L.; Sehon, C.A.; Stavenger, R.A.; Wang, G.Z.; Viet, A.Q.; Xu, W.; et al. Development of dihydropyridone indazole amides as selective Rho-kinase inhibitors. J. Med. Chem. 2007, 50, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Lamalice, L.; Le Boeuf, F.; Huot, J. Endothelial cell migration during angiogenesis. Circ. Res. 2007, 100, 782–794. [Google Scholar] [CrossRef]
- Zanivan, S.; Maione, F.; Hein, M.Y.; Hernandez-Fernaud, J.R.; Ostasiewicz, P.; Giraudo, E.; Mann, M. SILAC-based proteomics of human primary endothelial cell morphogenesis unveils tumor angiogenic markers. Mol. Cell. Proteom. 2013, 12, 3599–3611. [Google Scholar] [CrossRef]
- Mongiat, M.; Andreuzzi, E.; Tarticchio, G.; Paulitti, A. Extracellular Matrix, a Hard Player in Angiogenesis. Int. J. Mol. Sci. 2016, 17, 1822. [Google Scholar] [CrossRef]
- Dixelius, J.; Jakobsson, L.; Genersch, E.; Bohman, S.; Ekblom, P.; Claesson-Welsh, L. Laminin-1 promotes angiogenesis in synergy with fibroblast growth factor by distinct regulation of the gene and protein expression profile in endothelial cells. J. Biol. Chem. 2004, 279, 23766–23772. [Google Scholar] [CrossRef]
- Barbier, L.; Saez, P.J.; Attia, R.; Lennon-Dumenil, A.M.; Lavi, I.; Piel, M.; Vargas, P. Myosin II Activity Is Selectively Needed for Migration in Highly Confined Microenvironments in Mature Dendritic Cells. Front. Immunol. 2019, 10, 747. [Google Scholar] [CrossRef]
- Shi, J.; Wu, X.; Surma, M.; Vemula, S.; Zhang, L.; Yang, Y.; Kapur, R.; Wei, L. Distinct roles for ROCK1 and ROCK2 in the regulation of cell detachment. Cell Death Dis. 2013, 4, e483. [Google Scholar] [CrossRef]
- Hartmann, S.; Ridley, A.J.; Lutz, S. The Function of Rho-Associated Kinases ROCK1 and ROCK2 in the Pathogenesis of Cardiovascular Disease. Front. Pharmacol. 2015, 6, 276. [Google Scholar] [CrossRef]
- Al Haj Zen, A. In Vitro Models to Study the Regulatory Roles of Retinoids in Angiogenesis. Methods Mol. Biol. 2019, 2019, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Evensen, L.; Micklem, D.R.; Link, W.; Lorens, J.B. A novel imaging-based high-throughput screening approach to anti-angiogenic drug discovery. Cytometry A 2010, 77, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Liao, J.K. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J. Biol. Chem. 1998, 273, 24266–24271. [Google Scholar] [CrossRef] [PubMed]
- Ming, X.F.; Viswambharan, H.; Barandier, C.; Ruffieux, J.; Kaibuchi, K.; Rusconi, S.; Yang, Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol. Cell Biol. 2002, 22, 8467–8477. [Google Scholar] [CrossRef]
- Bode-Boger, S.M.; Boger, R.H.; Alfke, H.; Heinzel, D.; Tsikas, D.; Creutzig, A.; Alexander, K.; Frolich, J.C. L-arginine induces nitric oxide-dependent vasodilation in patients with critical limb ischemia. A randomized, controlled study. Circulation 1996, 93, 85–90. [Google Scholar] [CrossRef]
- Yu, J.; deMuinck, E.D.; Zhuang, Z.; Drinane, M.; Kauser, K.; Rubanyi, G.M.; Qian, H.S.; Murata, T.; Escalante, B.; Sessa, W.C. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proc. Natl. Acad. Sci. USA 2005, 102, 10999–11004. [Google Scholar] [CrossRef]
- Al Haj Zen, A.; Oikawa, A.; Bazan-Peregrino, M.; Meloni, M.; Emanueli, C.; Madeddu, P. Inhibition of delta-like-4-mediated signaling impairs reparative angiogenesis after ischemia. Circ. Res. 2010, 107, 283–293. [Google Scholar] [CrossRef]
- Arnaoutova, I.; George, J.; Kleinman, H.K.; Benton, G. The endothelial cell tube formation assay on basement membrane turns 20: State of the science and the art. Angiogenesis 2009, 12, 267–274. [Google Scholar] [CrossRef]
- Arnaoutova, I.; Kleinman, H.K. In vitro angiogenesis: Endothelial cell tube formation on gelled basement membrane extract. Nat. Protoc. 2010, 5, 628–635. [Google Scholar] [CrossRef]
- Al Haj Zen, A.; Nawrot, D.A.; Howarth, A.; Caporali, A.; Ebner, D.; Vernet, A.; Schneider, J.E.; Bhattacharya, S. The Retinoid Agonist Tazarotene Promotes Angiogenesis and Wound Healing. Mol. Ther. 2016, 24, 1745–1759. [Google Scholar] [CrossRef] [PubMed]
- Stavenger, R.A.; Cui, H.; Dowdell, S.E.; Franz, R.G.; Gaitanopoulos, D.E.; Goodman, K.B.; Hilfiker, M.A.; Ivy, R.L.; Leber, J.D.; Marino, J.P., Jr.; et al. Discovery of aminofurazan-azabenzimidazoles as inhibitors of Rho-kinase with high kinase selectivity and antihypertensive activity. J. Med. Chem. 2007, 50, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Tandon, N.; Luxami, V.; Kant, D.; Tandon, R.; Paul, K. Current progress, challenges and future prospects of indazoles as protein kinase inhibitors for the treatment of cancer. RSC Adv. 2021, 11, 25228–25257. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Epting, D.; Kern, K.; Dietz, C.T.; Feng, Y.; Hammes, H.P.; Wieland, T.; Augustin, H.G. Inhibition of Rho-dependent kinases ROCK I/II activates VEGF-driven retinal neovascularization and sprouting angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H893–H899. [Google Scholar] [CrossRef] [PubMed]
- Amano, M.; Nakayama, M.; Kaibuchi, K. Rho-kinase/ROCK: A key regulator of the cytoskeleton and cell polarity. Cytoskeleton 2010, 67, 545–554. [Google Scholar] [CrossRef]
- Pipparelli, A.; Arsenijevic, Y.; Thuret, G.; Gain, P.; Nicolas, M.; Majo, F. ROCK inhibitor enhances adhesion and wound healing of human corneal endothelial cells. PLoS ONE 2013, 8, e62095. [Google Scholar] [CrossRef]
- Fischer, R.S.; Gardel, M.; Ma, X.; Adelstein, R.S.; Waterman, C.M. Local cortical tension by myosin II guides 3D endothelial cell branching. Curr. Biol. 2009, 19, 260–265. [Google Scholar] [CrossRef]
- Elliott, H.; Fischer, R.S.; Myers, K.A.; Desai, R.A.; Gao, L.; Chen, C.S.; Adelstein, R.S.; Waterman, C.M.; Danuser, G. Myosin II controls cellular branching morphogenesis and migration in three dimensions by minimizing cell-surface curvature. Nat. Cell Biol. 2015, 17, 137–147. [Google Scholar] [CrossRef]
- Parsons, J.T.; Horwitz, A.R.; Schwartz, M.A. Cell adhesion: Integrating cytoskeletal dynamics and cellular tension. Nat. Rev. Mol. Cell Biol. 2010, 11, 633–643. [Google Scholar] [CrossRef]
- Bryan, B.A.; Dennstedt, E.; Mitchell, D.C.; Walshe, T.E.; Noma, K.; Loureiro, R.; Saint-Geniez, M.; Campaigniac, J.P.; Liao, J.K.; D’Amore, P.A. RhoA/ROCK signaling is essential for multiple aspects of VEGF-mediated angiogenesis. FASEB J. 2010, 24, 3186–3195. [Google Scholar] [CrossRef]
- Huang, L.; Dai, F.; Tang, L.; Bao, X.; Liu, Z.; Huang, C.; Zhang, T.; Yao, W. Distinct Roles For ROCK1 and ROCK2 in the Regulation of Oxldl-Mediated Endothelial Dysfunction. Cell Physiol. Biochem. 2018, 49, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Riento, K.; Ridley, A.J. Rocks: Multifunctional kinases in cell behaviour. Nat. Rev. Mol. Cell Biol. 2003, 4, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Rikitake, Y.; Liao, J.K. ROCKs as therapeutic targets in cardiovascular diseases. Expert Rev. Cardiovasc. Ther. 2005, 3, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Thumkeo, D.; Keel, J.; Ishizaki, T.; Hirose, M.; Nonomura, K.; Oshima, H.; Oshima, M.; Taketo, M.M.; Narumiya, S. Targeted disruption of the mouse rho-associated kinase 2 gene results in intrauterine growth retardation and fetal death. Mol. Cell Biol. 2003, 23, 5043–5055. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, Y.; Thumkeo, D.; Keel, J.; Ishizaki, T.; Oshima, H.; Oshima, M.; Noda, Y.; Matsumura, F.; Taketo, M.M.; Narumiya, S. ROCK-I regulates closure of the eyelids and ventral body wall by inducing assembly of actomyosin bundles. J. Cell Biol. 2005, 168, 941–953. [Google Scholar] [CrossRef]
- Shimokawa, H.; Seto, M.; Katsumata, N.; Amano, M.; Kozai, T.; Yamawaki, T.; Kuwata, K.; Kandabashi, T.; Egashira, K.; Ikegaki, I.; et al. Rho-kinase-mediated pathway induces enhanced myosin light chain phosphorylations in a swine model of coronary artery spasm. Cardiovasc. Res. 1999, 43, 1029–1039. [Google Scholar] [CrossRef]
- Sato, M.; Tani, E.; Fujikawa, H.; Kaibuchi, K. Involvement of Rho-kinase-mediated phosphorylation of myosin light chain in enhancement of cerebral vasospasm. Circ. Res. 2000, 87, 195–200. [Google Scholar] [CrossRef]
- Mukai, Y.; Shimokawa, H.; Matoba, T.; Kandabashi, T.; Satoh, S.; Hiroki, J.; Kaibuchi, K.; Takeshita, A. Involvement of Rho-kinase in hypertensive vascular disease: A novel therapeutic target in hypertension. FASEB J. 2001, 15, 1062–1064. [Google Scholar] [CrossRef]
- Abe, K.; Shimokawa, H.; Morikawa, K.; Uwatoku, T.; Oi, K.; Matsumoto, Y.; Hattori, T.; Nakashima, Y.; Kaibuchi, K.; Sueishi, K.; et al. Long-term treatment with a Rho-kinase inhibitor improves monocrotaline-induced fatal pulmonary hypertension in rats. Circ. Res. 2004, 94, 385–393. [Google Scholar] [CrossRef]
- Satoh, S.; Utsunomiya, T.; Tsurui, K.; Kobayashi, T.; Ikegaki, I.; Sasaki, Y.; Asano, T. Pharmacological profile of hydroxy fasudil as a selective rho kinase inhibitor on ischemic brain damage. Life Sci. 2001, 69, 1441–1453. [Google Scholar] [CrossRef]
- Zhao, J.; Zhou, D.; Guo, J.; Ren, Z.; Zhou, L.; Wang, S.; Xu, B.; Wang, R. Effect of fasudil hydrochloride, a protein kinase inhibitor, on cerebral vasospasm and delayed cerebral ischemic symptoms after aneurysmal subarachnoid hemorrhage. Neurol. Med. Chir. 2006, 46, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Seto, M.; Noma, K. Rho kinase (ROCK) inhibitors. J. Cardiovasc. Pharmacol. 2007, 50, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Barry, D.M.; Koo, Y.; Norden, P.R.; Wylie, L.A.; Xu, K.; Wichaidit, C.; Azizoglu, D.B.; Zheng, Y.; Cobb, M.H.; Davis, G.E.; et al. Rasip1-Mediated Rho GTPase Signaling Regulates Blood Vessel Tubulogenesis via Nonmuscle Myosin II. Circ. Res. 2016, 119, 810–826. [Google Scholar] [CrossRef] [PubMed]
- Sims, D.E. The pericyte—A review. Tissue Cell 1986, 18, 153–174. [Google Scholar] [CrossRef] [PubMed]
- Peppiatt, C.M.; Howarth, C.; Mobbs, P.; Attwell, D. Bidirectional control of CNS capillary diameter by pericytes. Nature 2006, 443, 700–704. [Google Scholar] [CrossRef]
- Hartmann, D.A.; Coelho-Santos, V.; Shih, A.Y. Pericyte Control of Blood Flow Across Microvascular Zones in the Central Nervous System. Annu. Rev. Physiol. 2022, 84, 331–354. [Google Scholar] [CrossRef]
- Wang, Y.; Zheng, X.R.; Riddick, N.; Bryden, M.; Baur, W.; Zhang, X.; Surks, H.K. ROCK isoform regulation of myosin phosphatase and contractility in vascular smooth muscle cells. Circ. Res. 2009, 104, 531–540. [Google Scholar] [CrossRef]
- Penzes, M.; Turos, D.; Mathe, D.; Szigeti, K.; Hegedus, N.; Rauscher, A.A.; Toth, P.; Ivic, I.; Padmanabhan, P.; Pal, G.; et al. Direct myosin-2 inhibition enhances cerebral perfusion resulting in functional improvement after ischemic stroke. Theranostics 2020, 10, 5341–5356. [Google Scholar] [CrossRef]
- Tang, L.; Dai, F.; Liu, Y.; Yu, X.; Huang, C.; Wang, Y.; Yao, W. RhoA/ROCK signaling regulates smooth muscle phenotypic modulation and vascular remodeling via the JNK pathway and vimentin cytoskeleton. Pharmacol. Res. 2018, 133, 201–212. [Google Scholar] [CrossRef]
- Ashraf, J.V.; Al Haj Zen, A. Role of Vascular Smooth Muscle Cell Phenotype Switching in Arteriogenesis. Int. J. Mol. Sci. 2021, 22, 585. [Google Scholar] [CrossRef]
- Doe, C.; Bentley, R.; Behm, D.J.; Lafferty, R.; Stavenger, R.; Jung, D.; Bamford, M.; Panchal, T.; Grygielko, E.; Wright, L.L.; et al. Novel Rho kinase inhibitors with anti-inflammatory and vasodilatory activities. J. Pharmacol. Exp. Ther. 2007, 320, 89–98. [Google Scholar] [CrossRef]
- Yao, L.; Chandra, S.; Toque, H.A.; Bhatta, A.; Rojas, M.; Caldwell, R.B.; Caldwell, R.W. Prevention of diabetes-induced arginase activation and vascular dysfunction by Rho kinase (ROCK) knockout. Cardiovasc. Res. 2013, 97, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Zhu, Y.; Zhang, S.; Jin, M.B.; Ishizaki, N.; Urakami, A.; Totsuka, E.; Kishida, A.; Lee, R.; Subbotin, V.; et al. Protective role of nitric oxide in ischemia and reperfusion injury of the liver. J. Am. Coll. Surg. 1999, 188, 43–52. [Google Scholar] [CrossRef]
- Cohen, P.; Alessi, D.R. Kinase drug discovery—What’s next in the field? ACS Chem. Biol. 2013, 8, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Aref, Z.; de Vries, M.R.; Quax, P.H.A. Variations in Surgical Procedures for Inducing Hind Limb Ischemia in Mice and the Impact of These Variations on Neovascularization Assessment. Int. J. Mol. Sci. 2019, 20, 3704. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Tang, G.; Yan, J.; Park, B.; Hoffman, A.; Tie, G.; Wang, R.; Messina, L.M. Cellular and molecular mechanism regulating blood flow recovery in acute versus gradual femoral artery occlusion are distinct in the mouse. J. Vasc. Surg. 2008, 48, 1546–1558. [Google Scholar] [CrossRef]
- Drewry, D.H.; Willson, T.M.; Zuercher, W.J. Seeding collaborations to advance kinase science with the GSK Published Kinase Inhibitor Set (PKIS). Curr. Top Med. Chem. 2014, 14, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Chung, T.D.; Oldenburg, K.R. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J. Biomol. Screen. 1999, 4, 67–73. [Google Scholar] [CrossRef]
- Pelz, O.; Gilsdorf, M.; Boutros, M. web cellHTS2: A web-application for the analysis of high-throughput screening data. BMC Bioinform. 2010, 11, 185. [Google Scholar] [CrossRef]
- Chen, Y.; Lu, B.; Yang, Q.; Fearns, C.; Yates, J.R., 3rd; Lee, J.D. Combined integrin phosphoproteomic analyses and small interfering RNA-based functional screening identify key regulators for cancer cell adhesion and migration. Cancer Res. 2009, 69, 3713–3720. [Google Scholar] [CrossRef]
- Kireeva, M.L.; Lam, S.C.; Lau, L.F. Adhesion of human umbilical vein endothelial cells to the immediate-early gene product Cyr61 is mediated through integrin alphavbeta3. J. Biol. Chem. 1998, 273, 3090–3096. [Google Scholar] [CrossRef] [PubMed]
- Abengozar, M.A.; de Frutos, S.; Ferreiro, S.; Soriano, J.; Perez-Martinez, M.; Olmeda, D.; Marenchino, M.; Canamero, M.; Ortega, S.; Megias, D.; et al. Blocking ephrinB2 with highly specific antibodies inhibits angiogenesis, lymphangiogenesis, and tumor growth. Blood 2012, 119, 4565–4576. [Google Scholar] [CrossRef] [PubMed]
- Stock, J.; Kazmar, T.; Schlumm, F.; Hannezo, E.; Pauli, A. A self-generated Toddler gradient guides mesodermal cell migration. Sci. Adv. 2022, 8, eadd2488. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, G.; Berndt, S.; Ferratge, S.; Rasband, W.; Cuendet, M.; Uzan, G.; Albanese, P. Angiogenesis Analyzer for ImageJ—A comparative morphometric analysis of “Endothelial Tube Formation Assay” and “Fibrin Bead Assay”. Sci. Rep. 2020, 10, 11568. [Google Scholar] [CrossRef]
- Couffinhal, T.; Silver, M.; Zheng, L.P.; Kearney, M.; Witzenbichler, B.; Isner, J.M. Mouse model of angiogenesis. Am. J. Pathol. 1998, 152, 1667–1679. [Google Scholar]
- Rikitake, Y.; Kim, H.H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef]
- Neale, J.P.H.; Pearson, J.T.; Thomas, K.N.; Tsuchimochi, H.; Hosoda, H.; Kojima, M.; Sato, T.; Jones, G.T.; Denny, A.P.; Daniels, L.J.; et al. Dysregulation of ghrelin in diabetes impairs the vascular reparative response to hindlimb ischemia in a mouse model; clinical relevance to peripheral artery disease. Sci. Rep. 2020, 10, 13651. [Google Scholar] [CrossRef]
- Martello, A.; Mellis, D.; Meloni, M.; Howarth, A.; Ebner, D.; Caporali, A.; Al Haj Zen, A. Phenotypic miRNA Screen Identifies miR-26b to Promote the Growth and Survival of Endothelial Cells. Mol. Ther. Nucleic Acids 2018, 13, 29–43. [Google Scholar] [CrossRef]
- Leikert, J.F.; Rathel, T.R.; Muller, C.; Vollmar, A.M.; Dirsch, V.M. Reliable in vitro measurement of nitric oxide released from endothelial cells using low concentrations of the fluorescent probe 4,5-diaminofluorescein. FEBS Lett. 2001, 506, 131–134. [Google Scholar] [CrossRef]
- Gu, L.; Lian, D.; Zheng, Y.; Zhou, W.; Gu, J.; Liu, X. Echinacoside-induced nitric oxide production in endothelial cells: Roles of androgen receptor and the PI3K-Akt pathway. Int. J. Mol. Med. 2020, 45, 1195–1202. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fayed, H.S.; Bakleh, M.Z.; Ashraf, J.V.; Howarth, A.; Ebner, D.; Al Haj Zen, A. Selective ROCK Inhibitor Enhances Blood Flow Recovery after Hindlimb Ischemia. Int. J. Mol. Sci. 2023, 24, 14410. https://doi.org/10.3390/ijms241914410
Fayed HS, Bakleh MZ, Ashraf JV, Howarth A, Ebner D, Al Haj Zen A. Selective ROCK Inhibitor Enhances Blood Flow Recovery after Hindlimb Ischemia. International Journal of Molecular Sciences. 2023; 24(19):14410. https://doi.org/10.3390/ijms241914410
Chicago/Turabian StyleFayed, Hend Salah, Mouayad Zuheir Bakleh, Jasni Viralippurath Ashraf, Alison Howarth, Daniel Ebner, and Ayman Al Haj Zen. 2023. "Selective ROCK Inhibitor Enhances Blood Flow Recovery after Hindlimb Ischemia" International Journal of Molecular Sciences 24, no. 19: 14410. https://doi.org/10.3390/ijms241914410
APA StyleFayed, H. S., Bakleh, M. Z., Ashraf, J. V., Howarth, A., Ebner, D., & Al Haj Zen, A. (2023). Selective ROCK Inhibitor Enhances Blood Flow Recovery after Hindlimb Ischemia. International Journal of Molecular Sciences, 24(19), 14410. https://doi.org/10.3390/ijms241914410