
Exploring the Influence of Spacers in EDTA–β-Cyclodextrin Dendrimers: Physicochemical Properties and In Vitro Biological Behavior

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
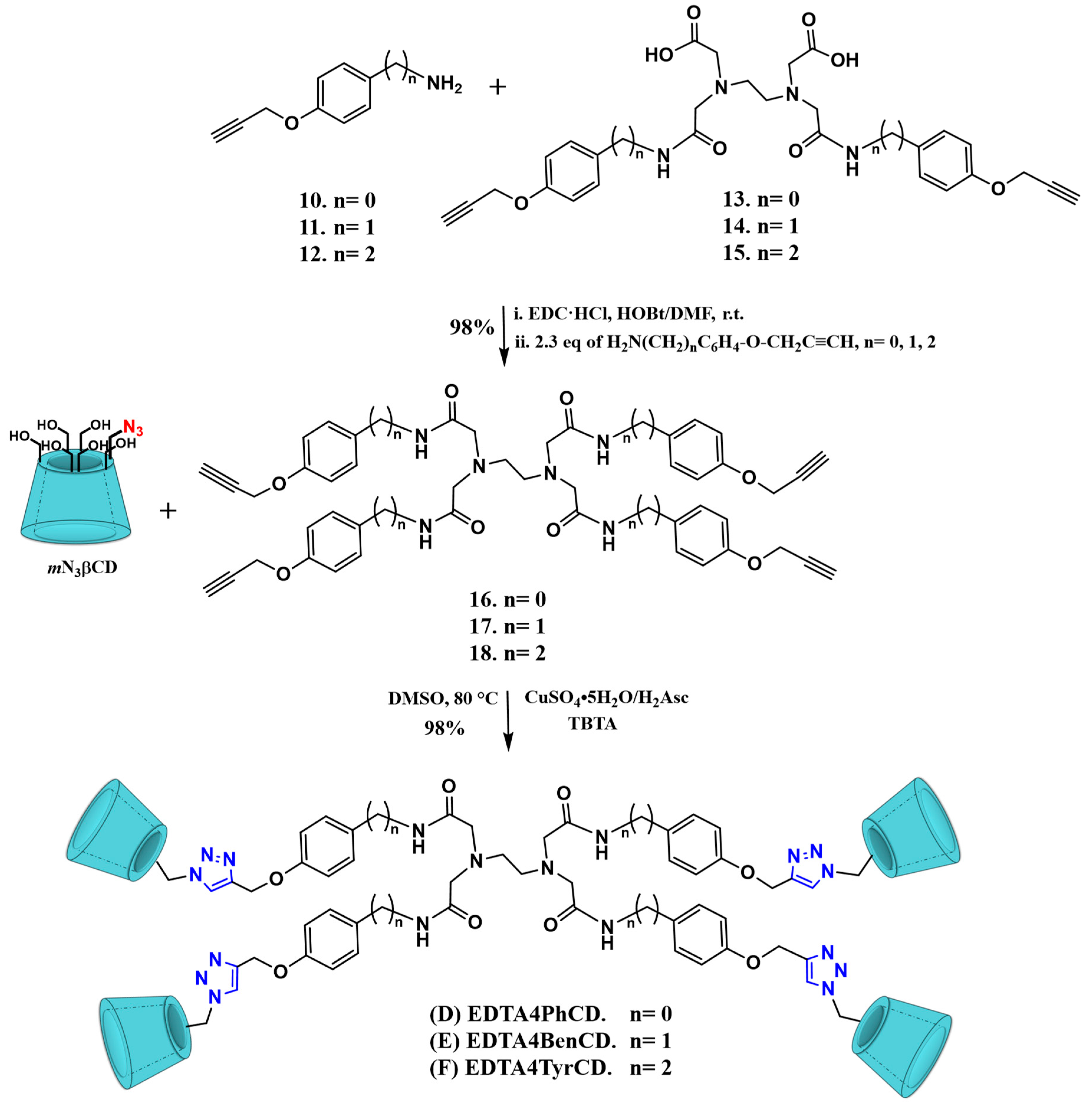
2.1. Synthesis
2.2. Characterization
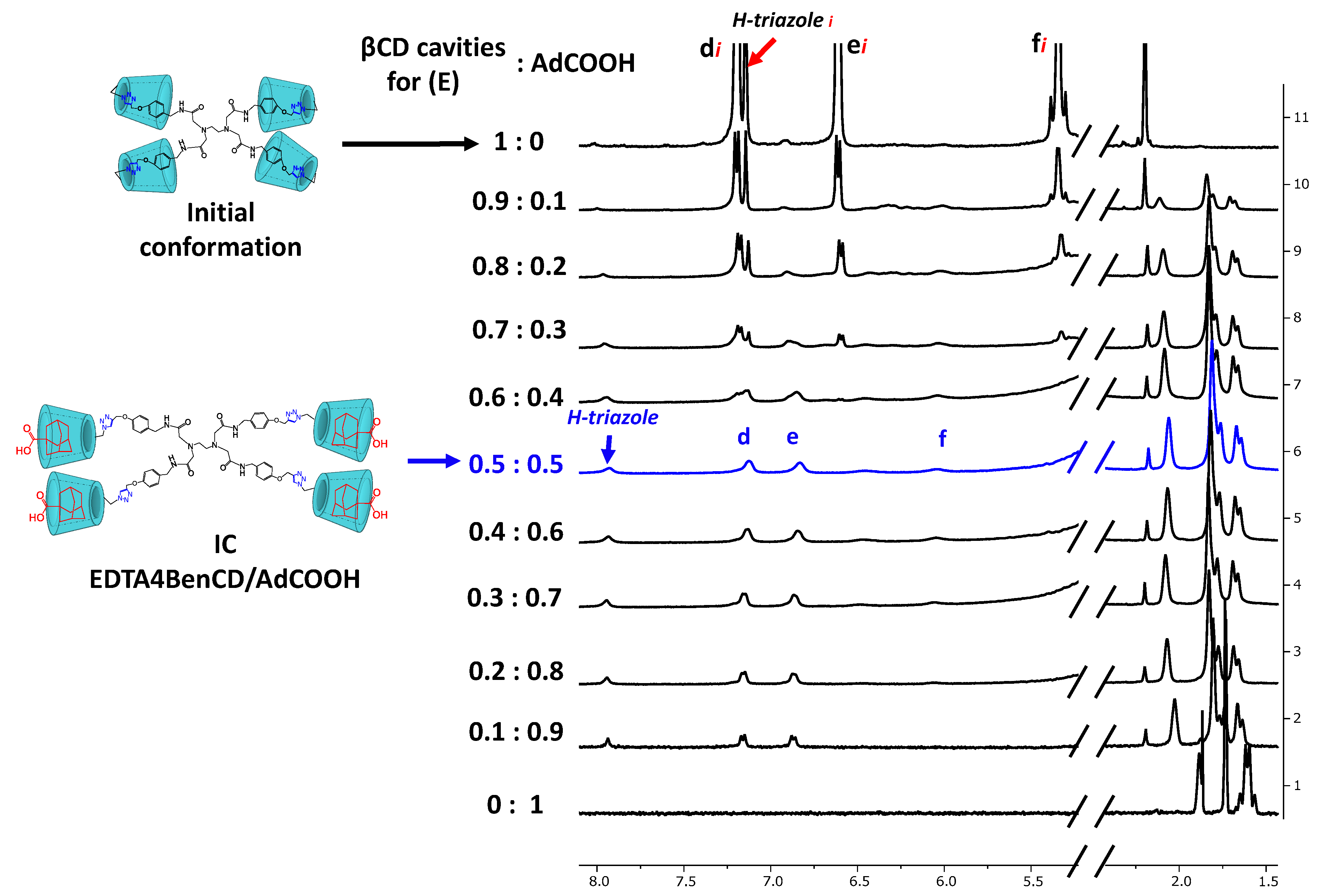
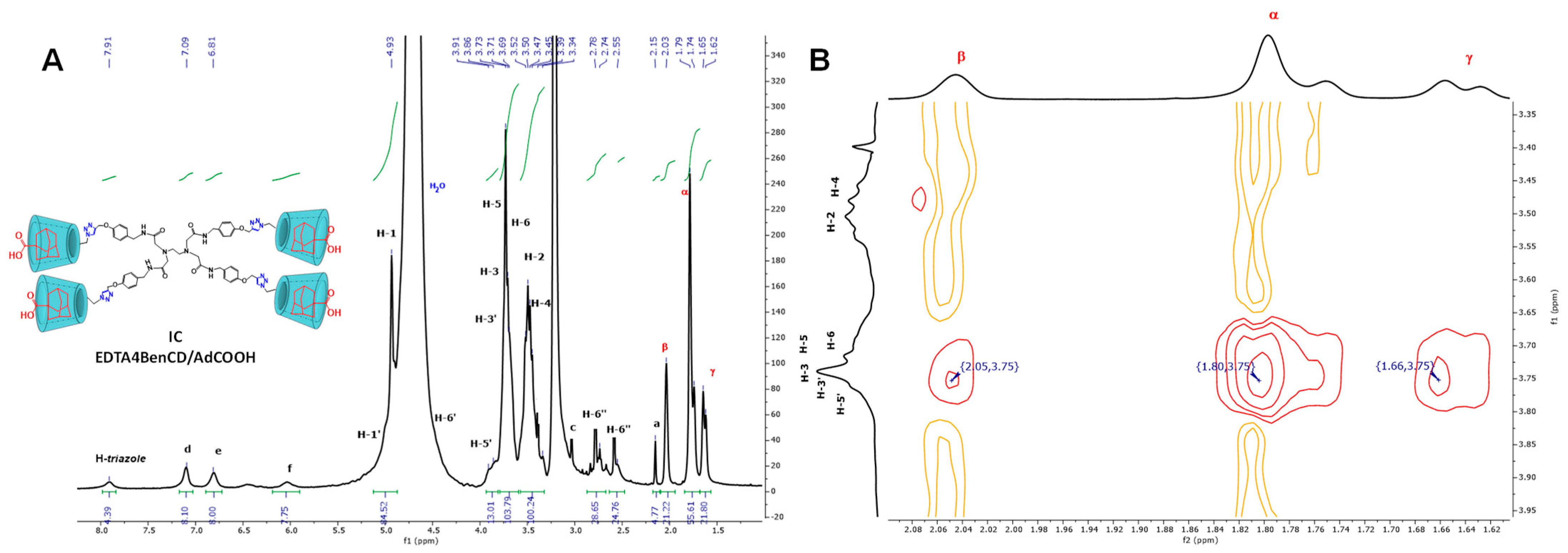
2.3. Availability of the βCD Cavities in EDTA di-βCD (A–C) and EDTA G0-βCD Dendrimers (D–F)
2.4. Determination of Quantitative Water Solubility for EDTA di-βCD (A–C) and EDTA G0-βCD Dendrimers (D–F)
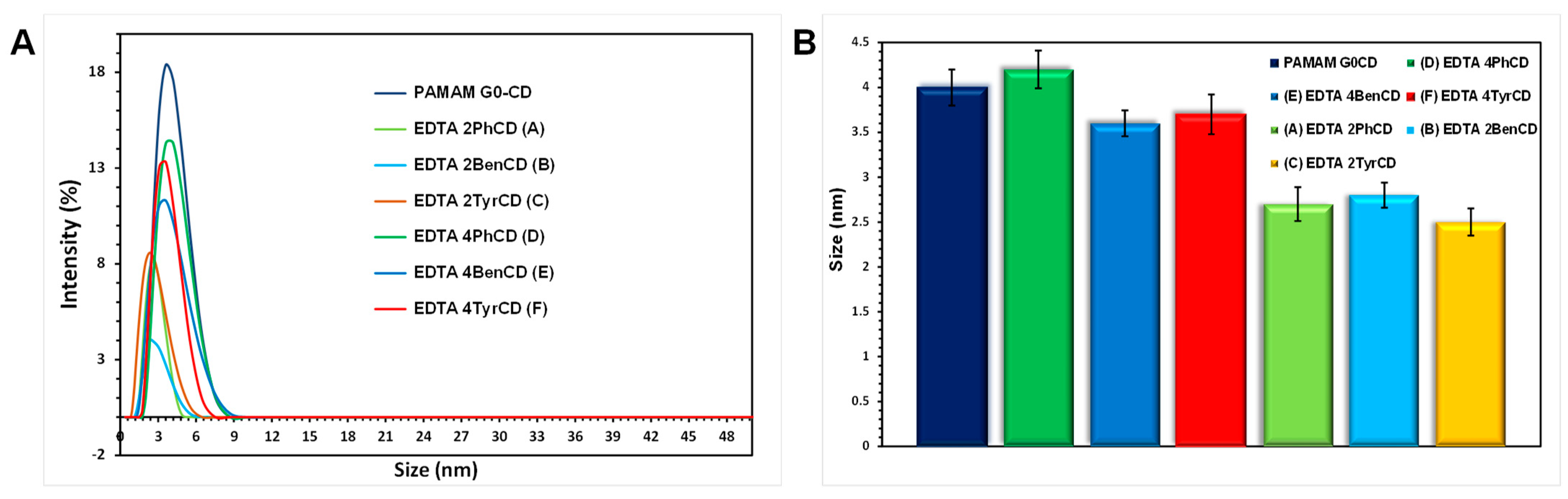
2.5. Dynamic Light Scattering Size Analysis of the EDTA di-βCD (A–C) and EDTA-βCD Dendrimers (D–F)
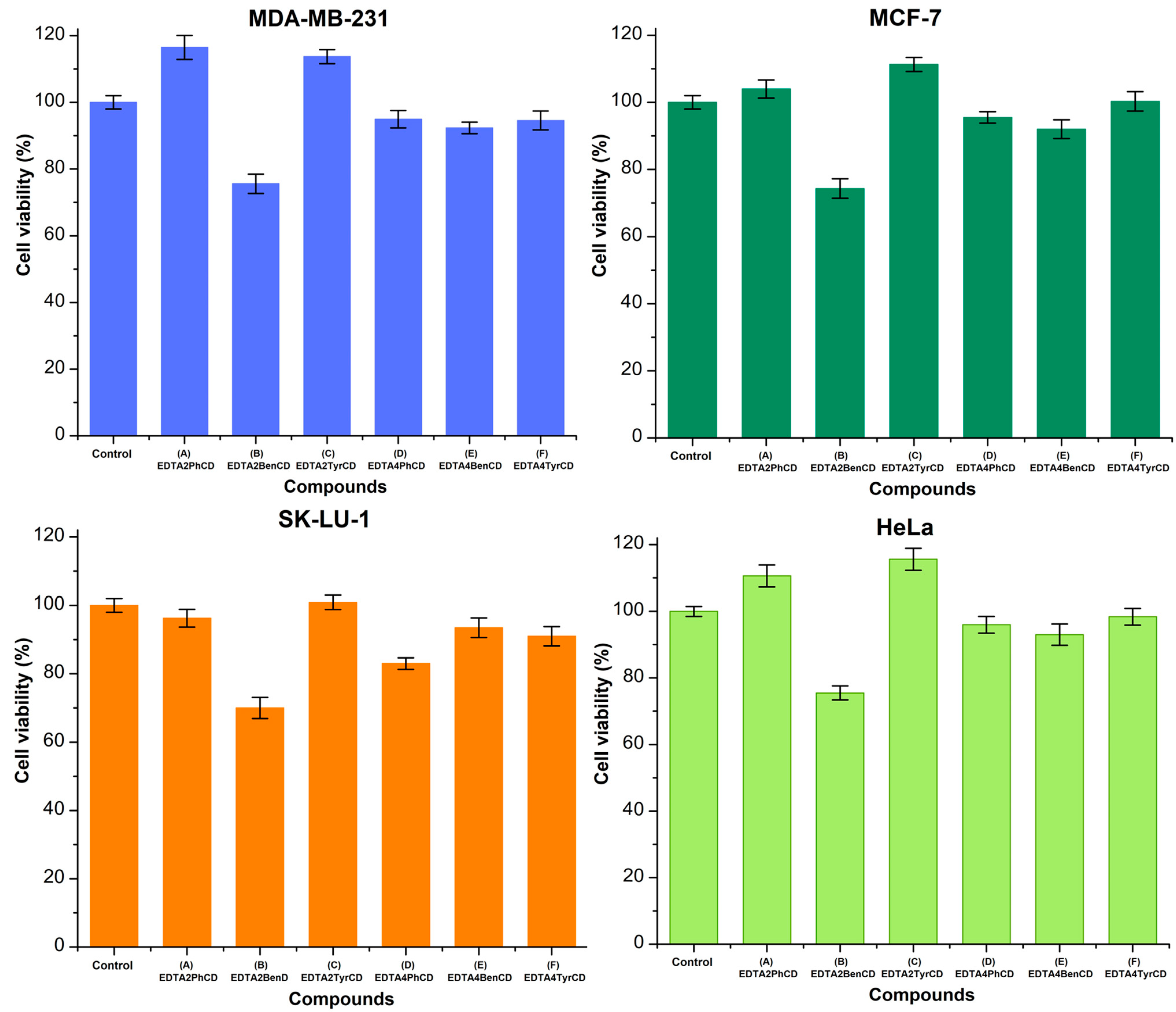
2.6. Viability Studies
3. Materials and Methods
3.1. General Notes
3.2. Synthetic Procedures
3.2.1. Mono-tosyl-βCD (mOTsβCD)
3.2.2. Mono-azide-βCD (mN3βCD)
3.2.3. General Procedure for the Synthesis of Tert-butyl Carbamate Intermediates (4–6)
Tert-butyl(4-hydroxyphenyl)carbamate (4)
Tert-butyl(4-hydroxybenzyl)carbamate (5)
Tert-butyl(4-hydroxyphenetyl)carbamate (6)
3.2.4. General Procedure for the Synthesis of tert-butyl 4-(Prop-2-yn-1-yloxy)carbamates Intermediaries (7–9)
Tert-butyl (4-(Prop-2-yn-1-yloxy)phenyl)carbamate (7)
Tert-butyl (4-(Prop-2-yn-1-yloxy)benzyl)carbamate (8)
Tert-butyl (4-(Prop-2-yn-1-yloxy)phenetyl)carbamate (9)
3.2.5. General Procedure for the Synthesis of 4-(Prop-2-yn-1-yloxy)amines Intermediaries (10–12)
4-(Prop-2-yn-1-yloxy)aniline (10)
[4-(Prop-2-yn-1-yloxy)phenyl]methanamine (11)
2-(4-(Prop-2-yn-1-yloxy)phenyl)ethan-1-amine (12)
3.2.6. General Procedure for the Synthesis of Disubstituted EDTA Alkynes (13–15)
Disubstituted EDTA Alkyne (13)
Disubstituted EDTA Alkyne (14)
Disubstituted EDTA Alkyne (15)
3.2.7. General Procedure for the Synthesis of Tetrasubstituted EDTA G0-alkyne (16–18)
Tetrasubstituted EDTA G0-Alkyne (16)
Tetrasubstituted EDTA G0-Alkyne (17)
Tetrasubstituted EDTA G0-Alkyne (18)
3.2.8. General Procedure for the Synthesis of Dendritic Compounds EDTA di-βCD (A, B, C)
EDTA2PhCD (A)
EDTA2BenCD (B)
EDTA2TyrCD (C)
3.2.9. General Procedure for the Synthesis of EDTA-βCD Dendrimers (D–F)
EDTA4PhCD Dendrimer (D)
EDTA4BenCD Dendrimer (E)
EDTA4TyrCD Dendrimer (F)
3.3. Job Plot Method
3.4. Determination of Water Solubility for EDTA di-βCD (A–C) and EDTA-βCD Dendrimers (D–F)
3.5. DLS Measurements
3.6. Biological Procedures
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanchi, S.; Suresh, G.; Priyakumar, U.D.; Ayappa, K.G.; Maiti, P.K. Molecular Dynamics Study of the Structure, Flexibility, and Hydrophilicity of PETIM Dendrimers: A Comparison with PAMAM Dendrimers. J. Phys. Chem. B 2015, 119, 12990–13001. [Google Scholar] [CrossRef] [PubMed]
- Al-Shdefat, R.; Kadhim, M.M.; Mahdi, A.B.; Lafta, H.A.; Kumar, A. Theoretical Evaluation of Poly(Amidoamine) Dendrimers with Different Peripheral Groups as a Purinethol Drug Delivery System in Aqueous Medium. Colloids Surf. B 2022, 216, 112534. [Google Scholar] [CrossRef] [PubMed]
- Esfand, R.; Tomalia, D.A. Poly(Amidoamine) (PAMAM) Dendrimers: From Biomimicry to Drug Delivery and Biomedical Applications. Drug Discov. Today 2001, 6, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Sherje, A.P.; Jadhav, M.; Dravyakar, B.R.; Kadam, D. Dendrimers: A Versatile Nanocarrier for Drug Delivery and Targeting. Int. J. Pharm. 2018, 548, 707–720. [Google Scholar] [CrossRef]
- Medina, S.H.; El-Sayed, M.E.H. Dendrimers as Carriers for Delivery of Chemotherapeutic Agents. Chem. Rev. 2009, 109, 3141–3157. [Google Scholar] [CrossRef]
- Ramírez-Palma, M.T.; Apolonio, V.M.; González, J.; Martínez-Barrera, G.; Corona, D.; Cuevas-Yañez, E. Synthesis of EDTA Core Dendrimers through a Consecutive Esterification-CuAAC Process. J. Macromol. Sci. Part A Pure Appl. Chem. 2017, 54, 908–914. [Google Scholar] [CrossRef]
- Jevprasesphant, R.F.J.; Jalal, R.; Attwood, D.; McKeown, N.B.; D’Emanuele, A. The influence of surface modification on the cytotoxicity of PAMAM dendrimers. Int. J. Pharm. 2003, 252, 263–266. [Google Scholar] [CrossRef]
- Malik, N.; Wiwattanapatapee, R.; Klopsch, R.; Lorenz, K.; Frey, H.; Weener, J.W.; Meijer, E.; Paulus, W.; Duncan, R. Dendrimers:: Relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J. Control. Release 2000, 65, 133–148. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Q.; Chang, H.; Cheng, Y. Surface-Engineered Dendrimers in Gene Delivery. Chem. Rev. 2015, 115, 5274–5300. [Google Scholar] [CrossRef]
- Mishra, V.; Gupta, U.; Jain, N.K. Surface-Engineered Dendrimers: A Solution for Toxicity Issues. J. Biomater. Sci. Polym. Ed. 2009, 20, 141–166. [Google Scholar] [CrossRef]
- Madaan, K.; Kumar, S.; Poonia, N.; Lather, V.; Pandita, D. Dendrimers in drug delivery and targeting: Drug-dendrimer interactions and toxicity issues. J. Pharm. Bioallied Sci. 2014, 6, 139. [Google Scholar] [CrossRef]
- Bandaru, R.; Sanket, A.S.; Rekha, S.; Kamble, O.; Dewangan, R.P.; Kesharwani, P.; Samal, S.K.; Dandela, R. Biological interaction of dendrimers. In Dendrimer-Based Nanotherapeutics; Elsevier: Amsterdam, The Netherlands, 2021; pp. 63–74. [Google Scholar] [CrossRef]
- Li, X.; Naeem, A.; Xiao, S.; Hu, L.; Zhang, J.; Zheng, Q. Safety Challenges and Application Strategies for the Use of Dendrimers in Medicine. Pharmaceutics 2022, 14, 1292. [Google Scholar] [CrossRef] [PubMed]
- Gajbhiye, V.; Kumar, P.V.; Tekade, R.K.; Jain, N. PEGylated PPI dendritic architectures for sustained delivery of H2 receptor antagonist. Eur. J. Med. Chem. 2009, 44, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Hidetoshi, A. Twenty Years of Research on Cyclodextrin Conjugates with PAMAM Dendrimers. Pharmaceutics 2021, 13, 697. [Google Scholar] [CrossRef]
- Szejtli, J. Introduction and General Overview of Cyclodextrin Chemistry. Chem. Rev. 1998, 98, 1743–1754. [Google Scholar] [CrossRef]
- Defaye, J.; Garcia Fernandez, J.M.; Ortiz Mellet, C. Les cyclodextrines en pharmacie: Perspectives pour le ciblage d’actifs thérapeutiques et le contrôle d’interactions membranaires. Ann. Pharm. Fr. 2007, 65, 33–49. [Google Scholar] [CrossRef]
- Malkoch, M.; Malmström, E.; Nyström, A.M. 6.04—Dendrimers: Properties and Applications; Elsevier B.V.: Amsterdam, The Netherlands, 2012; pp. 1–10. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise Huisgen Cycloaddition Process: Copper(I)-Catalyzed Regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chemie—Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Liu, Y.; Ke, C.-F.; Zhang, H.-Y.; Cui, J.; Ding, F. Complexation-Induced Transition of Nanorod to Network Aggregates: Alternate Porphyrin and Cyclodextrin Arrays. J. Am. Chem. Soc. 2008, 130, 600–605. [Google Scholar] [CrossRef]
- Yamauchi, K.; Miyawaki, A.; Takashima, Y.; Yamaguchi, H.; Harada, A. Switching from altro-α-Cyclodextrin Dimer to pseudo[1]Rotaxane Dimer through Tumbling. Org. Lett. 2010, 12, 1284–1286. [Google Scholar] [CrossRef]
- Yamauchi, K.; Miyawaki, A.; Takashima, Y.; Yamaguchi, H.; Harada, A.J. A Molecular Reel: Shuttling of a Rotor by Tumbling of a Macrocycle. Org. Chem. 2010, 75, 1040–1046. [Google Scholar] [CrossRef]
- Chmurski, K.; Stepniak, P.; Jurczak, J. Long-chain-linked β-cyclodextrin dimers: Synthesis and relationship between reactivity and inclusion complex formation. Carbohydr. Polym. 2016, 138, 8–15. [Google Scholar] [CrossRef]
- Zhong, N.; Byun, H.-S.; Bittman, R. An Improved Synthesis of 6-O-Monotosyl-6-Deoxy-β-Cyclodextrin. Tetrahedron Lett. 1998, 39, 2919–2920. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, Y.; Hu, J.; Li, C.; Liu, S. Multi-Responsive Supramolecular Double Hydrophilic Diblock Copolymer Driven by Host-Guest Inclusion Complexation between β-Cyclodextrin and Adamantyl Moieties. Macromol. Chem. Phys. 2009, 210, 2125–2137. [Google Scholar] [CrossRef]
- Sorroza-Martínez, K.; González-Méndez, I.; Vonlanthen, M.; Moineau-Chane Ching, K.I.; Caminade, A.-M.; Illescas, J.; Rivera, E. First Class of Phosphorus Dendritic Compounds Containing β-Cyclodextrin Units in the Periphery Prepared by CuAAC. Molecules 2020, 25, 4034. [Google Scholar] [CrossRef] [PubMed]
- Folgado, E.; Guerre, M.; Bijani, C.; Ladmiral, V.; Caminade, A.-M.; Ameduri, B.; Ouali, A. Well-defined poly(vinylidene fluoride) (PVDF) based-dendrimers synthesized by click chemistry: Enhanced crystallinity of PVDF and increased hydrophobicity of PVDF films. Polym. Chem. 2016, 7, 5625–5629. [Google Scholar] [CrossRef]
- Rahmani, S.; zadeh Mahani, A.S. Synthesis and characterization of novel poly(arylene ether)s containing 1,3,4-oxadiazole and triazole units through click chemistry. Macromol. Res. 2015, 23, 1018–1025. [Google Scholar] [CrossRef]
- González-Méndez, I.; Loera-Loera, E.; Sorroza-Martínez, K.; Vonlanthen, M.; Cuétara-Guadarrama, F.; Bernad-Bernad, M.-J.; Rivera, E.; Gracia-Mora, J. Synthesis of β-Cyclodextrin-Decorated Dendritic Compounds Based on EDTA Core: A New Class of PAMAM Dendrimer Analogs. Pharmaceutics 2022, 14, 2363. [Google Scholar] [CrossRef]
- Li, Z.; Huang, R.; Xu, H.; Chen, J.; Zhan, Y.; Zhou, X.; Chen, H.; Jiang, B. Divinylsulfonamides as Specific Linkers for Stapling Disulfide Bonds in Peptides. Org. Lett. 2017, 19, 4972–4975. [Google Scholar] [CrossRef]
- González-Méndez, I.; Hameau, A.; Laurent, R.; Bijani, C.; Bourdon, V.; Caminade, A.-M.; Rivera, E.; Moineau-Chane Ching, K.I. β-Cyclodextrin PAMAM Dendrimer: How to Overcome the Tumbling Process for Getting Fully Available Host Cavities. Eur. J. Org. Chem. 2020, 2020, 1114–1121. [Google Scholar] [CrossRef]
- Xi, W.; Scott, T.F.; Kloxin, C.J.; Bowman, C.N. Click Chemistry in Materials Science. Adv. Funct. Mater. 2014, 24, 2572–2590. [Google Scholar] [CrossRef]
- Iha, R.K.; Wooley, K.L.; Nyström, A.M.; Burke, D.J.; Kade, M.J.; Hawker, C.J. Applications of Orthogonal “Click” Chemistries in the Synthesis of Functional Soft Materials. Chem. Rev. 2009, 109, 5620–5686. [Google Scholar] [CrossRef] [PubMed]
- Such, G.K.; Johnston, A.P.R.; Liang, K.; Caruso, F. Synthesis and functionalization of nanoengineered materials using click chemistry. Prog. Polym. Sci. 2012, 37, 985–1003. [Google Scholar] [CrossRef]
- Huang, R.; Li, Z.; Ren, P.; Chen, W.; Kuang, Y.; Chen, J.; Zhan, Y.; Chen, H.; Jiang, B. N-Phenyl-N-aceto-vinylsulfonamides as Efficient and Chemoselective Handles for N-Terminal Modification of Peptides and Proteins. Eur. J. Org. Chem. 2018, 14, 829–836. [Google Scholar] [CrossRef]
- Hugenberg, V.; Wagner, S.; Kopka, K.; Schäfers, M.; Schuit, R.C.; Windhorst, A.D.; Hermann, S. Radiolabeled Selective Matrix Metalloproteinase 13 (MMP-13) Inhibitors: (Radio)Syntheses and in Vitro and First in Vivo Evaluation. J. Med. Chem. 2017, 60, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.H.Y.; Savoie, H.; Bryden, F.; Boyle, R.W. A Convenient Method for Multicolour Labelling of Proteins with BODIPY Fluorophores via Tyrosine Residues. Photochem. Photobiol. Sci. 2017, 16, 1260–1267. [Google Scholar] [CrossRef] [PubMed]
- Legros, V.; Vanhaverbeke, C.; Souard, F.; Len, C.; Désiré, J. β-Cyclodextrin-glycerol dimers: Synthesis and NMR conformational analysis. Eur. J. Org. Chem. 2013, 13, 2583–2590. [Google Scholar] [CrossRef]
- Menuel, S.; Azaroual, N.; Landy, D.; Six, N.; Hapiot, F.; Monflier, E. Unusual inversion phenomenon of β-cyclodextrin dimers in water. Chem. Eur. J. 2011, 17, 3949–3955. [Google Scholar] [CrossRef]
- Potier, J.; Menuel, S.; Azaroual, N.; Monflier, E.; Hapiot, F. Limits of the inversion phenomenon in triazolyl-substituted β-cyclodextrin dimers. Eur. J. Org. Chem. 2014, 2014, 1547–1556. [Google Scholar] [CrossRef]
- Gupta, U.; Agashe, H.; Asthana, A.; Jain, N. Dendrimers: Novel polymeric nanoarchitectures for solubility enhancement. Biomacromolecules 2006, 7, 649–658. [Google Scholar] [CrossRef]
- Shi, Y.; Porter, W.; Merdan, T.; Li, L. Recent advances in intravenous delivery of poorly water-soluble compounds. Expert Opin. Drug Deliv. 2009, 6, 1261–1282. [Google Scholar] [CrossRef]
- Svenson, S. Dendrimers as versatile platform in drug delivery applications. Eur. J. Pharm. Biopharm. 2009, 71, 445–462. [Google Scholar] [CrossRef] [PubMed]
- Svenson, S.; Chauhan, A. Dendrimers for enhanced drug solubilization. Nanomedicine 2008, 3, 679–702. [Google Scholar] [CrossRef] [PubMed]
- Jozwiakowski, M.J.; Connors, K.A. Aqueous solubility behavior of three cyclodextrins. Carbohydr. Res. 1985, 143, 51–59. [Google Scholar] [CrossRef]
- Brewster, M.E.; Loftsson, T. Cyclodextrins as pharmaceutical solubilizers. Adv. Drug Delivery Rev. 2007, 59, 645. [Google Scholar] [CrossRef]
- Renero-Lecuna, C.; Iturrioz-Rodríguez, N.; González-Lavado, E.; Padín-González, E.; Navarro-Palomares, E.; Valdivia-Fernández, L.; García-Hevia, L.; Fanarraga, M.L.; González-Legarreta, L. Effect of size, shape, and composition on the interaction of different nanomaterials with HeLa cells. J. Nanomater. 2019, 2019, 7518482. [Google Scholar] [CrossRef]
- Sorroza-Martínez, K.; González Méndez, I.; Martínez-Serrano, R.D.; Solano, J.D.; Ruiu, A.; Illescas, J.; Zhu, X.X.; Rivera, E. Efficient modification of PAMAM G1 dendrimer surface with β-cyclodextrin units by CuAAC: Impact on the water solubility and cytotoxicity. RSC Adv. 2020, 10, 25557–25566. [Google Scholar] [CrossRef]
- Dong, R.; Zhou, L.; Wu, J.; Tu, C.; Su, Y.; Zhu, B.; Gu, H.; Yan, D.; Zhu, X. A supramolecular approach to the preparation of charge-tunable dendritic polycations for efficient gene delivery. Chem. Commun. 2011, 47, 5473. [Google Scholar] [CrossRef]
- El-Sayed, M.; Rhodes, C.A.; Ginski, M.; Ghandehari, H. Transport mechanism(s) of poly (amidoamine) dendrimers across Caco-2 cell monolayers. Int. J. Pharm. 2003, 265, 151. [Google Scholar] [CrossRef]
- Tajarobi, F.; El-Sayed, M.; Rege, B.D.; Polli, J.E.; Ghandehari, H. Transport of poly amidoamine dendrimers across Madin–Darby canine kidney cells. Int. J. Pharm. 2001, 215, 263. [Google Scholar] [CrossRef]
- Duncan, R.; Izzo, L. Dendrimer biocompatibility and toxicity. Adv. Drug Deliv. Rev. 2005, 57, 2215. [Google Scholar] [CrossRef]
- Gräfenstein, A.; Rumancev, C.; Pollak, R.; Hämisch, B.; Galbierz, V.; Schroeder, W.H.; Garrevoet, J.; Falkenber, G.; Vöpel, T.; Huber, K.; et al. Spatial Distribution of Intracellular Ion Concentrations in Aggregate-Forming HeLa Cells Analyzed by μ-XRF Imaging. Chem. Open 2022, 11, e2022000. [Google Scholar] [CrossRef]
- Wyatt, H.V. Cation requirements of HeLa cells in tissue culture. Exp. Cell Res. 1961, 23, 97–117. [Google Scholar] [CrossRef] [PubMed]
- Hameau, A.; Fuchs, S.; Laurent, R.; Majoral, J.-P.; Caminade, A.-M. Synthesis of Dye/Fluorescent Functionalized Dendrons Based on Cyclotriphosphazene. Beilstein J. Org. Chem. 2011, 7, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Antelo, A.; Jover, A.; Galantini, L.; Meijide, F.; Alvarez Alcalde, M.; Viorel Pavel, N.; Vázquez Tato, J. Formation of Host-Guest and Sandwich Complexes by a β-Cyclodextrin Derivative. J. Incl. Phenom. Macrocycl. Chem. 2011, 69, 245–253. [Google Scholar] [CrossRef]
- Sorroza-Martínez, K.; González-Méndez, I.; Vonlanthen, M.; Cuétara-Guadarrama, F.; Illescas, J.; Zhu, X.X.; Rivera, E. Guest-Mediated Reversal of the Tumbling Process in Phosphorus-Dendritic Compounds Containing β-Cyclodextrin Units: An NMR Study. Pharmaceuticals 2021, 14, 556. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Solubility (g/mL) | Compound | Solubility (g/mL) |
|---|---|---|---|
| EDTA2PhCD (A) | 1.582 ± 0.002 | EDTA4PhCD (D) | 1.594 ± 0.003 |
| EDTA2BenCD (B) | 1.594 ± 0.001 | EDTA4BenCD (E) | 1.601 ± 0.002 |
| EDTA2TyrCD (C) | 1.591 ± 0.003 | EDTA4TyrCD (F) | 1.623 ± 0.002 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Méndez, I.; Sorroza-Martínez, K.; González-Sánchez, I.; Gracia-Mora, J.; Bernad-Bernad, M.J.; Cerbón, M.; Rivera, E.; Yatsimirsky, A.K. Exploring the Influence of Spacers in EDTA–β-Cyclodextrin Dendrimers: Physicochemical Properties and In Vitro Biological Behavior. Int. J. Mol. Sci. 2023, 24, 14422. https://doi.org/10.3390/ijms241914422
González-Méndez I, Sorroza-Martínez K, González-Sánchez I, Gracia-Mora J, Bernad-Bernad MJ, Cerbón M, Rivera E, Yatsimirsky AK. Exploring the Influence of Spacers in EDTA–β-Cyclodextrin Dendrimers: Physicochemical Properties and In Vitro Biological Behavior. International Journal of Molecular Sciences. 2023; 24(19):14422. https://doi.org/10.3390/ijms241914422
Chicago/Turabian StyleGonzález-Méndez, Israel, Kendra Sorroza-Martínez, Ignacio González-Sánchez, Jesús Gracia-Mora, María Josefa Bernad-Bernad, Marco Cerbón, Ernesto Rivera, and Anatoly K. Yatsimirsky. 2023. "Exploring the Influence of Spacers in EDTA–β-Cyclodextrin Dendrimers: Physicochemical Properties and In Vitro Biological Behavior" International Journal of Molecular Sciences 24, no. 19: 14422. https://doi.org/10.3390/ijms241914422