
Pervasive Sharing of Causal Genetic Risk Factors Contributes to Clinical and Molecular Overlap between Sjögren’s Disease and Systemic Lupus Erythematosus
 ,
,
Abstract
:1. Introduction
2. Results
2.1. Sharing of GWAS Identified Genes and Loci between Sjögren’s Disease and Systemic Lupus Erythematosus
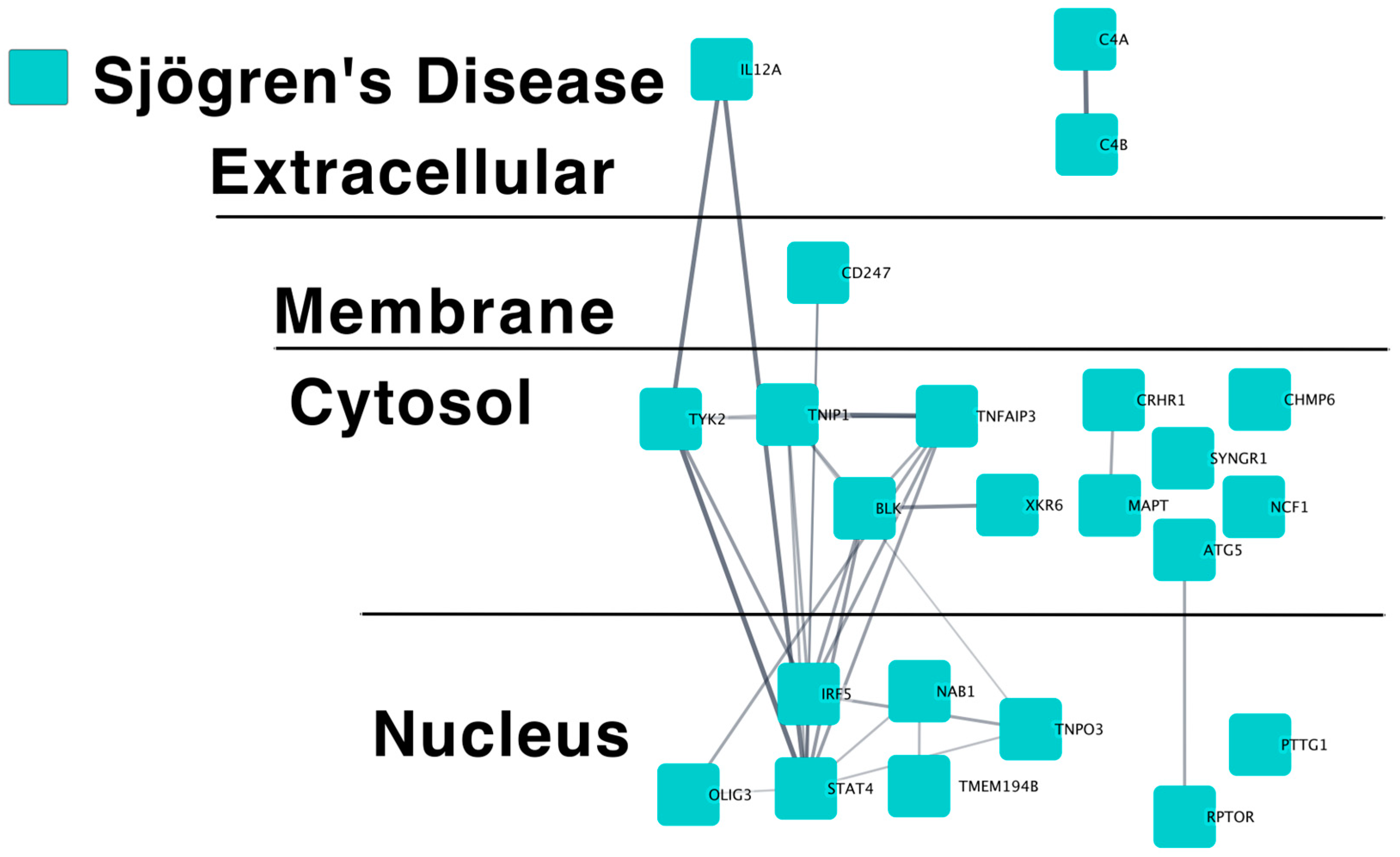
2.1.1. SjD Putative Causal Gene Predicted Protein–Protein Interaction Networks
2.1.2. Pathway Analysis of SjD Putative Causal Genes
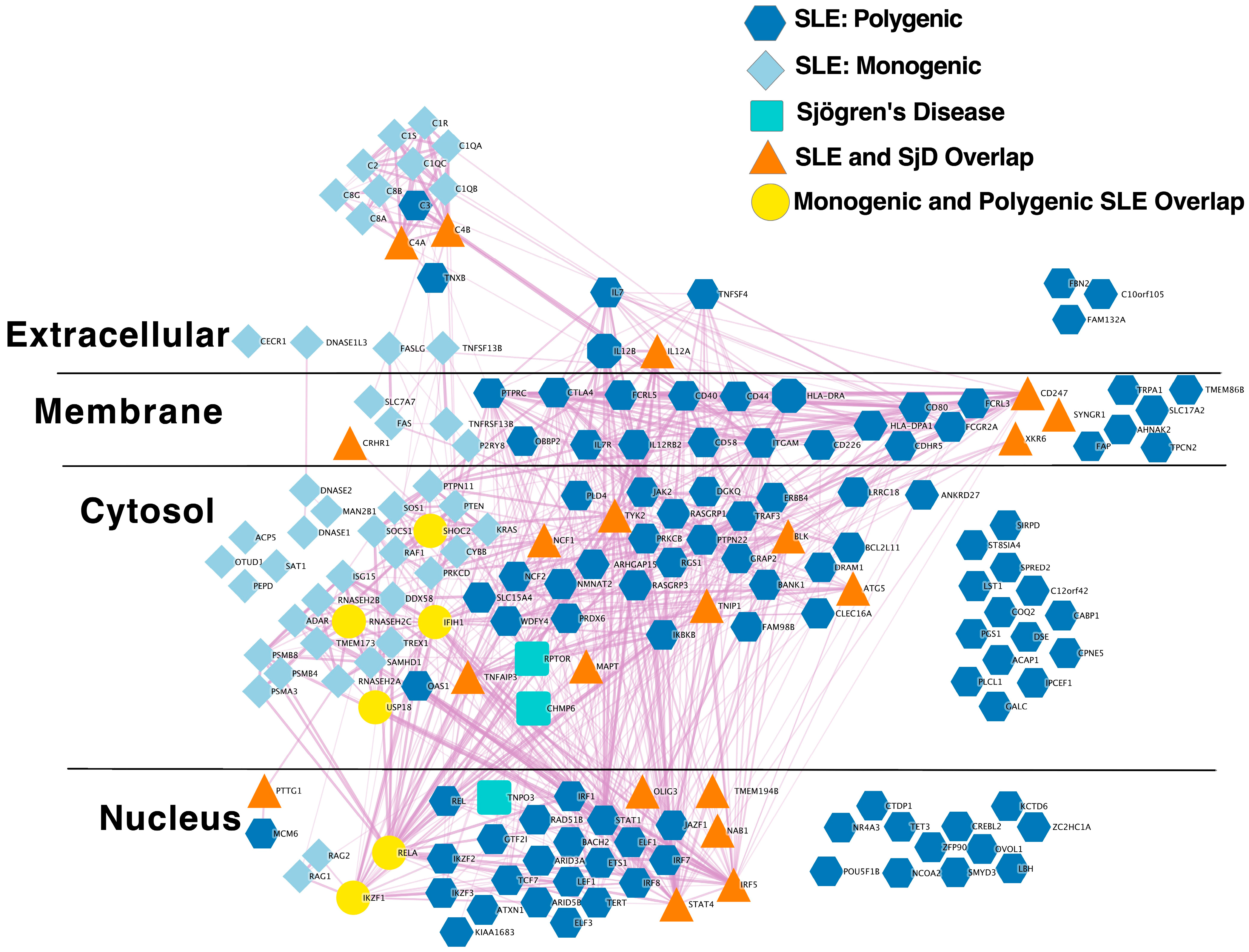
2.1.3. Extensive Overlap between SLE and SjD Putative Causal Gene Networks
2.1.4. Extensive Overlap between SLE and SjD GWAS Loci
2.2. Mendelian Randomization Analysis of Variants Associated with Sjögren’s Disease and Systemic Lupus Erythematosus
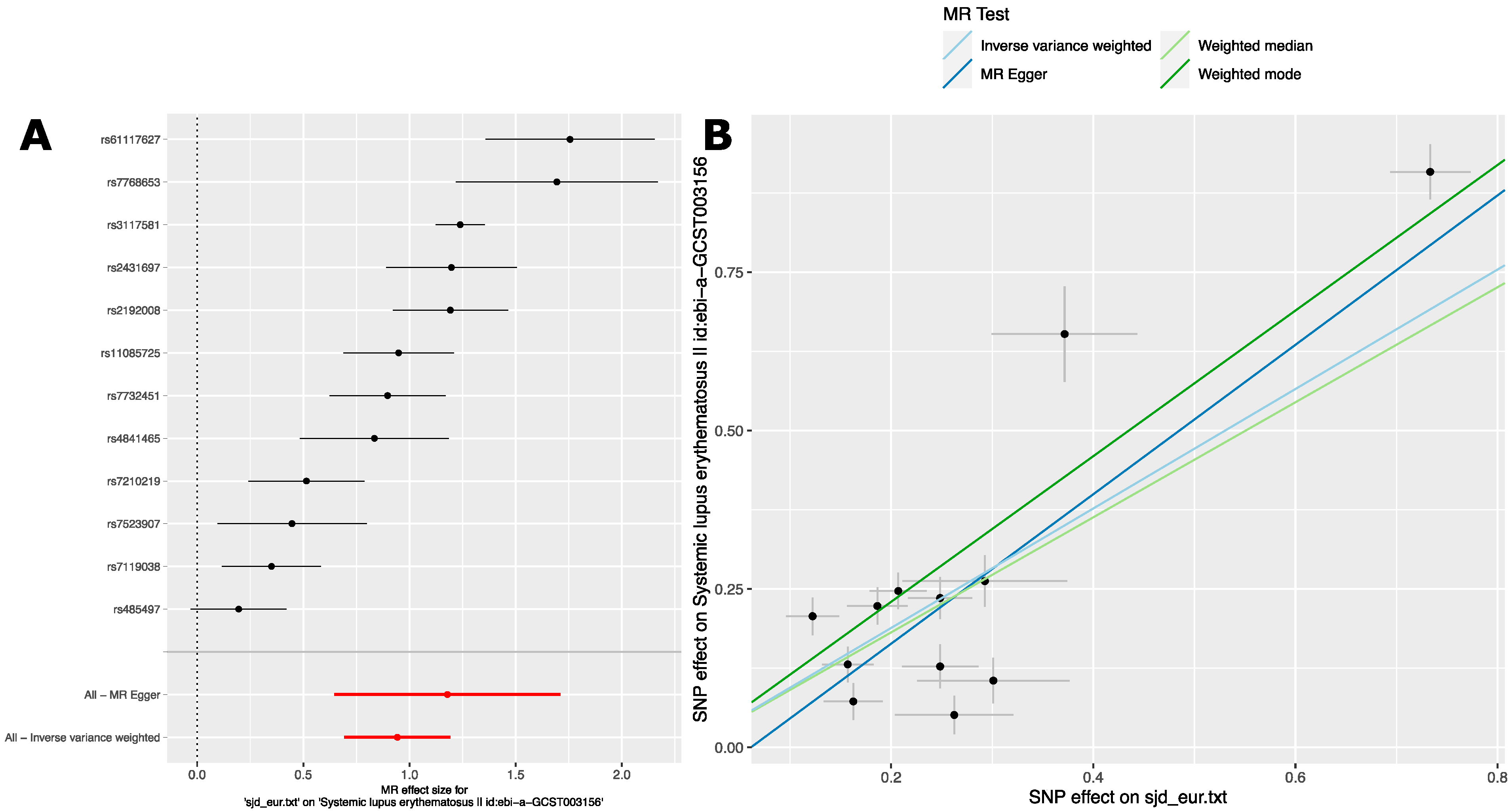
2.2.1. Analysis 1: The Causal Effect of SjD Variants on SLE in European Ancestry GWAS
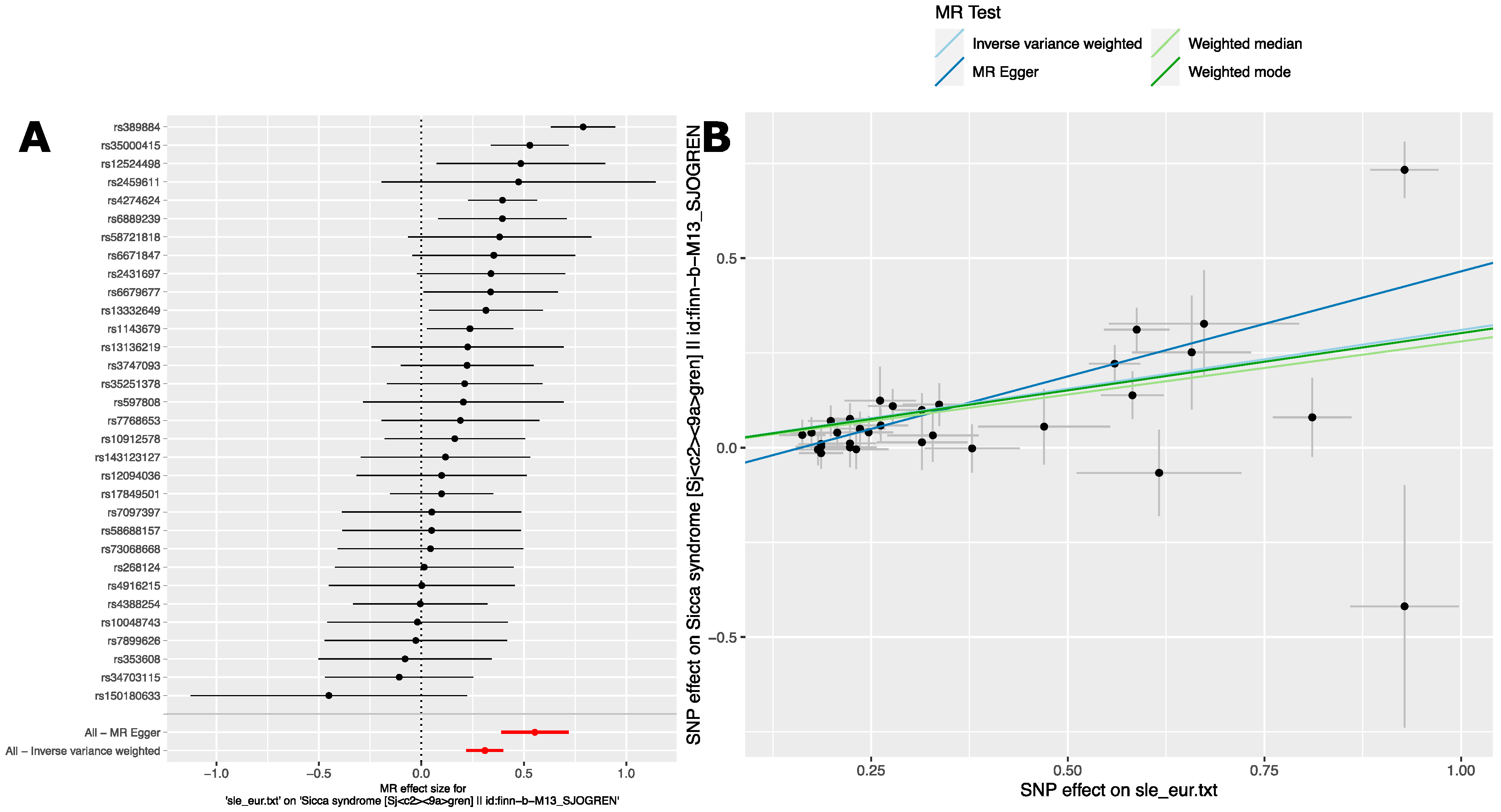
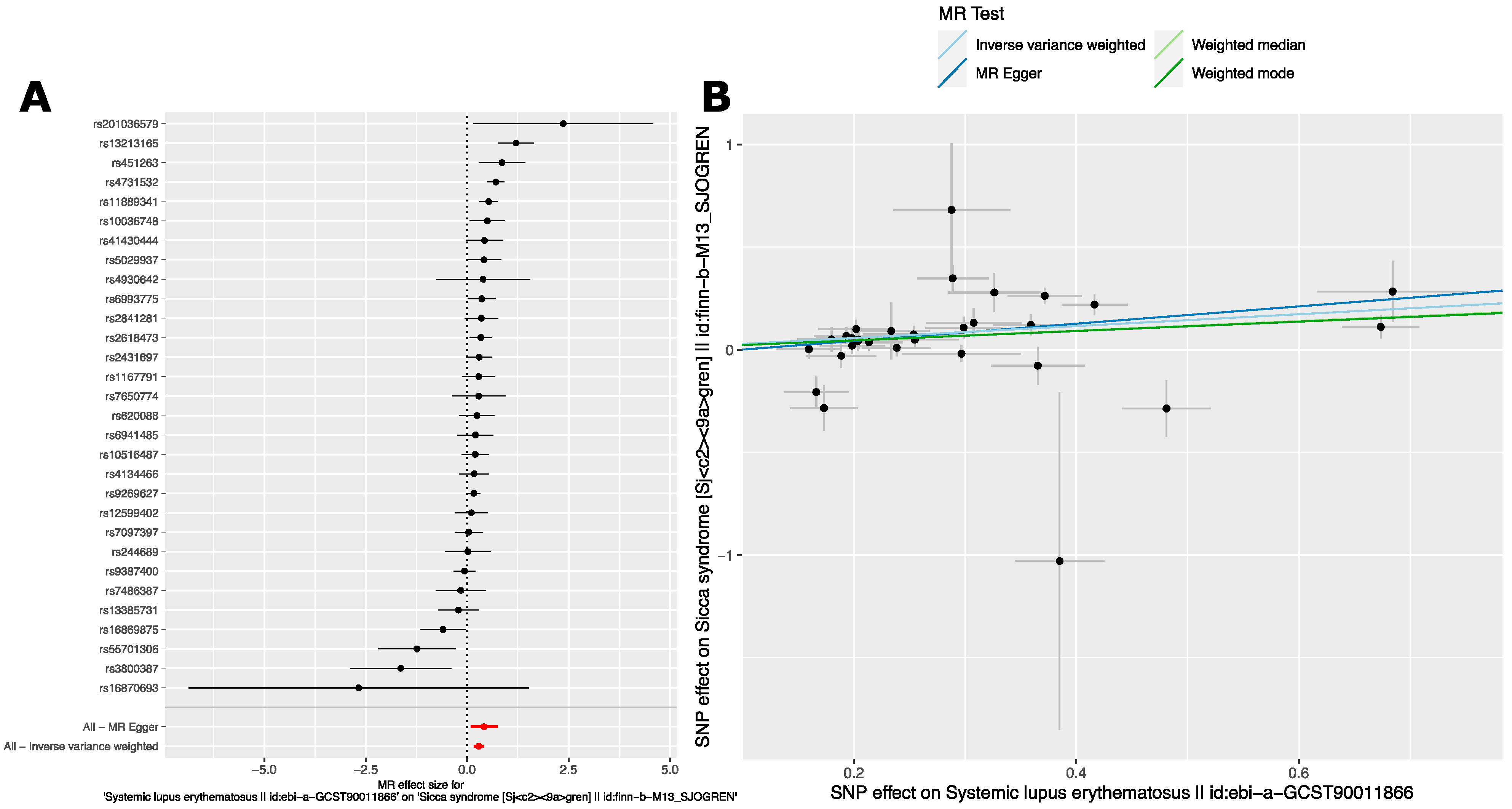
2.2.2. Analysis 2: The Causal Effect of SLE Variants on SjD in European Ancestry GWAS
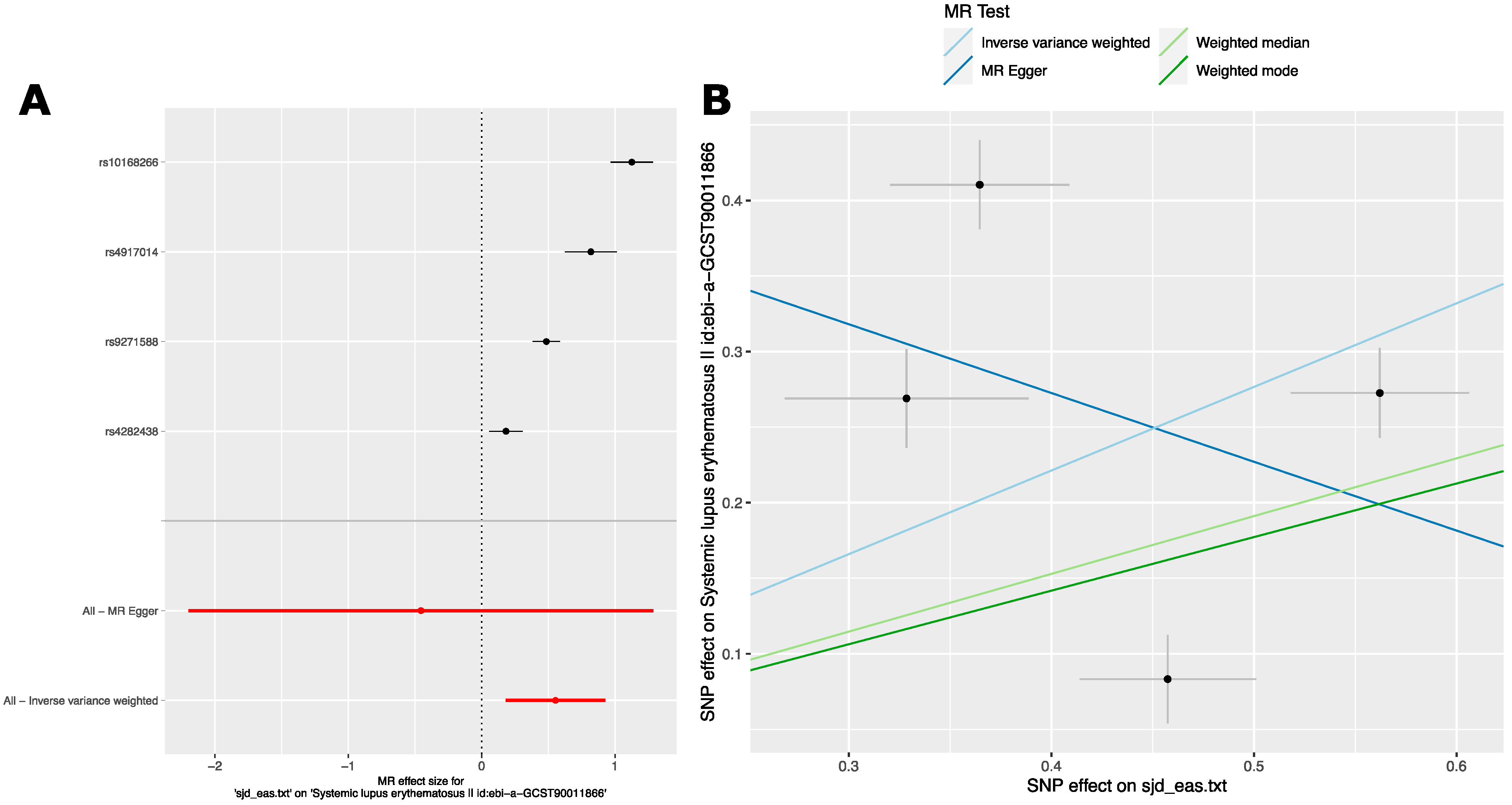
2.2.3. Analysis 3: The Causal Effect of SjD Variants on SLE in East Asian Ancestry GWAS
2.2.4. Analysis 4: The Causal Effect of SLE Variants from East Asian Ancestry GWAS on SjD in European Ancestry GWAS
3. Discussion
4. Materials and Methods
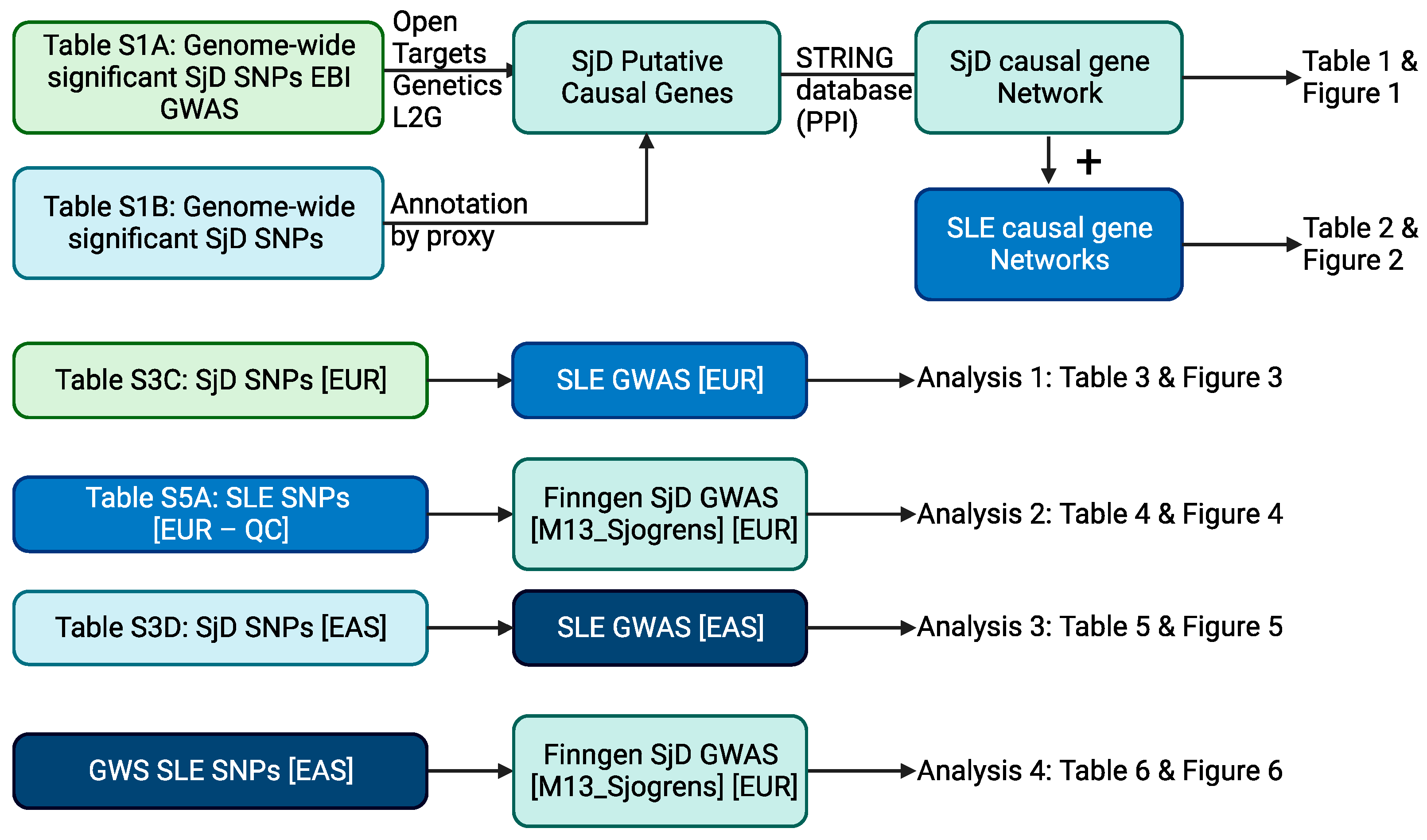
4.1. Overview of Analysis Scheme
4.2. Determining the Overlap of GWAS-Associated Loci in SLE and SjD
4.2.1. Defining SLE and SjD Loci
4.2.2. Pathway Analysis of Putatively Causal SjD Risk Genes
4.2.3. Calculating Locus-Level Overrepresentation between SLE and SjD
4.3. Predicted Causal Polygenic Risk Gene Annotation, Network Construction and Overlap
4.3.1. Calculating Locus-Level Overrepresentation between SLE and SjD
4.3.2. Building the Predicted Causal Gene Network for SjD
4.3.3. Calculating Overlap
4.4. Mendelian Randomization Analysis
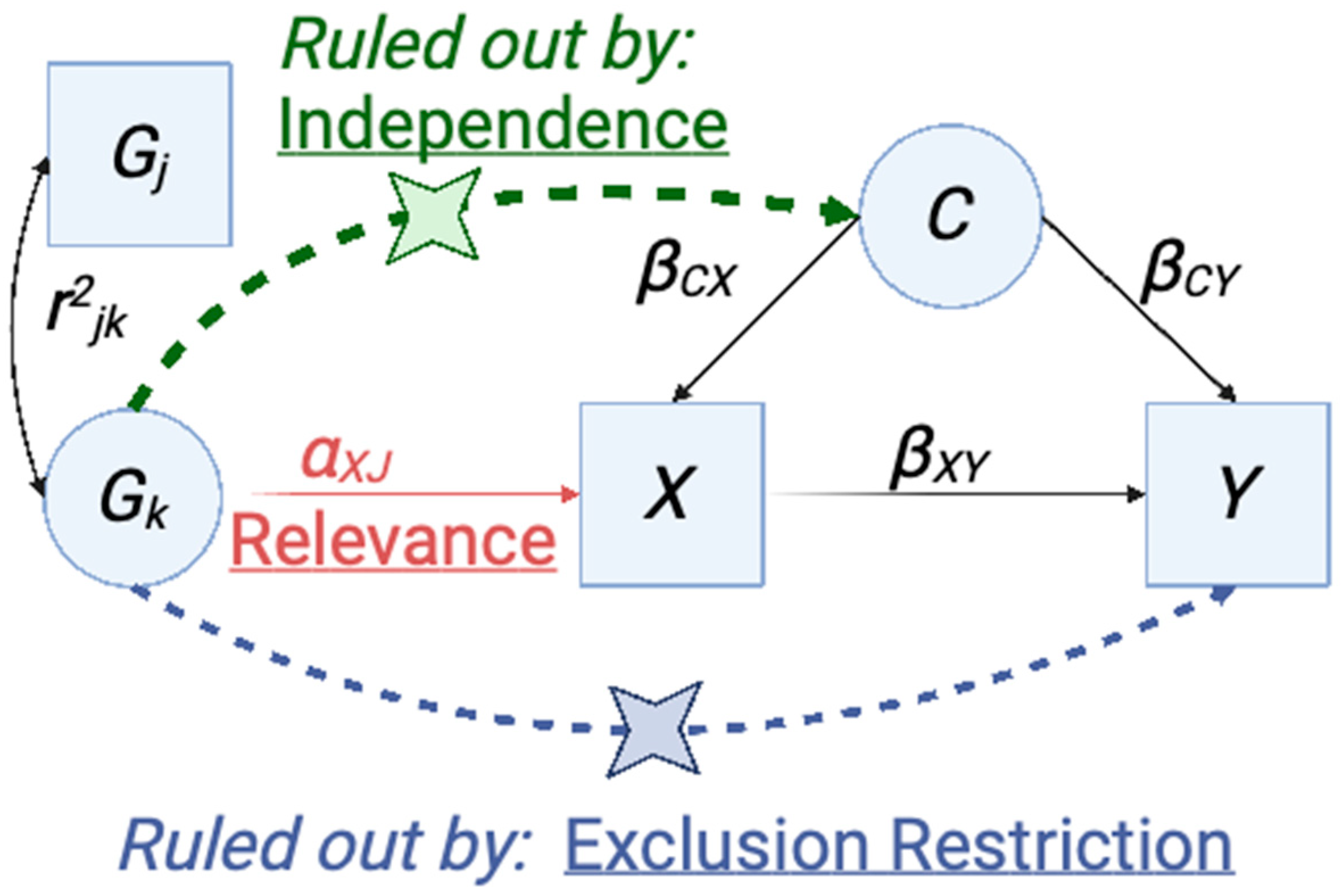
4.4.1. Overview of Mendelian Randomization and Application of Instrumental Variable Analysis to Genetic Studies
4.4.2. Evaluating the Causal Relationship between SjD and SLE
4.4.3. Selection of Instrumental Variables for SLE and SjD
4.4.4. Quality Control of Instrumental Variables for SLE and SjD
4.4.5. General Analysis Approach
4.4.6. MR Analysis Parameters
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moutsopoulos, H.M.; Mann, D.L.; Johnson, A.H.; Chused, T.M. Genetic Differences between Primary and Secondary Sicca Syndrome. N. Engl. J. Med. 1979, 301, 761–763. [Google Scholar] [CrossRef] [PubMed]
- Grennan, D.M.; Ferguson, M.; Ghobarey, A.E.; Williamson, J.; Dick, W.C.; Buchanan, W.W. Sjogren’s Syndrome in SLE: Part 2. An Examination of the Clinical Significance of Sjogren’s Syndrome by Comparison of Its Frequency in Typical and Atypical Forms of SLE, Overlap Syndromes and Scleroderma. N. Z. Med. J. 1977, 86, 376–379. [Google Scholar] [PubMed]
- Heaton, J.M. Sjögren’s Syndrome and Systemic Lupus Erythematosus. Br. Med. J. 1959, 1, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Provost, T.T.; Watson, R.; Simmons-O’Brien, E. Anti-Ro(SS-A) Antibody Positive Sjögren’s/Lupus Erythematosus Overlap Syndrome. Lupus 1997, 6, 105–111. [Google Scholar] [CrossRef]
- Alexander, E.L.; McNicholl, J.; Watson, R.M.; Bias, W.; Reichlin, M.; Provost, T.T. The Immunogenetic Relationship between Anti-Ro(SS-A)/La(SS-B) Antibody Positive Sjögren’s/Lupus Erythematosus Overlap Syndrome and the Neonatal Lupus Syndrome. J. Investig. Dermatol. 1989, 93, 751–756. [Google Scholar] [CrossRef]
- Gianordoli, A.P.E.; Laguardia, R.V.R.B.; Santos, M.C.F.S.; Jorge, F.C.; da Silva Salomão, A.; Caser, L.C.; Moulaz, I.R.; Serrano, É.V.; Miyamoto, S.T.; Machado, K.L.L.L.; et al. Prevalence of Sjögren’s Syndrome According to 2016 ACR-EULAR Classification Criteria in Patients with Systemic Lupus Erythematosus. Adv. Rheumatol. 2023, 63, 11. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.H.; Daikh, D.I.; Brinks, R.; Mosca, M.; Ramsey-Goldman, R.; Smolen, J.S.; Wofsy, D.; Boumpas, D.; Kamen, D.L.; et al. 2019 EULAR/ACR Classification Criteria for Systemic Lupus Erythematosus. Arthritis Rheumatol. 2019, 71, 1400–1412. [Google Scholar] [CrossRef]
- Aringer, M.; Costenbader, K.; Dörner, T.; Johnson, S.R. Response to: ‘New 2019 SLE EULAR/ACR Classification Criteria Are Valuable for Distinguishing Patients with SLE from Patients with PSS’ by Assan et Al. Ann. Rheum. Dis. 2021, 80, e123. [Google Scholar] [CrossRef]
- Pasoto, S.G.; Adriano de Oliveira Martins, V.; Bonfa, E. Sjögren’s Syndrome and Systemic Lupus Erythematosus: Links and Risks. Open Access Rheumatol. Res. Rev. 2019, 11, 33–45. [Google Scholar] [CrossRef]
- Pan, H.F.; Ye, D.Q.; Wang, Q.; Li, W.X.; Zhang, N.; Li, X.P.; Xu, J.H.; Dai, H. Clinical and Laboratory Profiles of Systemic Lupus Erythematosus Associated with Sjögren Syndrome in China: A Study of 542 Patients. Clin. Rheumatol. 2008, 27, 339–343. [Google Scholar] [CrossRef]
- Lockshin, M.D.; Levine, A.B.; Erkan, D. Patients with Overlap Autoimmune Disease Differ from Those with “pure” Disease. Lupus Sci. Med. 2015, 2, e000084. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, A.; Radfar, L.; Lewis, D.; Grundahl, K.; Stone, D.U.; Kaufman, C.E.; Rhodus, N.L.; Segal, B.; Wallace, D.J.; Weisman, M.H.; et al. Previous Diagnosis of Sjögren’s Syndrome as Rheumatoid Arthritis or Systemic Lupus Erythematosus. Rheumatology 2016, 55, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Kamitaki, N.; Sekar, A.; Handsaker, R.E.; de Rivera, H.; Tooley, K.; Morris, D.L.; Taylor, K.E.; Whelan, C.W.; Tombleson, P.; Loohuis, L.M.O.; et al. Complement Genes Contribute Sex-Biased Vulnerability in Diverse Disorders. Nature 2020, 582, 577–581. [Google Scholar] [CrossRef] [PubMed]
- Soret, P.; Le Dantec, C.; Desvaux, E.; Foulquier, N.; Chassagnol, B.; Hubert, S.; Jamin, C.; Barturen, G.; Desachy, G.; Devauchelle-Pensec, V.; et al. A New Molecular Classification to Drive Precision Treatment Strategies in Primary Sjögren’s Syndrome. Nat. Commun. 2021, 12, 3523. [Google Scholar] [CrossRef] [PubMed]
- Caielli, S.; Wan, Z.; Pascual, V. Systemic Lupus Erythematosus Pathogenesis: Interferon and Beyond. Annu. Rev. Immunol. 2023, 41, 533–560. [Google Scholar] [CrossRef]
- Rincon-Arevalo, H.; Wiedemann, A.; Stefanski, A.-L.; Lettau, M.; Szelinski, F.; Fuchs, S.; Frei, A.P.; Steinberg, M.; Kam-Thong, T.; Hatje, K.; et al. Deep Phenotyping of CD11c+ B Cells in Systemic Autoimmunity and Controls. Front. Immunol. 2021, 12, 635615. [Google Scholar] [CrossRef]
- Guthridge, J.M.; Lu, R.; Tran, L.T.-H.; Arriens, C.; Aberle, T.; Kamp, S.; Munroe, M.E.; Dominguez, N.; Gross, T.; DeJager, W.; et al. Adults with Systemic Lupus Exhibit Distinct Molecular Phenotypes in a Cross-Sectional Study. EClinicalMedicine 2020, 20, 100291. [Google Scholar] [CrossRef]
- Banchereau, R.; Hong, S.; Cantarel, B.; Baldwin, N.; Baisch, J.; Edens, M.; Cepika, A.-M.; Acs, P.; Turner, J.; Anguiano, E.; et al. Personalized Immunomonitoring Uncovers Molecular Networks That Stratify Lupus Patients. Cell 2016, 165, 551–565. [Google Scholar] [CrossRef]
- James, J.A.; Guthridge, J.M.; Chen, H.; Lu, R.; Bourn, R.L.; Bean, K.; Munroe, M.E.; Smith, M.; Chakravarty, E.; Baer, A.N.; et al. Unique Sjögren’s Syndrome Patient Subsets Defined by Molecular Features. Rheumatology 2020, 59, 860–868. [Google Scholar] [CrossRef]
- Teruel, M.; Barturen, G.; Martínez-Bueno, M.; Castellini-Pérez, O.; Barroso-Gil, M.; Povedano, E.; Kerick, M.; Català-Moll, F.; Makowska, Z.; Buttgereit, A.; et al. Integrative Epigenomics in Sjögren’s Syndrome Reveals Novel Pathways and a Strong Interaction between the HLA, Autoantibodies and the Interferon Signature. Sci. Rep. 2021, 11, 23292. [Google Scholar] [CrossRef]
- Barturen, G.; Babaei, S.; Català-Moll, F.; Martínez-Bueno, M.; Makowska, Z.; Martorell-Marugán, J.; Carmona-Sáez, P.; Toro-Domínguez, D.; Carnero-Montoro, E.; Teruel, M.; et al. Integrative Analysis Reveals a Molecular Stratification of Systemic Autoimmune Diseases. Arthritis Rheumatol. 2021, 73, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Foulquier, N.; Le Dantec, C.; Bettacchioli, E.; Jamin, C.; Alarcón-Riquelme, M.E.; Pers, J.-O. Machine Learning for the Identification of a Common Signature for Anti-SSA/Ro 60 Antibody Expression Across Autoimmune Diseases. Arthritis Rheumatol. 2022, 74, 1706–1719. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-F.; Grainge, M.J.; Valdes, A.M.; See, L.-C.; Luo, S.-F.; Yu, K.-H.; Zhang, W.; Doherty, M. Familial Risk of Sjögren’s Syndrome and Co-Aggregation of Autoimmune Diseases in Affected Families: A Nationwide Population Study. Arthritis Rheumatol. 2015, 67, 1904–1912. [Google Scholar] [CrossRef]
- Aggarwal, R.; Anaya, J.-M.; Koelsch, K.A.; Kurien, B.T.; Scofield, R.H. Association between Secondary and Primary Sjögren’s Syndrome in a Large Collection of Lupus Families. Autoimmune Dis. 2015, 2015, 298506. [Google Scholar] [CrossRef]
- Kuo, C.-F.; Grainge, M.J.; Valdes, A.M.; See, L.-C.; Luo, S.-F.; Yu, K.-H.; Zhang, W.; Doherty, M. Familial Aggregation of Systemic Lupus Erythematosus and Coaggregation of Autoimmune Diseases in Affected Families. JAMA Intern. Med. 2015, 175, 1518–1526. [Google Scholar] [CrossRef]
- Hou, G.; Harley, I.T.W.; Lu, X.; Zhou, T.; Xu, N.; Yao, C.; Qin, Y.; Ouyang, Y.; Ma, J.; Zhu, X.; et al. SLE Non-Coding Genetic Risk Variant Determines the Epigenetic Dysfunction of an Immune Cell Specific Enhancer That Controls Disease-Critical MicroRNA Expression. Nat. Commun. 2021, 12, 135. [Google Scholar] [CrossRef]
- Liu, K.; Kurien, B.T.; Zimmerman, S.L.; Kaufman, K.M.; Taft, D.H.; Kottyan, L.C.; Lazaro, S.; Weaver, C.A.; Ice, J.A.; Adler, A.J.; et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Arthritis Rheumatol. 2016, 68, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Barturen, G.; Beretta, L.; Cervera, R.; Van Vollenhoven, R.; Alarcón-Riquelme, M.E. Moving towards a Molecular Taxonomy of Autoimmune Rheumatic Diseases. Nat. Rev. Rheumatol. 2018, 14, 75–93. [Google Scholar] [CrossRef]
- Khatri, B.; Tessneer, K.L.; Rasmussen, A.; Aghakhanian, F.; Reksten, T.R.; Adler, A.; Alevizos, I.; Anaya, J.-M.; Aqrawi, L.A.; Baecklund, E.; et al. Genome-Wide Association Study Identifies Sjögren’s Risk Loci with Functional Implications in Immune and Glandular Cells. Nat. Commun. 2022, 13, 4287. [Google Scholar] [CrossRef]
- Harley, I.T.W.; Allison, K.; Scofield, R.H. Polygenic Autoimmune Disease Risk Alleles Impacting B Cell Tolerance Act in Concert across Shared Molecular Networks in Mouse and in Humans. Front. Immunol. 2022, 13, 953439. [Google Scholar] [CrossRef]
- Mountjoy, E.; Schmidt, E.M.; Carmona, M.; Schwartzentruber, J.; Peat, G.; Miranda, A.; Fumis, L.; Hayhurst, J.; Buniello, A.; Karim, M.A.; et al. An Open Approach to Systematically Prioritize Causal Variants and Genes at All Published Human GWAS Trait-Associated Loci. Nat. Genet. 2021, 53, 1527–1533. [Google Scholar] [CrossRef] [PubMed]
- Ghoussaini, M.; Mountjoy, E.; Carmona, M.; Peat, G.; Schmidt, E.M.; Hercules, A.; Fumis, L.; Miranda, A.; Carvalho-Silva, D.; Buniello, A.; et al. Open Targets Genetics: Systematic Identification of Trait-Associated Genes Using Large-Scale Genetics and Functional Genomics. Nucleic Acids Res. 2021, 49, D1311–D1320. [Google Scholar] [CrossRef] [PubMed]
- Harley, I.T.W. Monogenic SLE (8829bcb2-C7d9-11ec-B397-0ac135e8bacf). Available online: https://www.ndexbio.org/viewer/networks/8829bcb2-c7d9-11ec-b397-0ac135e8bacf (accessed on 6 June 2023).
- Harley, I.T.W. MonogenicPolygenicSLEUnion (Aa310899-C7de-11ec-B397-0ac135e8bacf). Available online: https://www.ndexbio.org/viewer/networks/aa310899-c7de-11ec-b397-0ac135e8bacf (accessed on 6 June 2023).
- Harley, I.T.W. Polygenic SLE (26855444-C7db-11ec-B397-0ac135e8bacf). Available online: https://www.ndexbio.org/viewer/networks/26855444-c7db-11ec-b397-0ac135e8bacf (accessed on 6 June 2023).
- Guo, J.; Gu, M.; Zhang, W.; Liu, Y.; Qian, C.; Deng, A. Aberrant IL-35 Levels in Patients with Primary Sjogren’s Syndrome. Scand. J. Immunol. 2018, 88, e12718. [Google Scholar] [CrossRef] [PubMed]
- Han, M.; Li, Y.; Liu, S.; Jiang, S.; Yuan, L.; Xia, L.; Shen, H.; Lu, J. Elevation of Serum IL-35 in Patients with Primary Sjögren’s Syndrome. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2018, 38, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Fogel, O.; Rivière, E.; Seror, R.; Nocturne, G.; Boudaoud, S.; Ly, B.; Gottenberg, J.-E.; Le Guern, V.; Dubost, J.-J.; Nititham, J.; et al. Role of the IL-12/IL-35 Balance in Patients with Sjögren Syndrome. J. Allergy Clin. Immunol. 2018, 142, 258–268.e5. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Kim, K.; Suetsugu, H.; Bang, S.-Y.; Wen, L.; Koido, M.; Ha, E.; Liu, L.; Sakamoto, Y.; Jo, S.; et al. Meta-Analysis of 208370 East Asians Identifies 113 Susceptibility Loci for Systemic Lupus Erythematosus. Ann. Rheum. Dis. 2021, 80, 632–640. [Google Scholar] [CrossRef]
- Bentham, J.; Morris, D.L.; Graham, D.S.C.; Pinder, C.L.; Tombleson, P.; Behrens, T.W.; Martín, J.; Fairfax, B.P.; Knight, J.C.; Chen, L.; et al. Genetic Association Analyses Implicate Aberrant Regulation of Innate and Adaptive Immunity Genes in the Pathogenesis of Systemic Lupus Erythematosus. Nat. Genet. 2015, 47, 1457–1464. [Google Scholar] [CrossRef]
- Burgess, S.; Thompson, S.G. Interpreting Findings from Mendelian Randomization Using the MR-Egger Method. Eur. J. Epidemiol. 2017, 32, 377–389. [Google Scholar] [CrossRef]
- Sterne, J.A.C.; Sutton, A.J.; Ioannidis, J.P.A.; Terrin, N.; Jones, D.R.; Lau, J.; Carpenter, J.; Rücker, G.; Harbord, R.M.; Schmid, C.H.; et al. Recommendations for Examining and Interpreting Funnel Plot Asymmetry in Meta-Analyses of Randomised Controlled Trials. BMJ 2011, 343, d4002. [Google Scholar] [CrossRef]
- Hemani, G.; Zheng, J.; Elsworth, B.; Wade, K.H.; Haberland, V.; Baird, D.; Laurin, C.; Burgess, S.; Bowden, J.; Langdon, R.; et al. The MR-Base Platform Supports Systematic Causal Inference across the Human Phenome. eLife 2018, 7, e34408. [Google Scholar] [CrossRef]
- Kurki, M.I.; Karjalainen, J.; Palta, P.; Sipilä, T.P.; Kristiansson, K.; Donner, K.M.; Reeve, M.P.; Laivuori, H.; Aavikko, M.; Kaunisto, M.A.; et al. FinnGen Provides Genetic Insights from a Well-Phenotyped Isolated Population. Nature 2023, 613, 508–518. [Google Scholar] [CrossRef]
- Lao, O.; Lu, T.T.; Nothnagel, M.; Junge, O.; Freitag-Wolf, S.; Caliebe, A.; Balascakova, M.; Bertranpetit, J.; Bindoff, L.A.; Comas, D.; et al. Correlation between Genetic and Geographic Structure in Europe. Curr. Biol. 2008, 18, 1241–1248. [Google Scholar] [CrossRef]
- Sherry, S.T.; Ward, M.H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-F.; Zhang, Y.; Lin, Z.; Zhang, H.; Wang, T.-Y.; Cao, Y.; Morris, D.L.; Sheng, Y.; Yin, X.; Zhong, S.-L.; et al. Identification of 38 Novel Loci for Systemic Lupus Erythematosus and Genetic Heterogeneity between Ancestral Groups. Nat. Commun. 2021, 12, 772. [Google Scholar] [CrossRef] [PubMed]
- Bowden, J.; Davey Smith, G.; Burgess, S. Mendelian Randomization with Invalid Instruments: Effect Estimation and Bias Detection through Egger Regression. Int. J. Epidemiol. 2015, 44, 512–525. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Abecasis, G.R.; Altshuler, D.M.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Donnelly, P.; Eichler, E.E.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Machiela, M.J.; Chanock, S.J. LDlink: A Web-Based Application for Exploring Population-Specific Haplotype Structure and Linking Correlated Alleles of Possible Functional Variants. Bioinformatics 2015, 31, 3555–3557. [Google Scholar] [CrossRef]
- Kottyan, L.C.; Zoller, E.E.; Bene, J.; Lu, X.; Kelly, J.A.; Rupert, A.M.; Lessard, C.J.; Vaughn, S.E.; Marion, M.; Weirauch, M.T.; et al. The IRF5-TNPO3 Association with Systemic Lupus Erythematosus Has Two Components That Other Autoimmune Disorders Variably Share. Hum. Mol. Genet. 2015, 24, 582–596. [Google Scholar] [CrossRef]
- Göring, H.H.; Terwilliger, J.D.; Blangero, J. Large Upward Bias in Estimation of Locus-Specific Effects from Genomewide Scans. Am. J. Hum. Genet. 2001, 69, 1357–1369. [Google Scholar] [CrossRef]
- Ioannidis, J.P.A. Why Most Discovered True Associations Are Inflated. Epidemiol. Camb. Mass 2008, 19, 640–648. [Google Scholar] [CrossRef]
- Harley, I.T.W.; Sawalha, A.H. Systemic Lupus Erythematosus as a Genetic Disease. Clin. Immunol. 2022, 236, 108953. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P.F. Tutorial: A Guide to Performing Polygenic Risk Score Analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef] [PubMed]
- Akatsu, C.; Shinagawa, K.; Numoto, N.; Liu, Z.; Ucar, A.K.; Aslam, M.; Phoon, S.; Adachi, T.; Furukawa, K.; Ito, N.; et al. CD72 Negatively Regulates B Lymphocyte Responses to the Lupus-Related Endogenous Toll-like Receptor 7 Ligand Sm/RNP. J. Exp. Med. 2016, 213, 2691–2706. [Google Scholar] [CrossRef] [PubMed]
- McKusick, V.A. On Lumpers and Splitters, or the Nosology of Genetic Disease. Perspect. Biol. Med. 1969, 12, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Eknoyan, G. A History of Edema and Its Management. Kidney Int. Suppl. 1997, 59, S118–S126. [Google Scholar] [PubMed]
- Peitzman, S.J. Framing Disease: Studies in Cultural History. From Bright’s Disease to End-Stage Renal Disease. Hosp. Pract. Off. Ed. 1992, 27, 192, 201–204, 207. [Google Scholar]
- CDC. Diseases Caused by Group A Strep. Available online: https://www.cdc.gov/groupastrep/diseases-public/index.html (accessed on 5 June 2023).
- Entry—#203100—ALBINISM, OCULOCUTANEOUS, TYPE IA; OCA1A–OMIM. Available online: https://omim.org/entry/203100?search=203100&highlight=203100 (accessed on 5 June 2023).
- Entry—#203500—ALKAPTONURIA; AKU–OMIM. Available online: https://omim.org/entry/203500?search=203500&highlight=203500 (accessed on 5 June 2023).
- Entry—#261600—PHENYLKETONURIA; PKU–OMIM. Available online: https://omim.org/entry/261600?search=261600&highlight=261600 (accessed on 5 June 2023).
- Entry—#276700—TYROSINEMIA, TYPE I; TYRSN1–OMIM. Available online: https://omim.org/entry/276700?search=276700&highlight=276700 (accessed on 5 June 2023).
- Rosenzwajg, M.; Lorenzon, R.; Cacoub, P.; Pham, H.P.; Pitoiset, F.; El Soufi, K.; RIbet, C.; Bernard, C.; Aractingi, S.; Banneville, B.; et al. Immunological and Clinical Effects of Low-Dose Interleukin-2 across 11 Autoimmune Diseases in a Single, Open Clinical Trial. Ann. Rheum. Dis. 2019, 78, 209–217. [Google Scholar] [CrossRef]
- Khunsriraksakul, C.; Li, Q.; Markus, H.; Patrick, M.T.; Sauteraud, R.; McGuire, D.; Wang, X.; Wang, C.; Wang, L.; Chen, S.; et al. Multi-Ancestry and Multi-Trait Genome-Wide Association Meta-Analyses Inform Clinical Risk Prediction for Systemic Lupus Erythematosus. Nat. Commun. 2023, 14, 668. [Google Scholar] [CrossRef]
- Shirai, Y.; Nakanishi, Y.; Suzuki, A.; Konaka, H.; Nishikawa, R.; Sonehara, K.; Namba, S.; Tanaka, H.; Masuda, T.; Yaga, M.; et al. Multi-Trait and Cross-Population Genome-Wide Association Studies across Autoimmune and Allergic Diseases Identify Shared and Distinct Genetic Component. Ann. Rheum. Dis. 2022, 81, 1301–1312. [Google Scholar] [CrossRef]
- Demela, P.; Pirastu, N.; Soskic, B. Cross-Disorder Genetic Analysis of Immune Diseases Reveals Distinct Gene Associations That Converge on Common Pathways. Nat. Commun. 2023, 14, 2743. [Google Scholar] [CrossRef]
- Turley, P.; Walters, R.K.; Maghzian, O.; Okbay, A.; Lee, J.J.; Fontana, M.A.; Nguyen-Viet, T.A.; Wedow, R.; Zacher, M.; Furlotte, N.A.; et al. Multi-Trait Analysis of Genome-Wide Association Summary Statistics Using MTAG. Nat. Genet. 2018, 50, 229–237. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, E.H.; Blokland, S.L.M.; Hillen, M.R.; Lopes, A.P.P.; van Vliet-Moret, F.M.; Rosenberg, A.J.W.P.; Janssen, N.G.; Welsing, P.M.J.; Iannizzotto, V.; Tao, W.; et al. Leflunomide–Hydroxychloroquine Combination Therapy in Patients with Primary Sjögren’s Syndrome (RepurpSS-I): A Placebo-Controlled, Double-Blinded, Randomised Clinical Trial. Lancet Rheumatol. 2020, 2, e260–e269. [Google Scholar] [CrossRef]
- Bowman, S.J.; Fox, R.; Dörner, T.; Mariette, X.; Papas, A.; Grader-Beck, T.; Fisher, B.A.; Barcelos, F.; De Vita, S.; Schulze-Koops, H.; et al. Safety and Efficacy of Subcutaneous Ianalumab (VAY736) in Patients with Primary Sjögren’s Syndrome: A Randomised, Double-Blind, Placebo-Controlled, Phase 2b Dose-Finding Trial. Lancet 2022, 399, 161–171. [Google Scholar] [CrossRef]
- He, J.; Chen, J.; Miao, M.; Zhang, R.; Cheng, G.; Wang, Y.; Feng, R.; Huang, B.; Luan, H.; Jia, Y.; et al. Efficacy and Safety of Low-Dose Interleukin 2 for Primary Sjögren Syndrome: A Randomized Clinical Trial. JAMA Netw. Open 2022, 5, e2241451. [Google Scholar] [CrossRef]
- Carsons, S.E.; Vivino, F.B.; Parke, A.; Carteron, N.; Sankar, V.; Brasington, R.; Brennan, M.T.; Ehlers, W.; Fox, R.; Scofield, H.; et al. Treatment Guidelines for Rheumatologic Manifestations of Sjögren’s Syndrome: Use of Biologic Agents, Management of Fatigue, and Inflammatory Musculoskeletal Pain. Arthritis Care Res. 2017, 69, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.N.; Vivino, F.B. Treatment of Sjögren’s Syndrome: Constitutional and Non-Sicca Organ-Based Manifestations—UpToDate. Available online: https://www.uptodate.com/contents/treatment-of-sjogrens-syndrome-constitutional-and-non-sicca-organ-based-manifestations (accessed on 6 June 2023).
- Harley, I.T.W. Repository Contains a Tutorial with Instructions for Synthesizing GWAS Results as a Putative Causal Gene Network. Available online: https://github.com/harleyi/gwas_catalog2polygenic_risk_network (accessed on 6 June 2023).
- Sollis, E.; Mosaku, A.; Abid, A.; Buniello, A.; Cerezo, M.; Gil, L.; Groza, T.; Güneş, O.; Hall, P.; Hayhurst, J.; et al. The NHGRI-EBI GWAS Catalog: Knowledgebase and Deposition Resource. Nucleic Acids Res. 2023, 51, D977–D985. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A Comprehensive Gene Set Enrichment Analysis Web Server 2016 Update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Chen, J.; Bardes, E.E.; Aronow, B.J.; Jegga, A.G. ToppGene Suite for Gene List Enrichment Analysis and Candidate Gene Prioritization. Nucleic Acids Res. 2009, 37, W305–W311. [Google Scholar] [CrossRef]
- Pillich, R.T.; Chen, J.; Churas, C.; Fong, D.; Gyori, B.M.; Ideker, T.; Karis, K.; Liu, S.N.; Ono, K.; Pico, A.; et al. NDEx IQuery: A Multi-Method Network Gene Set Analysis Leveraging the Network Data Exchange. Bioinformatics 2023, 39, btad118. [Google Scholar] [CrossRef]
- Zhao, J.; Ma, J.; Deng, Y.; Kelly, J.A.; Kim, K.; Bang, S.-Y.; Lee, H.-S.; Li, Q.-Z.; Wakeland, E.K.; Qiu, R.; et al. A Missense Variant in NCF1 Is Associated with Susceptibility to Multiple Autoimmune Diseases. Nat. Genet. 2017, 49, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.ebi.ac.uk/gwas/docs (accessed on 17 March 2023).
- Available online: https://genetics-docs.opentargets.org/our-approach/prioritising-causal-genes-at-gwas-loci-l2g (accessed on 17 March 2023).
- Available online: https://genetics.opentargets.org/ (accessed on 17 March 2023).
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Available online: https://string-db.org/ (accessed on 17 March 2023).
- Available online: https://www.youtube.com/c/LarsJuhlJensen (accessed on 17 March 2023).
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Available online: https://cytoscape.org/what_is_cytoscape.html (accessed on 17 March 2023).
- Available online: https://github.com/cytoscape/cytoscape-tutorials/wiki (accessed on 17 March 2023).
- Available online: https://manual.cytoscape.org/en/stable/ (accessed on 17 March 2023).
- Available online: https://systems.crump.ucla.edu/hypergeometric/index.php (accessed on 17 March 2023).
- Sanderson, E.; Glymour, M.M.; Holmes, M.V.; Kang, H.; Morrison, J.; Munafò, M.R.; Palmer, T.; Schooling, C.M.; Wallace, C.; Zhao, Q.; et al. Mendelian Randomization. Nat. Rev. Methods Primer 2022, 2, 6. [Google Scholar] [CrossRef] [PubMed]
- Davey Smith, G. Use of Genetic Markers and Gene-Diet Interactions for Interrogating Population-Level Causal Influences of Diet on Health. Genes Nutr. 2011, 6, 27–43. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.D.; Ebrahim, S. “Mendelian Randomization”: Can Genetic Epidemiology Contribute to Understanding Environmental Determinants of Disease? Int. J. Epidemiol. 2003, 32, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lassi, G.; Taylor, A.E.; Timpson, N.J.; Kenny, P.J.; Mather, R.J.; Eisen, T.; Munafò, M.R. The CHRNA5-A3-B4 Gene Cluster and Smoking: From Discovery to Therapeutics. Trends Neurosci. 2016, 39, 851–861. [Google Scholar] [CrossRef]
- de Leeuw, C.; Savage, J.; Bucur, I.G.; Heskes, T.; Posthuma, D. Understanding the Assumptions Underlying Mendelian Randomization. Eur. J. Hum. Genet. EJHG 2022, 30, 653–660. [Google Scholar] [CrossRef]
- Morrison, J.; Knoblauch, N.; Marcus, J.H.; Stephens, M.; He, X. Mendelian Randomization Accounting for Correlated and Uncorrelated Pleiotropic Effects Using Genome-Wide Summary Statistics. Nat. Genet. 2020, 52, 740–747. [Google Scholar] [CrossRef]
- Burgess, S.; Davey Smith, G.; Davies, N.M.; Dudbridge, F.; Gill, D.; Glymour, M.M.; Hartwig, F.P.; Holmes, M.V.; Minelli, C.; Relton, C.L.; et al. Guidelines for Performing Mendelian Randomization Investigations. Wellcome Open Res. 2019, 4, 186. [Google Scholar] [CrossRef]
- Hartwig, F.P.; Davey Smith, G.; Bowden, J. Robust Inference in Summary Data Mendelian Randomization via the Zero Modal Pleiotropy Assumption. Int. J. Epidemiol. 2017, 46, 1985–1998. [Google Scholar] [CrossRef]
- Bowden, J.; Del Greco, M.F.; Minelli, C.; Zhao, Q.; Lawlor, D.A.; Sheehan, N.A.; Thompson, J.; Davey Smith, G. Improving the Accuracy of Two-Sample Summary-Data Mendelian Randomization: Moving beyond the NOME Assumption. Int. J. Epidemiol. 2019, 48, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Elsworth, B.; Lyon, M.; Alexander, T.; Liu, Y.; Matthews, P.; Hallett, J.; Bates, P.; Palmer, T.; Haberland, V.; Smith, G.D.; et al. The MRC IEU OpenGWAS Data Infrastructure. bioRxiv 2020. [Google Scholar] [CrossRef]
- Hemani, G.; Tilling, K.; Smith, G.D. Orienting the Causal Relationship between Imprecisely Measured Traits Using GWAS Summary Data. PLoS Genet. 2017, 13, e1007081. [Google Scholar] [CrossRef]
- Lessard, C.J.; Li, H.; Adrianto, I.; Ice, J.A.; Rasmussen, A.; Grundahl, K.M.; Kelly, J.A.; Dozmorov, M.G.; Miceli-Richard, C.; Bowman, S.; et al. Variants at Multiple Loci Implicated in Both Innate and Adaptive Immune Responses Are Associated with Sjögren’s Syndrome. Nat. Genet. 2013, 45, 1284–1292. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, K.; Chen, H.; Sun, F.; Xu, J.; Wu, Z.; Li, P.; Zhang, L.; Du, Y.; Luan, H.; et al. A Genome-Wide Association Study in Han Chinese Identifies a Susceptibility Locus for Primary Sjögren’s Syndrome at 7q11.23. Nat. Genet. 2013, 45, 1361–1365. [Google Scholar] [CrossRef]
- Song, I.-W.; Chen, H.-C.; Lin, Y.-F.; Yang, J.-H.; Chang, C.-C.; Chou, C.-T.; Lee, M.-T.M.; Chou, Y.-C.; Chen, C.-H.; Chen, Y.-T.; et al. Identification of Susceptibility Gene Associated with Female Primary Sjögren’s Syndrome in Han Chinese by Genome-Wide Association Study. Hum. Genet. 2016, 135, 1287–1294. [Google Scholar] [CrossRef]
- Qu, S.; Du, Y.; Chang, S.; Guo, L.; Fang, K.; Li, Y.; Zhang, F.; Zhang, K.; Wang, J. Common Variants near IKZF1 Are Associated with Primary Sjögren’s Syndrome in Han Chinese. PLoS ONE 2017, 12, e0177320. [Google Scholar] [CrossRef]
- Sakaue, S.; Kanai, M.; Tanigawa, Y.; Karjalainen, J.; Kurki, M.; Koshiba, S.; Narita, A.; Konuma, T.; Yamamoto, K.; Akiyama, M.; et al. A Cross-Population Atlas of Genetic Associations for 220 Human Phenotypes. Nat. Genet. 2021, 53, 1415–1424. [Google Scholar] [CrossRef]
- Boughton, A.P.; Welch, R.P.; Flickinger, M.; VandeHaar, P.; Taliun, D.; Abecasis, G.R.; Boehnke, M. LocusZoom.Js: Interactive and Embeddable Visualization of Genetic Association Study Results. Bioinformatics 2021, 37, 3017–3018. [Google Scholar] [CrossRef]
- Chepelev, I.; Harley, I.T.W.; Harley, J.B. Modeling of Horizontal Pleiotropy Identifies Possible Causal Gene Expression in Systemic Lupus Erythematosus. Front. Lupus 2023, 1, 1234578. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Network 1 | Nodes 2 | Edges 3 | Degree 4 | Clustering 5 | Exp. Edges 6 | p 7 |
|---|---|---|---|---|---|---|
| Monogenic SLE | 54 | 169 | 6 | 0.65 | 33 | 1.0 × 10−16 |
| Polygenic SLE | 127 | 497 | 8 | 0.44 | 107 | 1.0 × 10−16 |
| Sjögren’s Disease | 23 | 29 | 3 | 0.68 | 4 | 5.5 × 10−16 |
| Network 1 | Exp. Overlap 2 | Fold Enriched 3 | p 4 |
|---|---|---|---|
| Monogenic SLE | 0.06 | 46 | 3.6 × 10−05 |
| Polygenic SLE | 0.15 | 130 | 1.1 × 10−41 |
| All SLE | 0.21 | 96 | 7.6 × 10−39 |
| Locus Overlap | 0.63 | 28 | 1.1 × 10−24 |
| Method 1 | #SNPs 2 | β 3 | se 4 | p 5 |
|---|---|---|---|---|
| Inverse-variance weight | 12 | 0.94 | 0.13 | 1.4 × 10−13 |
| Weighted median | 12 | 0.91 | 0.11 | 2.4 × 10−15 |
| Weighted mode | 12 | 1.15 | 0.12 | 9.7 × 10−07 |
| MR Egger | 12 | 1.18 | 0.27 | 0.0014 |
| Method 1 | #SNPs 2 | β 3 | se 4 | p 5 |
|---|---|---|---|---|
| Inverse-variance weight | 32 | 0.31 | 0.04 | 1.6 × 10−11 |
| Weighted median | 32 | 0.28 | 0.05 | 2.4 × 10−08 |
| Weighted mode | 32 | 0.30 | 0.10 | 0.0034 |
| MR Egger | 32 | 0.554 | 0.08 | 2.7 × 10−07 |
| Method 1 | #SNPs 2 | β 3 | se 4 | p 5 |
|---|---|---|---|---|
| Inverse-variance weight | 4 | 0.55 | 0.19 | 0.004 |
| Weighted median | 4 | 0.38 | 0.05 | 2.5 × 10−12 |
| Weighted mode | 4 | 0.35 | 0.05 | 0.003 |
| MR Egger | 4 | −0.45 | 0.89 | 0.911 |
| Method 1 | #SNPs 2 | β 3 | se 4 | p 5 |
|---|---|---|---|---|
| Inverse-variance weight | 30 | 0.29 | 0.06 | 3.5 × 10−05 |
| Weighted median | 30 | 0.23 | 0.06 | 5.6 × 10−06 |
| Weighted mode | 30 | 0.23 | 0.08 | 0.006 |
| MR Egger | 30 | 0.42 | 0.17 | 0.019 |
| 1st Author [Ref] 1 | Ncase:Nctl 2 | PMID 3 | Other IDs 4 | Note 5 |
|---|---|---|---|---|
| Lessard [104] | 1638:6754 | 24097067 | EUR SjD GWAS | |
| Li [105] | 1845:3757 | 24097066 | EAS SjD GWAS | |
| Khatri [29] | 3851:23652 | 35896530 | phs002723.v1.p1 | EUR SjD GWAS |
| Song [106] | 420:15876 | 27503288 | EAS SjD GWAS | |
| Qu [107] | 665:863 | 28552951 | EAS SjD GWAS | |
| Kurki [44] | 2735:416383 | 36653562 | M13_Sjogren finn-b-M13_SJOGREN | EUR SjD GWAS URL in ST3A |
| Sakaue [108] | 303:175599 | 34594039 | Sjogren_Syndrome | EAS SjD GWAS URL in ST3A |
| Bentham [40] | 5201:9066 | 26502338 | GCST003156 ebi-a-GCST003156 | EUR SLE GWAS |
| Wang [47] | 4222:8431 | 33536424 | GCST90011866 ebi-a-GCST90011866 | EAS SLE GWAS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chau, K.; Raksadawan, Y.; Allison, K.; Ice, J.A.; Scofield, R.H.; Chepelev, I.; Harley, I.T.W. Pervasive Sharing of Causal Genetic Risk Factors Contributes to Clinical and Molecular Overlap between Sjögren’s Disease and Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2023, 24, 14449. https://doi.org/10.3390/ijms241914449
Chau K, Raksadawan Y, Allison K, Ice JA, Scofield RH, Chepelev I, Harley ITW. Pervasive Sharing of Causal Genetic Risk Factors Contributes to Clinical and Molecular Overlap between Sjögren’s Disease and Systemic Lupus Erythematosus. International Journal of Molecular Sciences. 2023; 24(19):14449. https://doi.org/10.3390/ijms241914449
Chicago/Turabian StyleChau, Karen, Yanint Raksadawan, Kristen Allison, John A. Ice, Robert Hal Scofield, Iouri Chepelev, and Isaac T. W. Harley. 2023. "Pervasive Sharing of Causal Genetic Risk Factors Contributes to Clinical and Molecular Overlap between Sjögren’s Disease and Systemic Lupus Erythematosus" International Journal of Molecular Sciences 24, no. 19: 14449. https://doi.org/10.3390/ijms241914449