
Vanadium Pentoxide Exposure Causes Strain-Dependent Changes in Mitochondrial DNA Heteroplasmy, Copy Number, and Lesions, but Not Nuclear DNA Lesions



Abstract
:1. Introduction
2. Results
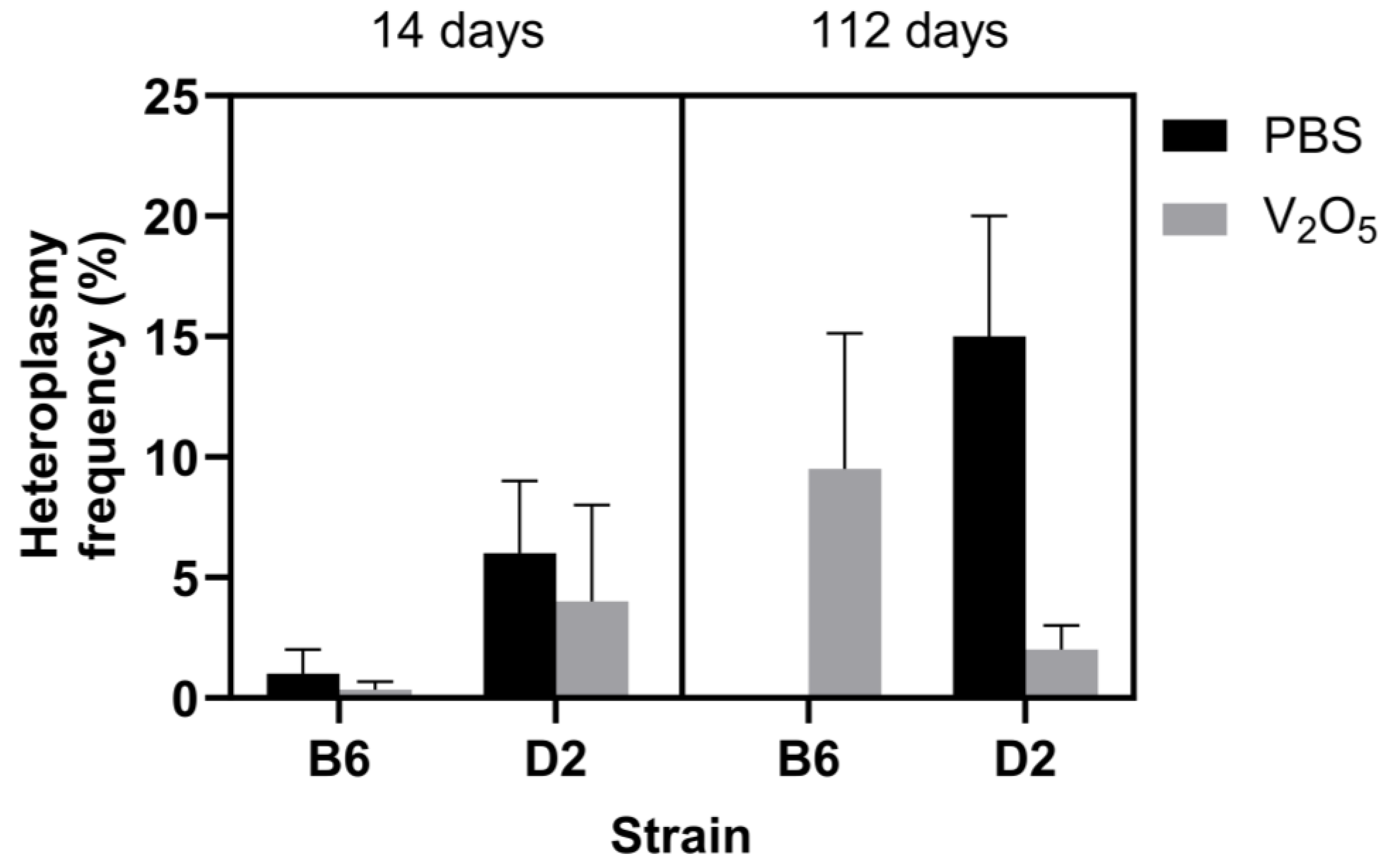
2.1. mtDNA Variants and Heteroplasmy Frequency
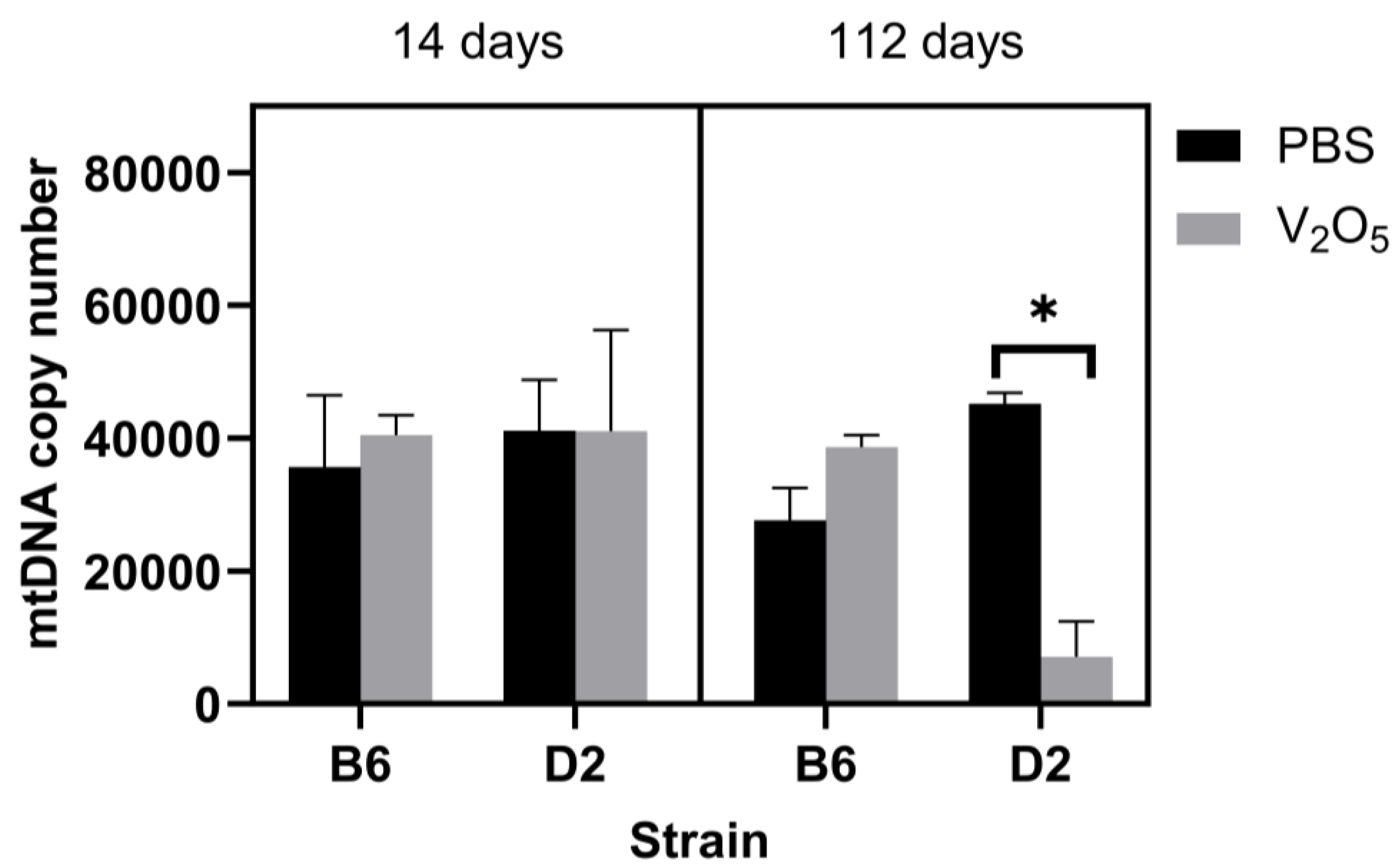
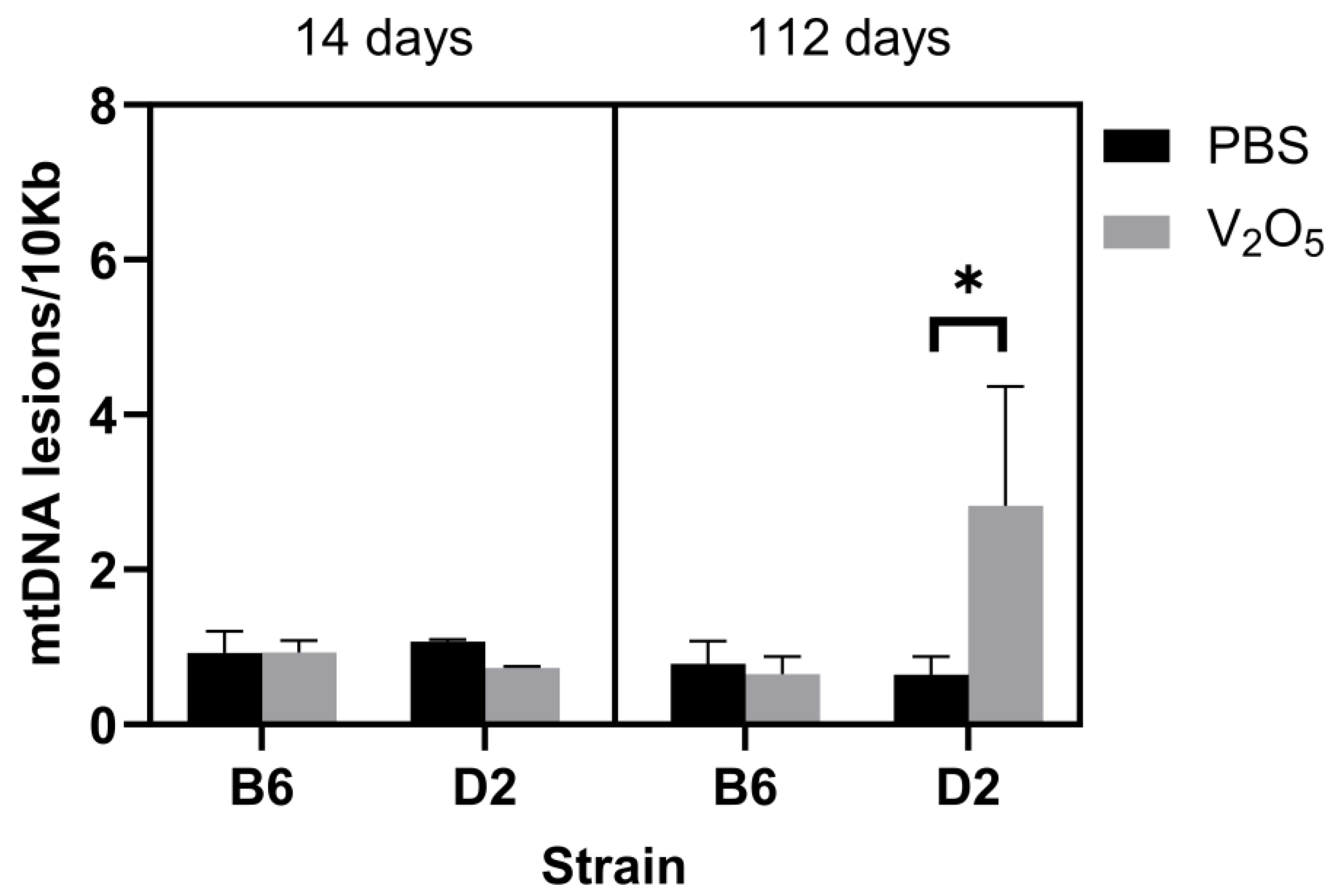
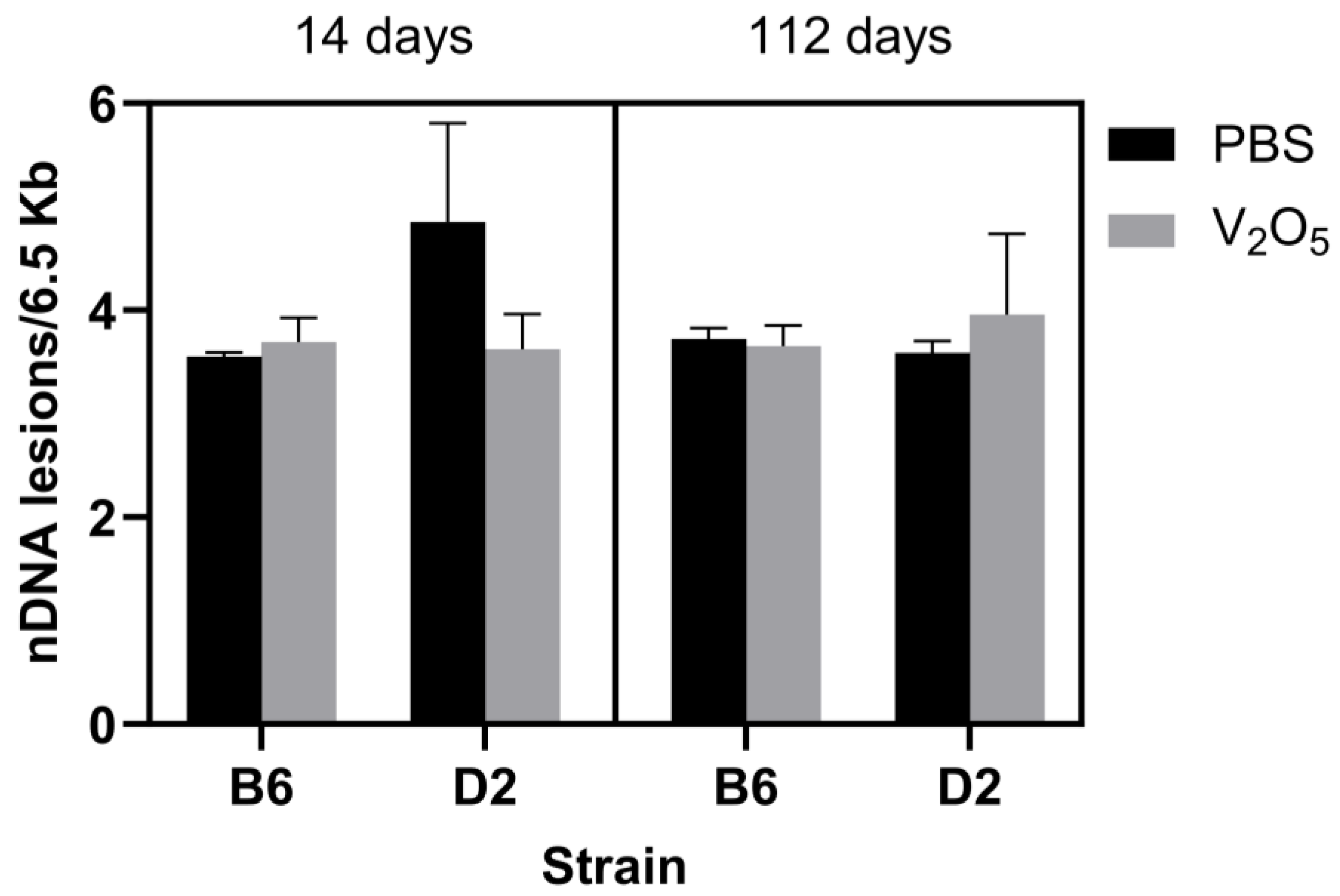
2.2. DNA Damage and mtDNA Copy Numbers
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. V2O5 Exposure
4.3. Mitochondrial Ultra-Deep DNA Sequencing
4.4. Nextera XT Mitochondrial DNA Library Preparation
4.5. Mitochondrial DNA Alignment and Variant Calling
4.6. Identification of Informative Mitochondrial DNA Variants and Heteroplasmy
4.7. Mouse DNA Lesions (Mitochondrial and Nuclear) and Mitochondrial Copy Number Assays
4.8. Statistical Analyses
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Podolanczuk, A.J.; Wong, A.W.; Saito, S.; Lasky, J.A.; Ryerson, C.J.; Eickelberg, O. Update in interstitial lung disease 2020. Am. J. Respir. Crit. Care Med. 2021, 203, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, P.; Kropski, J.A. Genome-Wide Association Studies in Idiopathic Pulmonary Fibrosis: Bridging the Gap between Sequence and Consequence; American Thoracic Society: New York, NY, USA, 2020; Volume 201, pp. 508–509. [Google Scholar]
- Allen, R.J.; Guillen-Guio, B.; Oldham, J.M.; Ma, S.-F.; Dressen, A.; Paynton, M.L.; Kraven, L.M.; Obeidat, M.; Li, X.; Ng, M.; et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2020, 201, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Moss, B.J.; Ryter, S.W.; Rosas, I.O. Pathogenic Mechanisms Underlying Idiopathic Pulmonary Fibrosis. Annu. Rev. Pathol. Mech. Dis. 2022, 17, 515–546. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Diseases of the mitochondrial DNA. Annu. Rev. Biochem. 1992, 61, 1175–1212. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial DNA mutations in diseases of energy metabolism. J. Bioenerg. Biomembr. 1994, 26, 241–250. [Google Scholar] [CrossRef]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Furda, A.M.; Marrangoni, A.M.; Lokshin, A.; Van Houten, B. Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair 2012, 11, 684–692. [Google Scholar] [CrossRef]
- Walters, D.M.; White, K.M.; Patel, U.; Davis, M.J.; Sunkesula, S.R.B.; Bonner, J.C.; Martin, J.R.; Gladwell, W.; Kleeberger, S.R.; Veluci-Marlow, R.M. Genetic susceptibility to interstitial pulmonary fibrosis in mice induced by vanadium pentoxide (V2O5). FASEB J. 2014, 28, 1098–1112. [Google Scholar] [CrossRef]
- Grabowski, G.M.; Paulauskis, J.D.; Godleski, J.J. Mediating phosphorylation events in the vanadium-induced respiratory burst of alveolar macrophages. Toxicol. Appl. Pharmacol. 1999, 156, 170–178. [Google Scholar] [CrossRef]
- Visalli, G.; Bertuccio, M.P.; Picerno, I.; Spataro, P.; Di Pietro, A. Mitochondrial dysfunction by pro-oxidant vanadium: Ex vivo assessment of individual susceptibility. Environ. Toxicol. Pharmacol. 2015, 39, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Medan, D.; Mercer, R.; Overmiller, D.; Leornard, S.; Castranova, V.; Shi, X.; Ding, M.; Huang, C.; Rojanasakul, Y. Vanadium-induced apoptosis and pulmonary inflammation in mice: Role of reactive oxygen species. J. Cell Physiol. 2003, 195, 99–107. [Google Scholar] [CrossRef]
- Wang, Y.-Z.; Ingram, J.L.; Walters, D.M.; Rice, A.B.; Santos, J.H.; Van Houten, B.; Bonner, J.C. Vanadium-induced STAT-1 activation in lung myofibroblasts requires H2O2 and P38 MAP kinase. Free Radic. Biol. Med. 2003, 35, 845–855. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, A.; Mabalirajan, U. Rejuvenating cellular respiration for optimizing respiratory function: Targeting mitochondria. Am. J. Physiol. Lung Cell. Mol. Physiol. 2016, 310, L103–L113. [Google Scholar] [CrossRef] [PubMed]
- Noël, A.; Ashbrook, D.G.; Xu, F.; Cormier, S.A.; Lu, L.; O’callaghan, J.P.; Menon, S.K.; Zhao, W.; Penn, A.L.; Jones, B.C. Genomic Basis for Individual Differences in Susceptibility to the Neurotoxic Effects of Diesel Exhaust. Int. J. Mol. Sci. 2022, 23, 12461. [Google Scholar] [CrossRef]
- Lechuga-Vieco, A.V.; Justo-Mendez, R.; Enriquez, J.A. Not all mitochondrial DNAs are made equal and the nucleus knows it. IUBMB Life 2021, 73, 511–529. [Google Scholar] [CrossRef]
- Wallace, D.C. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: A dawn for evolutionary medicine. Annu. Rev. Genet 2005, 39, 359–407. [Google Scholar] [CrossRef]
- Vellers, H.L.; Cho, H.-Y.; Gladwell, W.; Gerrish, K.; Santos, J.H.; Ofman, G.; Miller-DeGraff, L.; Mahler, T.B.; Kleeberger, S.R. NRF2 Alters Mitochondrial Gene Expression in Neonate Mice Exposed to Hyperoxia. Antioxidants 2022, 11, 760. [Google Scholar] [CrossRef]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Santos, J.H.; Meyer, J.N.; Mandavilli, B.S.; Van Houten, B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2006, 314, 183–199. [Google Scholar] [CrossRef]
- Furda, A.M.; Bess, A.S.; Meyer, J.N.; Van Houten, B. Analysis of DNA damage and repair in nuclear and mitochondrial DNA of animal cells using quantitative PCR. Methods Mol. Biol. 2012, 920, 111–132. [Google Scholar] [CrossRef] [PubMed]
- Vellers, H.L.; Verhein, K.C.; Burkholder, A.B.; Lee, J.; Kim, Y.; Lightfoot, J.T.; Shi, M.; Weinberg, C.R.; Sarzynski, M.A.; Bouchard, C.; et al. Association between mitochondrial DNA sequence variants and VO2 max trainability. Med. Sci. Sports Exerc. 2020, 52, 2303. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Perez-Amado, C.J.; Bazan-Cordoba, A.; Hidalgo-Miranda, A.; Jimenez-Morales, S. Mitochondrial Heteroplasmy Shifting as a Potential Biomarker of Cancer Progression. Int. J. Mol. Sci. 2021, 22, 7369. [Google Scholar] [CrossRef] [PubMed]
- Kopinski, P.K.; Janssen, K.A.; Schaefer, P.M.; Trefely, S.; Perry, C.E.; Potluri, P.; Tintos-Hernandez, J.A.; Singh, L.N.; Karch, K.R.; Campbell, S.L.; et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc. Natl. Acad. Sci. USA 2019, 116, 16028–16035. [Google Scholar] [CrossRef]
- Morin, A.L.; Win, P.W.; Lin, A.Z.; Castellani, C.A. Mitochondrial genomic integrity and the nuclear epigenome in health and disease. Front. Endocrinol. 2022, 13, 1059085. [Google Scholar] [CrossRef]
- Nadalutti, C.A.; Ayala-Peña, S.; Santos, J.H. Mitochondrial DNA damage as driver of cellular outcomes. Am. J. Physiol.-Cell Physiol. 2022, 322, C136–C150. [Google Scholar] [CrossRef]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Hong, Y.S.; Battle, S.L.; Puiu, D.; Shi, W.; Pankratz, N.; Zhao, D.; Arking, D.E.; Guallar, E. Long-Term Air Pollution Exposure and Mitochondrial DNA Copy Number: An Analysis of UK Biobank Data. Environ. Health Perspect. 2023, 131, 57703. [Google Scholar] [CrossRef]
- Hou, L.; Zhang, X.; Dioni, L.; Barretta, F.; Dou, C.; Zheng, Y.; Hoxha, M.; Bertazzi, P.A.; Schwartz, J.; Wu, S.; et al. Inhalable particulate matter and mitochondrial DNA copy number in highly exposed individuals in Beijing, China: A repeated-measure study. Part. Fibre Toxicol. 2013, 10, 17. [Google Scholar] [CrossRef]
- Kleinsasser, N.; Dirschedl, P.; Staudenmaier, R.; Harreus, U.; Wallner, B. Genotoxic effects of vanadium pentoxide on human peripheral lymphocytes and mucosal cells of the upper aerodigestive tract. Int. J. Environ. Health Res. 2003, 13, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, V.A.; Nersesyan, A.K.; Hoelzl, C.; Ferk, F.; Bichler, J.; Valic, E.; Schaffer, A.; Schulte-Hermann, R.; Fenech, M.; Wagner, K.-H.; et al. Inhalative exposure to vanadium pentoxide causes DNA damage in workers: Results of a multiple end point study. Environ. Health Perspect. 2008, 116, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Mercado, J.J.; Mateos-Nava, R.A.; Altamirano-Lozano, M.A. DNA damage induction in human cells exposed to vanadium oxides in vitro. Toxicol. In Vitro 2011, 25, 1996–2002. [Google Scholar] [CrossRef]
- Ivancsits, S.; Pilger, A.; Diem, E.; Schaffer, A.; Rudiger, H.W. Vanadate induces DNA strand breaks in cultured human fibroblasts at doses relevant to occupational exposure. Mutat. Res. 2002, 519, 25–35. [Google Scholar] [CrossRef]
- Zhong, B.Z.; Gu, Z.W.; Wallace, W.E.; Whong, W.Z.; Ong, T. Genotoxicity of vanadium pentoxide in Chinese hamster V79 cells. Mutat. Res. 1994, 321, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Hodgkinson, A.; Grenier, J.C.; Gbeha, E.; Awadalla, P. A haplotype-based normalization technique for the analysis and detection of allele specific expression. BMC Bioinform. 2016, 17, 364. [Google Scholar] [CrossRef] [PubMed]
- Minoche, A.E.; Dohm, J.C.; Himmelbauer, H. Evaluation of genomic high-throughput sequencing data generated on Illumina HiSeq and genome analyzer systems. Genome Biol. 2011, 12, R112. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Exposure | Time Point | Position | Reference Base | Gene Name | Heteroplasmy Frequency |
|---|---|---|---|---|---|---|
| D2 | PBS | 14 | 4405 | A | mt-Nd2 | 8.9% |
| D2 | PBS | 14 | 11,879 | T | mt-Nd5 | 2.8% |
| D2 | V2O5 | 14 | 11,879 | T | mt-Nd5 | 8.1% |
| B6 | PBS | 14 | 13,052 | T | mt-Nd5 | 3.0% |
| B6 | V2O5 | 14 | 14,992 * | C | mt-Cytb | 1.3% |
| D2 | V2O5 | 112 | 1054 | G | mt-Tv | 2.7% |
| D2 | PBS | 112 | 2781 * | G | mt-Nd1 | 10.3% |
| B6 | V2O5 | 112 | 5228 | G | mt-Tc | 7.2% |
| B6 | V2O5 | 112 | 9214 * | T | mt-Co3 | 4.1% |
| B6 | V2O5 | 112 | 9461 | T | mt-Nd3 | 26.0% |
| D2 | PBS | 112 | 11,879 | T | mt-Nd5 | 20.2% |
| B6 | V2O5 | 112 | 13,270 * | A | mt-Nd5 | 1.1% |
| D2 | V2O5 | 112 | 15,446 | A | D-loop | 1.4% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dobson, N.L.; Kleeberger, S.R.; Burkholder, A.B.; Walters, D.M.; Gladwell, W.; Gerrish, K.; Vellers, H.L. Vanadium Pentoxide Exposure Causes Strain-Dependent Changes in Mitochondrial DNA Heteroplasmy, Copy Number, and Lesions, but Not Nuclear DNA Lesions. Int. J. Mol. Sci. 2023, 24, 14507. https://doi.org/10.3390/ijms241914507
Dobson NL, Kleeberger SR, Burkholder AB, Walters DM, Gladwell W, Gerrish K, Vellers HL. Vanadium Pentoxide Exposure Causes Strain-Dependent Changes in Mitochondrial DNA Heteroplasmy, Copy Number, and Lesions, but Not Nuclear DNA Lesions. International Journal of Molecular Sciences. 2023; 24(19):14507. https://doi.org/10.3390/ijms241914507
Chicago/Turabian StyleDobson, Nick L., Steven R. Kleeberger, Adam B. Burkholder, Dianne M. Walters, Wesley Gladwell, Kevin Gerrish, and Heather L. Vellers. 2023. "Vanadium Pentoxide Exposure Causes Strain-Dependent Changes in Mitochondrial DNA Heteroplasmy, Copy Number, and Lesions, but Not Nuclear DNA Lesions" International Journal of Molecular Sciences 24, no. 19: 14507. https://doi.org/10.3390/ijms241914507
APA StyleDobson, N. L., Kleeberger, S. R., Burkholder, A. B., Walters, D. M., Gladwell, W., Gerrish, K., & Vellers, H. L. (2023). Vanadium Pentoxide Exposure Causes Strain-Dependent Changes in Mitochondrial DNA Heteroplasmy, Copy Number, and Lesions, but Not Nuclear DNA Lesions. International Journal of Molecular Sciences, 24(19), 14507. https://doi.org/10.3390/ijms241914507