
NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers


Abstract
:1. Introduction
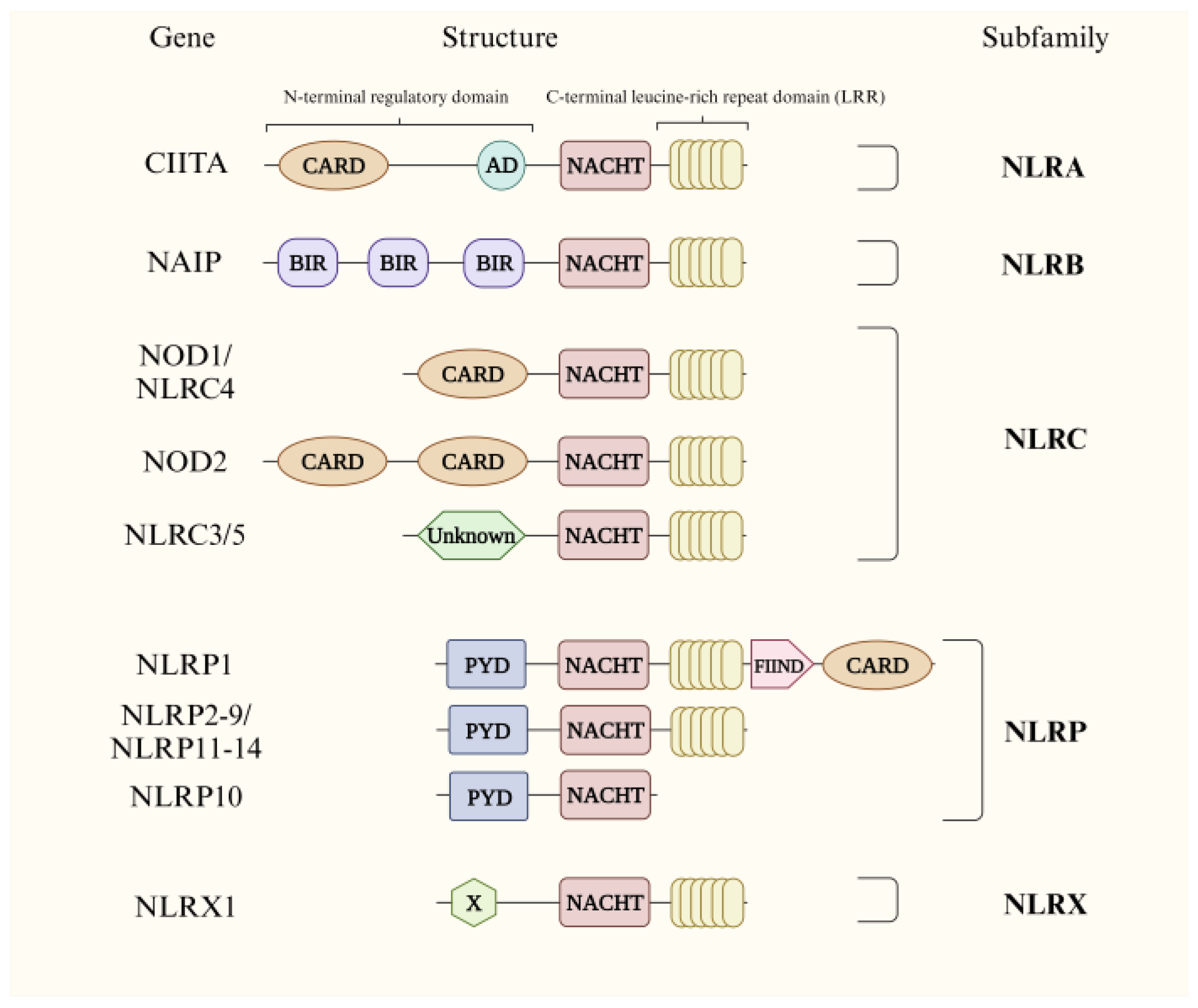
2. The NLR Family and Correlative Signaling Pathways
2.1. NLRA
2.2. NLRB
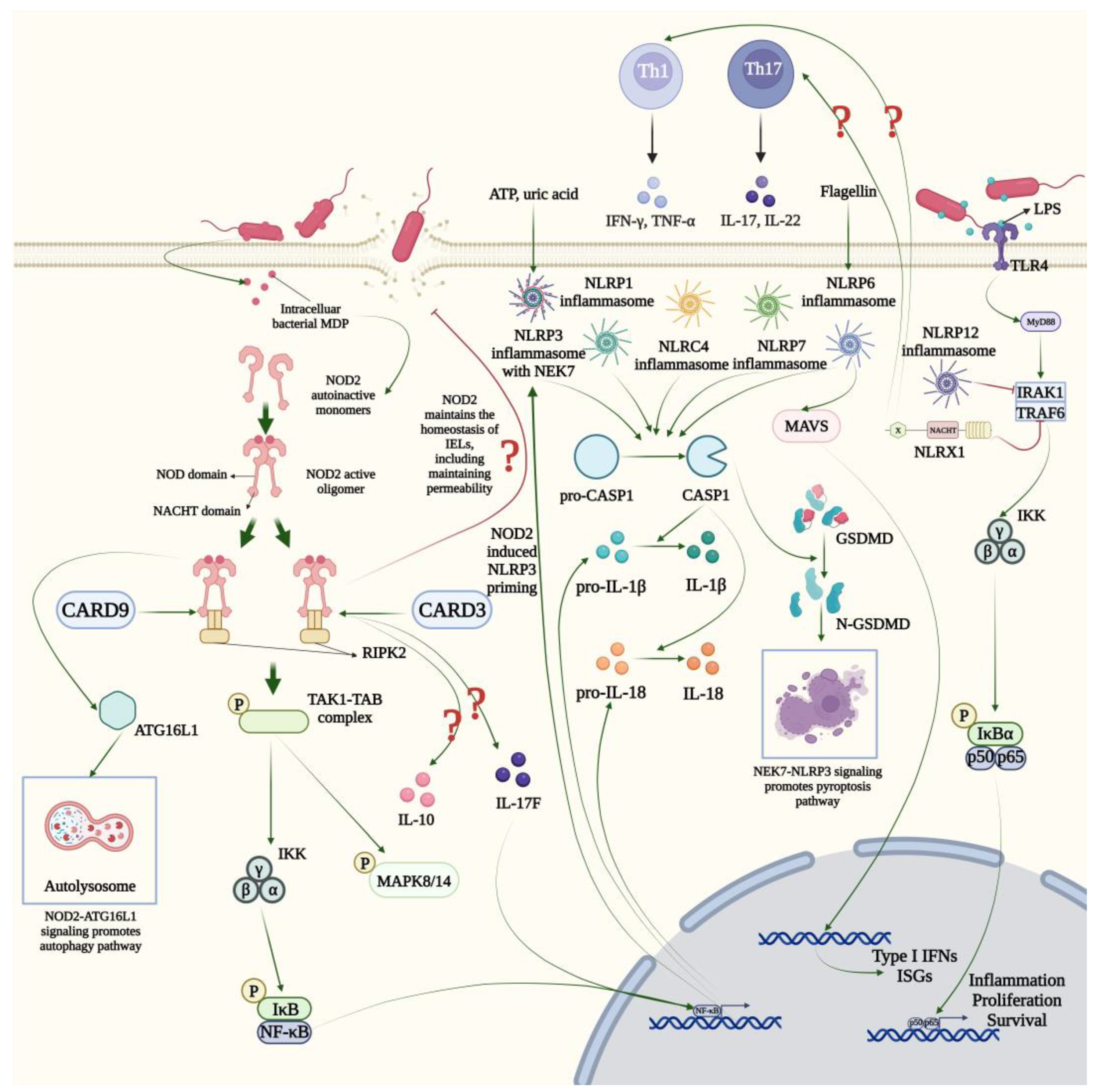
2.3. NLRC
2.4. NLRP
2.5. NLRX
3. NLRs in GI Inflammatory Diseases
3.1. Gastritis
3.2. IBD
3.2.1. Ulcerative Colitis
3.2.2. Crohn’s Disease
3.3. Celiac Disease
4. NLRs in GI Cancers
4.1. Esophageal Cancer
4.2. Gastric Cancer
4.3. Colorectal Cancer
5. Therapies Targeting the NLR Signaling Pathway
5.1. Treatment Strategies in Inflammatory Bowel Disease
5.2. Treatment Strategies in Colorectal Cancer

{kind=link}
{kind=link}
| Targets Related to NLR Signaling Pathways | Disease | Compounds | Authors | Findings | Reference |
|---|---|---|---|---|---|
| NOD2 | IBD 1 | SB203580 | Hollenbach et al. | Inhibiting NOD2/RIPK2/NF-κB signaling pathway in DSS-induced colitis | [165] |
| IBD | GSK583 | Haile et al. | Inhibiting NOD2 signaling in IBD patients | [166] | |
| NLRP3 | IBD | Curcumin | Gong et al. | Inhibiting the activation of NF-κB and NLRP3 inflammasome in DSS-induced colitis | [167] |
| IBD | Apigenin | Márquez-Flores et al. | Inhibiting the activation of CASP1 and NLRP3 inflammasome in DSS-induced colitis | [168] | |
| IBD | Alpinetin | He et al. | Inhibiting the activation of NF-κB and NLRP3 inflammasome in DSS-induced colitis | [169] | |
| IBD | Wogonoside | Sun et al. | Inhibiting the activation of NF-κB and NLRP3 inflammasome in DSS-induced colitis | [171] | |
| IBD | Naringin | Cao et al. | Inhibiting the activation of MAPK and NLRP3 inflammasome in DSS-induced ulcerative colitis | [170] | |
| IBD | Cardamonin | Wang et al. | Inhibiting Nrf2/NQO1 signals and NLRP3 inflammasome in in DSS-induced colitis | [172] | |
| IBD | MCC950 | Wang et al. | Inhibiting the activation of NLRP3 inflammasome in DSS-induced colitis | [189] | |
| CRC 2 | Galloflavin | Guo et al. | Reducing NLRP3 expression and inflammatory factors level, while reducing the expression of c-Myc and p21 | [190] | |
| CRC | Arctigenin | Qiao et al. | Downregulating fatty acid oxidation to inhibit NLRP3 inflammasome assembly in macrophages, which leads to a decrease in IL-1β | [191] | |
| CRC | Caffeic acid phenethyl ester | Dai et al. | Inhibited CRC by inhibiting the production of reactive oxygen species to promote the ubiquitination of NLRP3 | [192] | |
| CRC | Docosahexaenoic acid | Dumont et al. | Inhibiting NLRP3 assembly and JNK-mediated IL-1β secretion to improve 5-FU chemotherapy | [178] | |
| CRC | Andrographolide sulfonate | Xu et al. | Inhibiting the activation of NLRP3 inflammasome in myeloid-derived suppressor cells, followed by the increase in IL-17 produced by CD4+ T cells and reduction in IL-1β to sensitize 5-FU treatment | [177] | |
| CRC | FL118 | Tang et al. | Inhibiting the growth and metastasis of CRC by inducing NLRP3-ASC-CASP1 mediated pyroptosis | [193] | |
| CRC | Atractylenolide I | Qin et al. | Inhibiting NLRP3 inflammasome activation by suppressing Dynamin-related protein 1-mediated mitochondrial fission | [194] | |
| CRC | Fermented quercetin | Lee et al. | Downregulating expression of NLRP3 and phosphorylation of ERK to improve cell sensitivity to 5-FU | [179] | |
| CRC | Ginsenoside Rh3 | Wu et al. | Triggering pyroptotic cell death and ferroptotic cell death in CRC cells via the Stat3/p53/Nrf2 axis | [195] | |
| CRC | Huoxiang Zhengqi | Dong et al. | Regulating the composition of intestinal microbiome and metabolism; activated Nrf2 mediated antioxidant response and inhibited NF-κB mediated NLRP3 activation | [196] | |
| CRC | Oxymatrine | Liang et al. | Reducing mitophagy-activated NLRP3 inflammasome through LRPPPRC inhibition | [197] | |
| NLRP6 | IBD | Apigenin | Radulovic et al. | Regulating NLRP6 signaling pathway in DSS-induced colitis | [173] |
| NLRP1 | IBD | Secoisolariciresinol diglucoside | Wang et al. | Inhibiting NLRP1 inflammasome in DSS-induced colitis | [174] |
| NLRX1 | IBD | NX-13 | Leber et al. | Activating NLRX1 signaling in DSS-induced colitis | [175] |
| NLRC3 | CRC | Dihydromethysticin | Pan et al. | Affecting cell proliferation, migration, invasion, apoptosis, cell cycle and angiogenesis via NLRC3/PI3K pathway to inhibit CRC | [198] |
| CRC | The Green Walnut Husks | Chen et al. | NLRC3/PI3K/AKT pathway to regulate the levels of mTOR, Bcl-2 and Bax to promote apoptosis of tumor cells and inhibit cell proliferation, invasion and migration to prevent CRC progression | [199] |
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Toubi, E.; Vadasz, Z. Innate immune-responses and their role in driving autoimmunity. Autoimmun. Rev. 2019, 18, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef]
- Michallet, M.-C.; Rota, G.; Maslowski, K.; Guarda, G. Innate receptors for adaptive immunity. Curr. Opin. Microbiol. 2013, 16, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, G.G. The global burden of IBD: From 2015 to 2025. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 720–727. [Google Scholar] [CrossRef] [PubMed]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; van Grieken, N.C.; Lordick, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Bentham, A.; Burdett, H.; Anderson, P.A.; Williams, S.J.; Kobe, B. Animal NLRs provide structural insights into plant NLR function. Ann. Bot. 2017, 119, 698–702. [Google Scholar] [CrossRef] [PubMed]
- Meunier, E.; Broz, P. Evolutionary Convergence and Divergence in NLR Function and Structure. Trends Immunol. 2017, 38, 744–757. [Google Scholar] [CrossRef]
- Chen, L.; Cao, S.-Q.; Lin, Z.-M.; He, S.-J.; Zuo, J.-P. NOD-like receptors in autoimmune diseases. Acta Pharmacol. Sin. 2021, 42, 1742–1756. [Google Scholar] [CrossRef]
- Chou, W.-C.; Jha, S.; Linhoff, M.W.; Ting, J.P.Y. The NLR gene family: From discovery to present day. Nat. Rev. Immunol. 2023, 23, 635–654. [Google Scholar] [CrossRef]
- Cressman, D.E.; Chin, K.C.; Taxman, D.J.; Ting, J.P. A defect in the nuclear translocation of CIITA causes a form of type II bare lymphocyte syndrome. Immunity 1999, 10, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Linhoff, M.W.; Harton, J.A.; Cressman, D.E.; Martin, B.K.; Ting, J.P. Two distinct domains within CIITA mediate self-association: Involvement of the GTP-binding and leucine-rich repeat domains. Mol. Cell. Biol. 2001, 21, 3001–3011. [Google Scholar] [CrossRef] [PubMed]
- Sisk, T.J.; Roys, S.; Chang, C.H. Self-association of CIITA and its transactivation potential. Mol. Cell. Biol. 2001, 21, 4919–4928. [Google Scholar] [CrossRef] [PubMed]
- Kretsovali, A.; Spilianakis, C.; Dimakopoulos, A.; Makatounakis, T.; Papamatheakis, J. Self-association of class II transactivator correlates with its intracellular localization and transactivation. J. Biol. Chem. 2001, 276, 32191–32197. [Google Scholar] [CrossRef] [PubMed]
- Tosi, G.; Jabrane-Ferrat, N.; Peterlin, B.M. Phosphorylation of CIITA directs its oligomerization, accumulation and increased activity on MHCII promoters. EMBO J. 2002, 21, 5467–5476. [Google Scholar] [CrossRef]
- Choi, N.M.; Majumder, P.; Boss, J.M. Regulation of major histocompatibility complex class II genes. Curr. Opin. Immunol. 2011, 23, 81–87. [Google Scholar] [CrossRef]
- Masternak, K.; Muhlethaler-Mottet, A.; Villard, J.; Zufferey, M.; Steimle, V.; Reith, W. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000, 14, 1156–1166. [Google Scholar] [CrossRef]
- Reith, W.; LeibundGut-Landmann, S.; Waldburger, J.-M. Regulation of MHC class II gene expression by the class II transactivator. Nat. Rev. Immunol. 2005, 5, 793–806. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Singer, D.S. CIITA and Its Dual Roles in MHC Gene Transcription. Front. Immunol. 2013, 4, 476. [Google Scholar] [CrossRef]
- Tenthorey, J.L.; Haloupek, N.; López-Blanco, J.R.; Grob, P.; Adamson, E.; Hartenian, E.; Lind, N.A.; Bourgeois, N.M.; Chacón, P.; Nogales, E.; et al. The structural basis of flagellin detection by NAIP5: A strategy to limit pathogen immune evasion. Science 2017, 358, 888–893. [Google Scholar] [CrossRef]
- Grandjean, T.; Boucher, A.; Thepaut, M.; Monlezun, L.; Guery, B.; Faudry, E.; Kipnis, E.; Dessein, R. The human NAIP-NLRC4-inflammasome senses the Pseudomonas aeruginosa T3SS inner-rod protein. Int. Immunol. 2017, 29, 377–384. [Google Scholar] [CrossRef]
- Yang, J.; Zhao, Y.; Shi, J.; Shao, F. Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc. Natl. Acad. Sci. USA 2013, 110, 14408–14413. [Google Scholar] [CrossRef]
- Reyes Ruiz, V.M.; Ramirez, J.; Naseer, N.; Palacio, N.M.; Siddarthan, I.J.; Yan, B.M.; Boyer, M.A.; Pensinger, D.A.; Sauer, J.-D.; Shin, S. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 2017, 114, 13242–13247. [Google Scholar] [CrossRef]
- Halff, E.F.; Diebolder, C.A.; Versteeg, M.; Schouten, A.; Brondijk, T.H.C.; Huizinga, E.G. Formation and structure of a NAIP5-NLRC4 inflammasome induced by direct interactions with conserved N- and C-terminal regions of flagellin. J. Biol. Chem. 2012, 287, 38460–38472. [Google Scholar] [CrossRef] [PubMed]
- Davoodi, J.; Ghahremani, M.-H.; Es-Haghi, A.; Mohammad-Gholi, A.; Mackenzie, A. Neuronal apoptosis inhibitory protein, NAIP, is an inhibitor of procaspase-9. Int. J. Biochem. Cell Biol. 2010, 42, 958–964. [Google Scholar] [CrossRef]
- Motta, V.; Soares, F.; Sun, T.; Philpott, D.J. NOD-like receptors: Versatile cytosolic sentinels. Physiol. Rev. 2015, 95, 149–178. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Caruso, R.; Warner, N.; Inohara, N.; Núñez, G. NOD1 and NOD2: Signaling, host defense, and inflammatory disease. Immunity 2014, 41, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-Y.; Omori, E.; Matsumoto, K.; Núñez, G.; Ninomiya-Tsuji, J. TAK1 is a central mediator of NOD2 signaling in epidermal cells. J. Biol. Chem. 2008, 283, 137–144. [Google Scholar] [CrossRef]
- Pashenkov, M.V.; Dagil, Y.A.; Pinegin, B.V. NOD1 and NOD2: Molecular targets in prevention and treatment of infectious diseases. Int. Immunopharmacol. 2018, 54, 385–400. [Google Scholar] [CrossRef]
- Conti, B.J.; Davis, B.K.; Zhang, J.; O’Connor, W.; Williams, K.L.; Ting, J.P.Y. CATERPILLER 16.2 (CLR16.2), a novel NBD/LRR family member that negatively regulates T cell function. J. Biol. Chem. 2005, 280, 18375–18385. [Google Scholar] [CrossRef]
- Guo, L.; Kong, Q.; Dong, Z.; Dong, W.; Fu, X.; Su, L.; Tan, X. NLRC3 promotes host resistance against Pseudomonas aeruginosa-induced keratitis by promoting the degradation of IRAK1. Int. J. Mol. Med. 2017, 40, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Oyama, Y.; Deng, M.; Guo, H.; Wilson, J.E.; Rampanelli, E.; Cook, K.D.; Misumi, I.; Tan, X.; Chen, L.; et al. The Innate Immune Sensor NLRC3 Acts as a Rheostat that Fine-Tunes T Cell Responses in Infection and Autoimmunity. Immunity 2018, 49, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mo, J.; Swanson, K.V.; Wen, H.; Petrucelli, A.; Gregory, S.M.; Zhang, Z.; Schneider, M.; Jiang, Y.; Fitzgerald, K.A.; et al. NLRC3, a member of the NLR family of proteins, is a negative regulator of innate immune signaling induced by the DNA sensor STING. Immunity 2014, 40, 329–341. [Google Scholar] [CrossRef]
- Eren, E.; Berber, M.; Özören, N. NLRC3 protein inhibits inflammation by disrupting NALP3 inflammasome assembly via competition with the adaptor protein ASC for pro-caspase-1 binding. J. Biol. Chem. 2017, 292, 12691–12701. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Man, S.M.; Malireddi, R.K.S.; Kesavardhana, S.; Zhu, Q.; Burton, A.R.; Sharma, B.R.; Qi, X.; Pelletier, S.; Vogel, P.; et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature 2016, 540, 583–587. [Google Scholar] [CrossRef]
- Diebolder, C.A.; Halff, E.F.; Koster, A.J.; Huizinga, E.G.; Koning, R.I. Cryoelectron Tomography of the NAIP5/NLRC4 Inflammasome: Implications for NLR Activation. Structure 2015, 23, 2349–2357. [Google Scholar] [CrossRef]
- Bauer, R.; Rauch, I. The NAIP/NLRC4 inflammasome in infection and pathology. Mol. Asp. Med. 2020, 76, 100863. [Google Scholar] [CrossRef]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- Ball, D.P.; Taabazuing, C.Y.; Griswold, A.R.; Orth, E.L.; Rao, S.D.; Kotliar, I.B.; Vostal, L.E.; Johnson, D.C.; Bachovchin, D.A. Caspase-1 interdomain linker cleavage is required for pyroptosis. Life Sci. Alliance 2020, 3, e202000664. [Google Scholar] [CrossRef]
- Frew, B.C.; Joag, V.R.; Mogridge, J. Proteolytic processing of Nlrp1b is required for inflammasome activity. PLoS Pathog. 2012, 8, e1002659. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Wilson, J.E.; Schneider, M.; Lich, J.D.; Roberts, R.A.; Arthur, J.C.; Woodford, R.-M.T.; Davis, B.K.; Uronis, J.M.; Herfarth, H.H.; et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-κB signaling. Immunity 2012, 36, 742–754. [Google Scholar] [CrossRef]
- Lu, W.L.; Zhang, L.; Song, D.Z.; Yi, X.W.; Xu, W.Z.; Ye, L.; Huang, D.M. NLRP6 suppresses the inflammatory response of human periodontal ligament cells by inhibiting NF-κB and ERK signal pathways. Int. Endod. J. 2019, 52, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Malireddi, R.K.S.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.-D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef]
- Levy, M.; Shapiro, H.; Thaiss, C.A.; Elinav, E. NLRP6: A Multifaceted Innate Immune Sensor. Trends Immunol. 2017, 38, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Silveira, T.N.; Gomes, M.T.R.; Oliveira, L.S.; Campos, P.C.; Machado, G.G.; Oliveira, S.C. NLRP12 negatively regulates proinflammatory cytokine production and host defense against Brucella abortus. Eur. J. Immunol. 2017, 47, 51–59. [Google Scholar] [CrossRef]
- Hornick, E.E.; Banoth, B.; Miller, A.M.; Zacharias, Z.R.; Jain, N.; Wilson, M.E.; Gibson-Corley, K.N.; Legge, K.L.; Bishop, G.A.; Sutterwala, F.S.; et al. Nlrp12 Mediates Adverse Neutrophil Recruitment during Influenza Virus Infection. J. Immunol. 2018, 200, 1188–1197. [Google Scholar] [CrossRef]
- Tsao, Y.-P.; Tseng, F.-Y.; Chao, C.-W.; Chen, M.-H.; Yeh, Y.-C.; Abdulkareem, B.O.; Chen, S.-Y.; Chuang, W.-T.; Chang, P.-C.; Chen, I.C.; et al. NLRP12 is an innate immune checkpoint for repressing IFN signatures and attenuating lupus nephritis progression. J. Clin. Investig. 2023, 133, e157272. [Google Scholar] [CrossRef] [PubMed]
- Pudla, M.; Onsoi, P.; Utaisincharoen, P. NLRP12 attenuates tumor necrosis factor-α production in Burkholderia pseudomallei-infected RAW264.7 macrophages. Asian Pac. J. Allergy Immunol. 2022. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Wang, J.; Deng, H.; Luo, X.; Shen, X.; Huan, Y.; Huang, G.; Ye, H. Meta-analysis identifies candidate key genes in endometrium as predictive biomarkers for clinical pregnancy in IVF. Oncotarget 2017, 8, 102428–102436. [Google Scholar] [CrossRef] [PubMed]
- Fontalba, A.; Gutierrez, O.; Fernandez-Luna, J.L. NLRP2, an inhibitor of the NF-kappaB pathway, is transcriptionally activated by NF-kappaB and exhibits a nonfunctional allelic variant. J. Immunol. 2007, 179, 8519–8524. [Google Scholar] [CrossRef]
- Rossi, M.N.; Pascarella, A.; Licursi, V.; Caiello, I.; Taranta, A.; Rega, L.R.; Levtchenko, E.; Emma, F.; De Benedetti, F.; Prencipe, G. NLRP2 Regulates Proinflammatory and Antiapoptotic Responses in Proximal Tubular Epithelial Cells. Front. Cell Dev. Biol. 2019, 7, 252. [Google Scholar] [CrossRef]
- Tsai, P.-Y.; Chen, K.-R.; Li, Y.-C.; Kuo, P.-L. NLRP7 Is Involved in the Differentiation of the Decidual Macrophages. Int. J. Mol. Sci. 2019, 20, 5994. [Google Scholar] [CrossRef] [PubMed]
- Próchnicki, T.; Vasconcelos, M.B.; Robinson, K.S.; Mangan, M.S.J.; De Graaf, D.; Shkarina, K.; Lovotti, M.; Standke, L.; Kaiser, R.; Stahl, R.; et al. Mitochondrial damage activates the NLRP10 inflammasome. Nat. Immunol. 2023, 24, 595–603. [Google Scholar] [CrossRef]
- Zheng, D.; Mohapatra, G.; Kern, L.; He, Y.; Shmueli, M.D.; Valdés-Mas, R.; Kolodziejczyk, A.A.; Próchnicki, T.; Vasconcelos, M.B.; Schorr, L.; et al. Epithelial Nlrp10 inflammasome mediates protection against intestinal autoinflammation. Nat. Immunol. 2023, 24, 585–594. [Google Scholar] [CrossRef]
- Arnoult, D.; Soares, F.; Tattoli, I.; Castanier, C.; Philpott, D.J.; Girardin, S.E. An N-terminal addressing sequence targets NLRX1 to the mitochondrial matrix. J. Cell Sci. 2009, 122, 3161–3168. [Google Scholar] [CrossRef]
- Tattoli, I.; Carneiro, L.A.; Jéhanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-kappaB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef]
- Peek, R.M.; Crabtree, J.E. Helicobacter infection and gastric neoplasia. J. Pathol. 2006, 208, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Odenbreit, S.; Püls, J.; Sedlmaier, B.; Gerland, E.; Fischer, W.; Haas, R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 2000, 287, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- El-Omar, E.M.; Carrington, M.; Chow, W.H.; McColl, K.E.; Bream, J.H.; Young, H.A.; Herrera, J.; Lissowska, J.; Yuan, C.C.; Rothman, N.; et al. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 2000, 404, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, K.; Choi, I.-J.; Lu, H.; Ogiwara, H.; Graham, D.Y.; Yamaoka, Y. Regulation of IL-18 in Helicobacter pylori infection. J. Immunol. 2008, 180, 1207–1216. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Koch, K.N.; Müller, A. Helicobacter pylori activates the TLR2/NLRP3/caspase-1/IL-18 axis to induce regulatory T-cells, establish persistent infection and promote tolerance to allergens. Gut Microbes 2015, 6, 382–387. [Google Scholar] [CrossRef]
- Ng, G.Z.; Menheniott, T.R.; Every, A.L.; Stent, A.; Judd, L.M.; Chionh, Y.T.; Dhar, P.; Komen, J.C.; Giraud, A.S.; Wang, T.C.; et al. The MUC1 mucin protects against Helicobacter pylori pathogenesis in mice by regulation of the NLRP3 inflammasome. Gut 2016, 65, 1087–1099. [Google Scholar] [CrossRef]
- Semper, R.P.; Vieth, M.; Gerhard, M.; Mejías-Luque, R. Helicobacter pylori Exploits the NLRC4 Inflammasome to Dampen Host Defenses. J. Immunol. 2019, 203, 2183–2193. [Google Scholar] [CrossRef]
- Koch, K.N.; Hartung, M.L.; Urban, S.; Kyburz, A.; Bahlmann, A.S.; Lind, J.; Backert, S.; Taube, C.; Müller, A. Helicobacter urease-induced activation of the TLR2/NLRP3/IL-18 axis protects against asthma. J. Clin. Investig. 2015, 125, 3297–3302. [Google Scholar] [CrossRef]
- Suarez, G.; Romero-Gallo, J.; Piazuelo, M.B.; Sierra, J.C.; Delgado, A.G.; Washington, M.K.; Shah, S.C.; Wilson, K.T.; Peek, R.M. Nod1 Imprints Inflammatory and Carcinogenic Responses toward the Gastric Pathogen Helicobacter pylori. Cancer Res. 2019, 79, 1600–1611. [Google Scholar] [CrossRef]
- Grubman, A.; Kaparakis, M.; Viala, J.; Allison, C.; Badea, L.; Karrar, A.; Boneca, I.G.; Le Bourhis, L.; Reeve, S.; Smith, I.A.; et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell. Microbiol. 2010, 12, 626–639. [Google Scholar] [CrossRef]
- Castaño-Rodríguez, N.; Kaakoush, N.O.; Goh, K.-L.; Fock, K.M.; Mitchell, H.M. The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: A case-control study and gene expression analyses. PLoS ONE 2014, 9, e98899. [Google Scholar] [CrossRef]
- Davari, F.; Shokri-Shirvani, J.; Sepidarkish, M.; Nouri, H.R. Elevated expression of the AIM2 gene in response to Helicobacter pylori along with the decrease of NLRC4 inflammasome is associated with peptic ulcer development. APMIS 2023, 131, 339–350. [Google Scholar] [CrossRef]
- Lewis, J.D.; Parlett, L.E.; Jonsson-Funk, M.L.; Brensinger, C.; Pate, V.; Wu, Q.; Dawwas, G.K.; Weiss, A.; Constant, B.D.; McCauley, M.; et al. Incidence, Prevalence, and Racial and Ethnic Distribution of Inflammatory Bowel Disease in the United States. Gastroenterology 2023. [Google Scholar] [CrossRef] [PubMed]
- Jamontt, J.; Petit, S.; Clark, N.; Parkinson, S.J.; Smith, P. Nucleotide-binding oligomerization domain 2 signaling promotes hyperresponsive macrophages and colitis in IL-10-deficient mice. J. Immunol. 2013, 190, 2948–2958. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Cao, P.; Su, W.; Zhan, N.; Dong, W. Fusobacterium nucleatum facilitates ulcerative colitis through activating IL-17F signaling to NF-κB via the upregulation of CARD3 expression. J. Pathol. 2020, 250, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Natividad, J.M.M.; Petit, V.; Huang, X.; de Palma, G.; Jury, J.; Sanz, Y.; Philpott, D.; Garcia Rodenas, C.L.; McCoy, K.D.; Verdu, E.F. Commensal and probiotic bacteria influence intestinal barrier function and susceptibility to colitis in Nod1−/−; Nod2−/− mice. Inflamm. Bowel Dis. 2012, 18, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Couturier-Maillard, A.; Secher, T.; Rehman, A.; Normand, S.; De Arcangelis, A.; Haesler, R.; Huot, L.; Grandjean, T.; Bressenot, A.; Delanoye-Crespin, A.; et al. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J. Clin. Investig. 2013, 123, 700–711. [Google Scholar] [CrossRef]
- Ranson, N.; Veldhuis, M.; Mitchell, B.; Fanning, S.; Cook, A.L.; Kunde, D.; Eri, R. NLRP3-Dependent and -Independent Processing of Interleukin (IL)-1β in Active Ulcerative Colitis. Int. J. Mol. Sci. 2018, 20, 57. [Google Scholar] [CrossRef]
- Chen, X.; Liu, G.; Yuan, Y.; Wu, G.; Wang, S.; Yuan, L. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019, 10, 906. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Boyd, K.L.; Vogel, P.; Kastan, M.B.; Lamkanfi, M.; Kanneganti, T.-D. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 2010, 32, 379–391. [Google Scholar] [CrossRef] [PubMed]
- Hirota, S.A.; Ng, J.; Lueng, A.; Khajah, M.; Parhar, K.; Li, Y.; Lam, V.; Potentier, M.S.; Ng, K.; Bawa, M.; et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 2011, 17, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Zhang, C.; Xing, Y.; Xue, G.; Zhang, Q.; Pan, F.; Wu, G.; Hu, Y.; Guo, Q.; Lu, A.; et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat. Commun. 2017, 8, 1896. [Google Scholar] [CrossRef]
- Steiner, A.; Reygaerts, T.; Pontillo, A.; Ceccherini, I.; Moecking, J.; Moghaddas, F.; Davidson, S.; Caroli, F.; Grossi, A.; Castro, F.F.M.; et al. Recessive NLRC4-Autoinflammatory Disease Reveals an Ulcerative Colitis Locus. J. Clin. Immunol. 2022, 42, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Tye, H.; Yu, C.-H.; Simms, L.A.; de Zoete, M.R.; Kim, M.L.; Zakrzewski, M.; Penington, J.S.; Harapas, C.R.; Souza-Fonseca-Guimaraes, F.; Wockner, L.F.; et al. NLRP1 restricts butyrate producing commensals to exacerbate inflammatory bowel disease. Nat. Commun. 2018, 9, 3728. [Google Scholar] [CrossRef]
- Chen, L.; Wilson, J.E.; Koenigsknecht, M.J.; Chou, W.-C.; Montgomery, S.A.; Truax, A.D.; Brickey, W.J.; Packey, C.D.; Maharshak, N.; Matsushima, G.K.; et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat. Immunol. 2017, 18, 541–551. [Google Scholar] [CrossRef]
- Onoufriadis, A.; Stone, K.; Katsiamides, A.; Amar, A.; Omar, Y.; de Lange, K.M.; Taylor, K.; Barrett, J.C.; Pollok, R.; Hayee, B.H.; et al. Exome Sequencing and Genotyping Identify a Rare Variant in NLRP7 Gene Associated With Ulcerative Colitis. J. Crohn’s Colitis 2018, 12, 321–326. [Google Scholar] [CrossRef]
- Morrison, H.A.; Trusiano, B.; Rowe, A.J.; Allen, I.C. Negative Regulatory NLRs Mitigate Inflammation via NF-κB Pathway Signaling in Inflammatory Bowel Disease. Biomed. J. 2023, 46, 100616. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Abedi, V.; Bassaganya-Riera, J. NLRX1 Modulates Immunometabolic Mechanisms Controlling the Host-Gut Microbiota Interactions during Inflammatory Bowel Disease. Front. Immunol. 2018, 9, 363. [Google Scholar] [CrossRef]
- Leber, A.; Hontecillas, R.; Tubau-Juni, N.; Zoccoli-Rodriguez, V.; Hulver, M.; McMillan, R.; Eden, K.; Allen, I.C.; Bassaganya-Riera, J. NLRX1 Regulates Effector and Metabolic Functions of CD4+ T Cells. J. Immunol. 2017, 198, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, J.; Li, H.; Li, P.; Xu, C. Serum exosomes derived from Hp-positive gastritis patients inhibit MCP-1 and MIP-1α expression via NLRP12-Notch signaling pathway in intestinal epithelial cells and improve DSS-induced colitis in mice. Int. Immunopharmacol. 2020, 88, 107012. [Google Scholar] [CrossRef] [PubMed]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Ellison, L.K.; Ramjeet, M.; Travassos, L.H.; Jones, N.L.; Girardin, S.E.; Philpott, D.J. The protein ATG16L1 suppresses inflammatory cytokines induced by the intracellular sensors Nod1 and Nod2 in an autophagy-independent manner. Immunity 2013, 39, 858–873. [Google Scholar] [CrossRef] [PubMed]
- Ashton, J.J.; Seaby, E.G.; Beattie, R.M.; Ennis, S. NOD2 in Crohn’s Disease-Unfinished Business. J. Crohn’s Colitis 2023, 17, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Zhen, Y.; Zhang, H. NLRP3 Inflammasome and Inflammatory Bowel Disease. Front. Immunol. 2019, 10, 276. [Google Scholar] [CrossRef]
- Mao, L.; Kitani, A.; Similuk, M.; Oler, A.J.; Albenberg, L.; Kelsen, J.; Aktay, A.; Quezado, M.; Yao, M.; Montgomery-Recht, K.; et al. Loss-of-function CARD8 mutation causes NLRP3 inflammasome activation and Crohn’s disease. J. Clin. Investig. 2018, 128, 1793–1806. [Google Scholar] [CrossRef]
- Gao, S.-J.; Zhang, L.; Lu, W.; Wang, L.; Chen, L.; Zhu, Z.; Zhu, H.-H. Interleukin-18 genetic polymorphisms contribute differentially to the susceptibility to Crohn’s disease. World J. Gastroenterol. 2015, 21, 8711–8722. [Google Scholar] [CrossRef]
- Ranson, N.; Veldhuis, M.; Mitchell, B.; Fanning, S.; Cook, A.L.; Kunde, D.; Eri, R. Nod-Like Receptor Pyrin-Containing Protein 6 (NLRP6) Is Up-regulated in Ileal Crohn’s Disease and Differentially Expressed in Goblet Cells. Cell. Mol. Gastroenterol. Hepatol. 2018, 6, 110. [Google Scholar] [CrossRef]
- Catassi, C.; Verdu, E.F.; Bai, J.C.; Lionetti, E. Coeliac disease. Lancet 2022, 399, 2413–2426. [Google Scholar] [CrossRef]
- Gómez Castro, M.F.; Miculán, E.; Herrera, M.G.; Ruera, C.; Perez, F.; Prieto, E.D.; Barrera, E.; Pantano, S.; Carasi, P.; Chirdo, F.G. p31-43 Gliadin Peptide Forms Oligomers and Induces NLRP3 Inflammasome/Caspase 1- Dependent Mucosal Damage in Small Intestine. Front. Immunol. 2019, 10, 31. [Google Scholar] [CrossRef] [PubMed]
- Pietz, G.; De, R.; Hedberg, M.; Sjöberg, V.; Sandström, O.; Hernell, O.; Hammarström, S.; Hammarström, M.-L. Immunopathology of childhood celiac disease-Key role of intestinal epithelial cells. PLoS ONE 2017, 12, e0185025. [Google Scholar] [CrossRef] [PubMed]
- Morrison, H.A.; Liu, Y.; Eden, K.; Nagai-Singer, M.A.; Wade, P.A.; Allen, I.C. NLRX1 Deficiency Alters the Gut Microbiome and Is Further Exacerbated by Adherence to a Gluten-Free Diet. Front. Immunol. 2022, 13, 882521. [Google Scholar] [CrossRef]
- Morgan, E.; Soerjomataram, I.; Rumgay, H.; Coleman, H.G.; Thrift, A.P.; Vignat, J.; Laversanne, M.; Ferlay, J.; Arnold, M. The Global Landscape of Esophageal Squamous Cell Carcinoma and Esophageal Adenocarcinoma Incidence and Mortality in 2020 and Projections to 2040: New Estimates From GLOBOCAN 2020. Gastroenterology 2022, 163, 649–658. [Google Scholar] [CrossRef]
- Yu, S.; Yin, J.-J.; Miao, J.-X.; Li, S.-G.; Huang, C.-Z.; Huang, N.; Fan, T.-L.; Li, X.-N.; Wang, Y.-H.; Han, S.-N.; et al. Activation of NLRP3 inflammasome promotes the proliferation and migration of esophageal squamous cell carcinoma. Oncol. Rep. 2020, 43, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Liu, Y.; Zhang, Z.; Yang, H.; Dai, N.; Zhang, N.; Sun, W.; Guo, Y.; Kong, J.; Wang, X.; et al. Fusobacterium nucleatum induces MDSCs enrichment via activation the NLRP3 inflammosome in ESCC cells, leading to cisplatin resistance. Ann. Med. 2022, 54, 989–1003. [Google Scholar] [CrossRef]
- Nomoto, D.; Baba, Y.; Liu, Y.; Tsutsuki, H.; Okadome, K.; Harada, K.; Ishimoto, T.; Iwatsuki, M.; Iwagami, S.; Miyamoto, Y.; et al. Fusobacterium nucleatum promotes esophageal squamous cell carcinoma progression via the NOD1/RIPK2/NF-κB pathway. Cancer Lett. 2022, 530, 59–67. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, C.; Zhou, J.; Zhang, H.; Zhang, Y.; Wang, S.; Pu, Y.; Yin, L. Reactive oxygen species-mediated activation of NLRP3 inflammasome associated with pyroptosis in Het-1A cells induced by the co-exposure of nitrosamines. J. Appl. Toxicol. 2022, 42, 1651–1661. [Google Scholar] [CrossRef]
- Allison, C.C.; Kufer, T.A.; Kremmer, E.; Kaparakis, M.; Ferrero, R.L. Helicobacter pylori induces MAPK phosphorylation and AP-1 activation via a NOD1-dependent mechanism. J. Immunol. 2009, 183, 8099–8109. [Google Scholar] [CrossRef]
- Mommersteeg, M.C.; Yu, J.; Peppelenbosch, M.P.; Fuhler, G.M. Genetic host factors in Helicobacter pylori-induced carcinogenesis: Emerging new paradigms. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 42–52. [Google Scholar] [CrossRef]
- Kim, D.-J.; Park, J.-H.; Franchi, L.; Backert, S.; Núñez, G. The Cag pathogenicity island and interaction between TLR2/NOD2 and NLRP3 regulate IL-1β production in Helicobacter pylori infected dendritic cells. Eur. J. Immunol. 2013, 43, 2650–2658. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, C.; Chen, D.; He, X.; Zhao, Y.; Bao, L.; Wang, Q.; Zhou, J.; Xie, Y.H. pylori CagA activates the NLRP3 inflammasome to promote gastric cancer cell migration and invasion. Inflamm. Res. 2022, 71, 141–155. [Google Scholar] [CrossRef]
- Li, X.; Liu, S.; Luo, J.; Liu, A.; Tang, S.; Liu, S.; Yu, M.; Zhang, Y. Helicobacter pylori induces IL-1β and IL-18 production in human monocytic cell line through activation of NLRP3 inflammasome via ROS signaling pathway. Pathog. Dis. 2015, 73, ftu024. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Monack, D.M. Inflammasome adaptors and sensors: Intracellular regulators of infection and inflammation. Nat. Rev. Immunol. 2007, 7, 31–40. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunological and inflammatory functions of the interleukin-1 family. Annu. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Filaly, H.E.; Outlioua, A.; Medyouf, H.; Guessous, F.; Akarid, K. Targeting IL-1β in patients with advanced Helicobacter pylori infection: A potential therapy for gastric cancer. Future Microbiol. 2022, 17, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.; Bhagat, G.; Cui, G.; Takaishi, S.; Kurt-Jones, E.A.; Rickman, B.; Betz, K.S.; Penz-Oesterreicher, M.; Bjorkdahl, O.; Fox, J.G.; et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 2008, 14, 408–419. [Google Scholar] [CrossRef]
- Terme, M.; Ullrich, E.; Aymeric, L.; Meinhardt, K.; Desbois, M.; Delahaye, N.; Viaud, S.; Ryffel, B.; Yagita, H.; Kaplanski, G.; et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res. 2011, 71, 5393–5399. [Google Scholar] [CrossRef]
- Kang, J.S.; Bae, S.Y.; Kim, H.R.; Kim, Y.S.; Kim, D.J.; Cho, B.J.; Yang, H.-K.; Hwang, Y.-I.; Kim, K.J.; Park, H.S.; et al. Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis 2009, 30, 1987–1996. [Google Scholar] [CrossRef]
- Deswaerte, V.; Nguyen, P.; West, A.; Browning, A.F.; Yu, L.; Ruwanpura, S.M.; Balic, J.; Livis, T.; Girard, C.; Preaudet, A.; et al. Inflammasome Adaptor ASC Suppresses Apoptosis of Gastric Cancer Cells by an IL18-Mediated Inflammation-Independent Mechanism. Cancer Res. 2018, 78, 1293–1307. [Google Scholar] [CrossRef]
- Yu, Q.; Shi, H.; Ding, Z.; Wang, Z.; Yao, H.; Lin, R. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation in Helicobacter pylori-associated gastritis by regulating ROS and autophagy. Cell Commun. Signal 2023, 21, 1. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.-D. Inflammasome signaling in colorectal cancer. Transl. Res. 2023, 252, 45–52. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Pandey, A.; Shen, C.; Man, S.M. Inflammasomes in Colitis and Colorectal Cancer: Mechanism of Action and Therapies. Yale J. Biol. Med. 2019, 92, 481–498. [Google Scholar] [PubMed]
- Herrinton, L.J.; Liu, L.; Levin, T.R.; Allison, J.E.; Lewis, J.D.; Velayos, F. Incidence and mortality of colorectal adenocarcinoma in persons with inflammatory bowel disease from 1998 to 2010. Gastroenterology 2012, 143, 382–389. [Google Scholar] [CrossRef]
- Karki, R.; Malireddi, R.K.S.; Zhu, Q.; Kanneganti, T.-D. NLRC3 regulates cellular proliferation and apoptosis to attenuate the development of colorectal cancer. Cell Cycle 2017, 16, 1243–1251. [Google Scholar] [CrossRef]
- Hu, B.; Elinav, E.; Huber, S.; Booth, C.J.; Strowig, T.; Jin, C.; Eisenbarth, S.C.; Flavell, R.A. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 2010, 107, 21635–21640. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Dong, W.; Wang, S.; Zhang, Y.; Liu, T.; Xie, R.; Wang, B.; Cao, H. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. 2018, 9, 5588–5597. [Google Scholar] [CrossRef] [PubMed]
- Cambui, R.A.G.; Fernandes, F.P.; Leal, V.N.C.; Reis, E.C.; de Lima, D.S.; do Espírito Santo, G.F.; Elias, R.M.; Pontillo, A. The Ala134Thr variant in TMEM176B exerts a beneficial role in colorectal cancer prognosis by increasing NLRP3 inflammasome activation. J. Cancer Res. Clin. Oncol. 2023, 149, 3729–3738. [Google Scholar] [CrossRef]
- Tezcan, G.; Garanina, E.E.; Zhuravleva, M.N.; Hamza, S.; Rizvanov, A.A.; Khaiboullina, S.F. Rab GTPase Mediating Regulation of NALP3 in Colorectal Cancer. Molecules 2020, 25, 4834. [Google Scholar] [CrossRef]
- Frühbeck, G.; Mentxaka, A.; Ahechu, P.; Gómez-Ambrosi, J.; Ramírez, B.; Becerril, S.; Rodríguez, A.; Unamuno, X.; Cienfuegos, J.A.; Casado, M.; et al. The Differential Expression of the Inflammasomes in Adipose Tissue and Colon Influences the Development of Colon Cancer in a Context of Obesity by Regulating Intestinal Inflammation. J. Inflamm. Res. 2021, 14, 6431–6446. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Qi, Z.-P.; He, D.-L.; Chen, Z.-H.; Liu, J.-Y.; Wong, M.-W.; Zhang, J.-W.; Xu, E.-P.; Shi, Q.; Cai, S.-L.; et al. NLRP7 deubiquitination by USP10 promotes tumor progression and tumor-associated macrophage polarization in colorectal cancer. J. Exp. Clin. Cancer Res. 2021, 40, 126. [Google Scholar] [CrossRef] [PubMed]
- Zaki, M.H.; Vogel, P.; Malireddi, R.K.S.; Body-Malapel, M.; Anand, P.K.; Bertin, J.; Green, D.R.; Lamkanfi, M.; Kanneganti, T.-D. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 2011, 20, 649–660. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Kwak, Y.-T.; Peng, L.; Hu, S.; Cantarel, B.L.; Lewis, C.M.; Gao, Y.; Mani, R.S.; Kanneganti, T.-D.; Zaki, H. NLRP12 downregulates the Wnt/β-catenin pathway via interaction with STK38 to suppress colorectal cancer. J. Clin. Investig. 2023, e166295. [Google Scholar] [CrossRef]
- Tattoli, I.; Killackey, S.A.; Foerster, E.G.; Molinaro, R.; Maisonneuve, C.; Rahman, M.A.; Winer, S.; Winer, D.A.; Streutker, C.J.; Philpott, D.J.; et al. NLRX1 Acts as an Epithelial-Intrinsic Tumor Suppressor through the Modulation of TNF-Mediated Proliferation. Cell Rep. 2016, 14, 2576–2586. [Google Scholar] [CrossRef]
- Koblansky, A.A.; Truax, A.D.; Liu, R.; Montgomery, S.A.; Ding, S.; Wilson, J.E.; Brickey, W.J.; Mühlbauer, M.; McFadden, R.-M.T.; Hu, P.; et al. The Innate Immune Receptor NLRX1 Functions as a Tumor Suppressor by Reducing Colon Tumorigenesis and Key Tumor-Promoting Signals. Cell Rep. 2016, 14, 2562–2575. [Google Scholar] [CrossRef]
- Zhan, Y.; Seregin, S.S.; Chen, J.; Chen, G.Y. Nod1 Limits Colitis-Associated Tumorigenesis by Regulating IFN-γ Production. J. Immunol. 2016, 196, 5121–5129. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Najmeh, S.; Martel, G.; MacFadden-Murphy, E.; Farias, R.; Savage, P.; Leone, A.; Roussel, L.; Cools-Lartigue, J.; Gowing, S.; et al. Activation of the pattern recognition receptor NOD1 augments colon cancer metastasis. Protein Cell 2020, 11, 187–201. [Google Scholar] [CrossRef]
- Maisonneuve, C.; Tsang, D.K.L.; Foerster, E.G.; Robert, L.M.; Mukherjee, T.; Prescott, D.; Tattoli, I.; Lemire, P.; Winer, D.A.; Winer, S.; et al. Nod1 promotes colorectal carcinogenesis by regulating the immunosuppressive functions of tumor-infiltrating myeloid cells. Cell Rep. 2021, 34, 108677. [Google Scholar] [CrossRef]
- Wei, X.; Ye, J.; Pei, Y.; Wang, C.; Yang, H.; Tian, J.; Si, G.; Ma, Y.; Wang, K.; Liu, G. Extracellular vesicles from colorectal cancer cells promote metastasis via the NOD1 signalling pathway. J. Extracell. Vesicles 2022, 11, e12264. [Google Scholar] [CrossRef]
- Udden, S.M.N.; Peng, L.; Gan, J.-L.; Shelton, J.M.; Malter, J.S.; Hooper, L.V.; Zaki, M.H. NOD2 Suppresses Colorectal Tumorigenesis via Downregulation of the TLR Pathways. Cell Rep. 2017, 19, 2756–2770. [Google Scholar] [CrossRef]
- Williams, T.M.; Leeth, R.A.; Rothschild, D.E.; Coutermarsh-Ott, S.L.; McDaniel, D.K.; Simmons, A.E.; Heid, B.; Cecere, T.E.; Allen, I.C. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J. Immunol. 2015, 194, 3369–3380. [Google Scholar] [CrossRef]
- Allen, I.C.; TeKippe, E.M.; Woodford, R.-M.T.; Uronis, J.M.; Holl, E.K.; Rogers, A.B.; Herfarth, H.H.; Jobin, C.; Ting, J.P.Y. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 2010, 207, 1045–1056. [Google Scholar] [CrossRef]
- Chen, G.Y.; Liu, M.; Wang, F.; Bertin, J.; Núñez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 2011, 186, 7187–7194. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Wei, B.; Lan, T.; Xiao, Y.; Quan, X.; Chen, J.; Zhao, C.; Gao, J. Low NLRP3 expression predicts a better prognosis of colorectal cancer. Biosci. Rep. 2021, 41, BSR20210280. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.; Geng, Y.; Zhao, L.; Li, R.; Zhang, Z.; Li, K.; Liang, R.; Shao, X.; Huang, M.; Zuo, D.; et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett. 2019, 442, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Y.; Liu, X.; Zhang, Y. NLRP3 Inflammasome Activation in MΦs-CRC Crosstalk Promotes Colorectal Cancer Metastasis. Ann. Clin. Lab. Sci. 2022, 52, 571–579. [Google Scholar]
- Shao, X.; Lei, Z.; Zhou, C. NLRP3 Promotes Colorectal Cancer Cell Proliferation and Metastasis via Regulating Epithelial Mesenchymal Transformation. Anticancer Agents Med. Chem. 2020, 20, 820–827. [Google Scholar] [CrossRef]
- Marandi, Y.; Hashemzade, S.; Tayebinia, H.; Karimi, J.; Zamani, A.; Khodadadi, I. NLRP3-inflammasome activation is associated with epithelial-mesenchymal transition and progression of colorectal cancer. Iran. J. Basic Med. Sci. 2021, 24, 483–492. [Google Scholar] [CrossRef]
- Liu, X.; Fang, Y.; Lv, X.; Hu, C.; Chen, G.; Zhang, L.; Jin, B.; Huang, L.; Luo, W.; Liang, G.; et al. Deubiquitinase OTUD6A in macrophages promotes intestinal inflammation and colitis via deubiquitination of NLRP3. Cell Death Differ. 2023, 30, 1457–1471. [Google Scholar] [CrossRef]
- Li, T.; Fu, B.; Zhang, X.; Zhou, Y.; Yang, M.; Cao, M.; Chen, Y.; Tan, Y.; Hu, R. Overproduction of Gastrointestinal 5-HT Promotes Colitis-Associated Colorectal Cancer Progression via Enhancing NLRP3 Inflammasome Activation. Cancer Immunol. Res. 2021, 9, 1008–1023. [Google Scholar] [CrossRef]
- Iida, T.; Hirayama, D.; Minami, N.; Matsuura, M.; Wagatsuma, K.; Kawakami, K.; Nagaishi, K.; Nojima, M.; Ikeuchi, H.; Hirota, S.; et al. Down-regulation of RalGTPase-Activating Protein Promotes Colitis-Associated Cancer via NLRP3 Inflammasome Activation. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ding, C.-L.; Wu, M.-X.; Guo, W.; Hu, R.; Liu, Y.; Qi, Z.-T.; Jia, X.-M. RAI16 maintains intestinal homeostasis and inhibits NLRP3-dependent IL-18/CXCL16-induced colitis and the progression of colitis-associated colorectal cancer. Clin. Transl. Med. 2022, 12, e993. [Google Scholar] [CrossRef] [PubMed]
- Fabbi, M.; Carbotti, G.; Ferrini, S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J. Leukoc. Biol. 2015, 97, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, H.; Wang, X.; Zhu, X. The association of aberrant expression of NLRP3 and p-S6K1 in colorectal cancer. Pathol. Res. Pract. 2020, 216, 152737. [Google Scholar] [CrossRef]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Son, H.J.; Sohn, S.H.; Kim, N.; Lee, H.-N.; Lee, S.M.; Nam, R.H.; Park, J.H.; Song, C.-H.; Shin, E.; Na, H.Y.; et al. Effect of Estradiol in an Azoxymethane/Dextran Sulfate Sodium-Treated Mouse Model of Colorectal Cancer: Implication for Sex Difference in Colorectal Cancer Development. Cancer Res. Treat. 2019, 51, 632–648. [Google Scholar] [CrossRef]
- Fan, W.; Gao, X.; Ding, C.; Lv, Y.; Shen, T.; Ma, G.; Yan, L.; Song, S. Estrogen receptors participate in carcinogenesis signaling pathways by directly regulating NOD-like receptors. Biochem. Biophys. Res. Commun. 2019, 511, 468–475. [Google Scholar] [CrossRef]
- Liu, S.; Fan, W.; Gao, X.; Huang, K.; Ding, C.; Ma, G.; Yan, L.; Song, S. Estrogen receptor alpha regulates the Wnt/β-catenin signaling pathway in colon cancer by targeting the NOD-like receptors. Cell. Signal. 2019, 61, 86–92. [Google Scholar] [CrossRef]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, Y.; Yin, Y.; Chen, Y.; Chen, Q.; Bing, Z.; Zheng, Y.; Hou, Y.; Shen, S.; Chen, Y.; et al. Candida tropicalis induces NLRP3 inflammasome activation via glycogen metabolism-dependent glycolysis and JAK-STAT1 signaling pathway in myeloid-derived suppressor cells to promote colorectal carcinogenesis. Int. Immunopharmacol. 2022, 113, 109430. [Google Scholar] [CrossRef]
- Wang, X.; Jia, Y.; Wen, L.; Mu, W.; Wu, X.; Liu, T.; Liu, X.; Fang, J.; Luan, Y.; Chen, P.; et al. Porphyromonas gingivalis Promotes Colorectal Carcinoma by Activating the Hematopoietic NLRP3 Inflammasome. Cancer Res. 2021, 81, 2745–2759. [Google Scholar] [CrossRef]
- Fortoul, M.C.; Kim, E.; Ardeljan, A.D.; Frankel, L.; Takabe, K.; Rashid, O.M. The Role of Hemophilus influenzae Infection and Its Relationship With Colorectal Cancer. World J. Oncol. 2023, 14, 188–194. [Google Scholar] [CrossRef]
- Hollenbach, E.; Neumann, M.; Vieth, M.; Roessner, A.; Malfertheiner, P.; Naumann, M. Inhibition of p38 MAP kinase- and RICK/NF-kappaB-signaling suppresses inflammatory bowel disease. FASEB J. 2004, 18, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Haile, P.A.; Casillas, L.N.; Bury, M.J.; Mehlmann, J.F.; Singhaus, R., Jr.; Charnley, A.K.; Hughes, T.V.; DeMartino, M.P.; Wang, G.Z.; Romano, J.J.; et al. Identification of Quinoline-Based RIP2 Kinase Inhibitors with an Improved Therapeutic Index to the hERG Ion Channel. ACS Med. Chem. Lett. 2018, 9, 1039–1044. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Zhao, S.; Zhou, J.; Yan, J.; Wang, L.; Du, X.; Li, H.; Chen, Y.; Cai, W.; Wu, J. Curcumin alleviates DSS-induced colitis via inhibiting NLRP3 inflammsome activation and IL-1β production. Mol. Immunol. 2018, 104, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Flores, Y.K.; Villegas, I.; Cárdeno, A.; Rosillo, M.; Alarcón-de-la-Lastra, C. Apigenin supplementation protects the development of dextran sulfate sodium-induced murine experimental colitis by inhibiting canonical and non-canonical inflammasome signaling pathways. J. Nutr. Biochem. 2016, 30, 143–152. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Wei, Z.; Wang, J.; Kou, J.; Liu, W.; Fu, Y.; Yang, Z. Alpinetin attenuates inflammatory responses by suppressing TLR4 and NLRP3 signaling pathways in DSS-induced acute colitis. Sci. Rep. 2016, 6, 28370. [Google Scholar] [CrossRef]
- Cao, H.; Liu, J.; Shen, P.; Cai, J.; Han, Y.; Zhu, K.; Fu, Y.; Zhang, N.; Zhang, Z.; Cao, Y. Protective Effect of Naringin on DSS-Induced Ulcerative Colitis in Mice. J. Agric. Food Chem. 2018, 66, 13133–13140. [Google Scholar] [CrossRef]
- Sun, Y.; Zhao, Y.; Yao, J.; Zhao, L.; Wu, Z.; Wang, Y.; Pan, D.; Miao, H.; Guo, Q.; Lu, N. Wogonoside protects against dextran sulfate sodium-induced experimental colitis in mice by inhibiting NF-κB and NLRP3 inflammasome activation. Biochem. Pharmacol. 2015, 94, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Lv, Q.; Miao, Y.M.; Qiao, S.M.; Dai, Y.; Wei, Z.F. Cardamonin, a natural flavone, alleviates inflammatory bowel disease by the inhibition of NLRP3 inflammasome activation via an AhR/Nrf2/NQO1 pathway. Biochem. Pharmacol. 2018, 155, 494–509. [Google Scholar] [CrossRef] [PubMed]
- Radulovic, K.; Normand, S.; Rehman, A.; Delanoye-Crespin, A.; Chatagnon, J.; Delacre, M.; Waldschmitt, N.; Poulin, L.F.; Iovanna, J.; Ryffel, B.; et al. A dietary flavone confers communicable protection against colitis through NLRP6 signaling independently of inflammasome activation. Mucosal Immunol. 2018, 11, 811–819. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, T.; Yang, C.; Bao, T.; Yang, X.; He, F.; Zhang, Y.; Zhu, L.; Chen, H.; Rong, S.; et al. Secoisolariciresinol diglucoside suppresses Dextran sulfate sodium salt-induced colitis through inhibiting NLRP1 inflammasome. Int. Immunopharmacol. 2020, 78, 105931. [Google Scholar] [CrossRef] [PubMed]
- Leber, A.; Hontecillas, R.; Zoccoli-Rodriguez, V.; Bienert, C.; Chauhan, J.; Bassaganya-Riera, J. Activation of NLRX1 by NX-13 Alleviates Inflammatory Bowel Disease through Immunometabolic Mechanisms in CD4+ T Cells. J. Immunol. 2019, 203, 3407–3415. [Google Scholar] [CrossRef]
- Bruchard, M.; Mignot, G.; Derangere, V.; Chalmin, F.; Chevriaux, A.; Vegran, F.; Boireau, W.; Simon, B.; Ryffel, B.; Connat, J.L.; et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat. Med. 2013, 19, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Cai, P.; Li, X.; Wu, X.; Gao, J.; Liu, W.; Yang, J.; Xu, Q.; Guo, W.; Gu, Y. Inhibition of NLRP3 inflammasome activation in myeloid-derived suppressor cells by andrographolide sulfonate contributes to 5-FU sensitization in mice. Toxicol. Appl. Pharmacol. 2021, 428, 115672. [Google Scholar] [CrossRef]
- Dumont, A.; de Rosny, C.; Kieu, T.-L.-V.; Perrey, S.; Berger, H.; Fluckiger, A.; Muller, T.; Pais de Barros, J.-P.; Pichon, L.; Hichami, A.; et al. Docosahexaenoic acid inhibits both NLRP3 inflammasome assembly and JNK-mediated mature IL-1β secretion in 5-fluorouracil-treated MDSC: Implication in cancer treatment. Cell Death Dis. 2019, 10, 485. [Google Scholar] [CrossRef]
- Lee, K.-C.; Wu, K.-L.; Yen, C.-K.; Chang, S.-F.; Chen, C.-N.; Lu, Y.-C. Inhibition of NLRP3 by Fermented Quercetin Decreases Resistin-Induced Chemoresistance to 5-Fluorouracil in Human Colorectal Cancer Cells. Pharmaceuticals 2022, 15, 798. [Google Scholar] [CrossRef]
- Weng, W.; Goel, A. Curcumin and colorectal cancer: An update and current perspective on this natural medicine. Semin. Cancer Biol. 2022, 80, 73–86. [Google Scholar] [CrossRef]
- Hasanzadeh, S.; Read, M.I.; Bland, A.R.; Majeed, M.; Jamialahmadi, T.; Sahebkar, A. Curcumin: An inflammasome silencer. Pharmacol. Res. 2020, 159, 104921. [Google Scholar] [CrossRef]
- Goel, A.; Aggarwal, B.B. Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr. Cancer 2010, 62, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Sun, S.; Zhou, Y.; Wang, H.; Yu, Y.; Hu, T.; Yao, Y.; Zhou, C. Bacteroides fragilis restricts colitis-associated cancer via negative regulation of the NLRP3 axis. Cancer Lett. 2021, 523, 170–181. [Google Scholar] [CrossRef]
- Kerry, R.G.; Patra, J.K.; Gouda, S.; Park, Y.; Shin, H.-S.; Das, G. Benefaction of probiotics for human health: A review. J. Food Drug Anal. 2018, 26, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Xu, C.; Ge, Q.; Lin, Y.; Wong, C.C.; Qi, Y.; Ye, B.; Lian, Q.; Zhuo, W.; Si, J.; et al. A. Muciniphila Suppresses Colorectal Tumorigenesis by Inducing TLR2/NLRP3-Mediated M1-Like TAMs. Cancer Immunol. Res. 2021, 9, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Chung, I.C.; OuYang, C.-N.; Yuan, S.-N.; Lin, H.-C.; Huang, K.-Y.; Wu, P.-S.; Liu, C.-Y.; Tsai, K.-J.; Loi, L.-K.; Chen, Y.-J.; et al. Pretreatment with a Heat-Killed Probiotic Modulates the NLRP3 Inflammasome and Attenuates Colitis-Associated Colorectal Cancer in Mice. Nutrients 2019, 11, 516. [Google Scholar] [CrossRef]
- Li, J.; Qu, C.; Li, F.; Chen, Y.; Zheng, J.; Xiao, Y.; Jin, Q.; Jin, G.; Huang, X.; Jin, D. Inonotus obliquus Polysaccharide Ameliorates Azoxymethane/Dextran Sulfate Sodium-Induced Colitis-Associated Cancer in Mice via Activation of the NLRP3 Inflammasome. Front. Pharmacol. 2020, 11, 621835. [Google Scholar] [CrossRef]
- Gao, J.; Wang, L.; Jiang, J.; Xu, Q.; Zeng, N.; Lu, B.; Yuan, P.; Sun, K.; Zhou, H.; He, X. A probiotic bi-functional peptidoglycan hydrolase sheds NOD2 ligands to regulate gut homeostasis in female mice. Nat. Commun. 2023, 14, 3338. [Google Scholar] [CrossRef]
- Wang, S.-L.; Zhang, M.-M.; Zhou, H.; Su, G.-Q.; Ding, Y.; Xu, G.-H.; Wang, X.; Li, C.-F.; Huang, W.-F.; Yi, L.-T. Inhibition of NLRP3 attenuates sodium dextran sulfate-induced inflammatory bowel disease through gut microbiota regulation. Biomed. J. 2023, 46, 100580. [Google Scholar] [CrossRef]
- Guo, L.; Yang, Y.; Sheng, Y.; Wang, J.; Li, W.; Zhou, X.; Ruan, S.; Han, C. Galloflavin Relieves the Malignant Behavior of Colorectal Cancer Cells in the Inflammatory Tumor Microenvironment. Front. Pharmacol. 2021, 12, 752118. [Google Scholar] [CrossRef]
- Qiao, S.; Lv, C.; Tao, Y.; Miao, Y.; Zhu, Y.; Zhang, W.; Sun, D.; Yun, X.; Xia, Y.; Wei, Z.; et al. Arctigenin disrupts NLRP3 inflammasome assembly in colonic macrophages via downregulating fatty acid oxidation to prevent colitis-associated cancer. Cancer Lett. 2020, 491, 162–179. [Google Scholar] [CrossRef] [PubMed]
- Dai, G.; Jiang, Z.; Sun, B.; Liu, C.; Meng, Q.; Ding, K.; Jing, W.; Ju, W. Caffeic Acid Phenethyl Ester Prevents Colitis-Associated Cancer by Inhibiting NLRP3 Inflammasome. Front. Oncol. 2020, 10, 721. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.; Ji, L.; Han, M.; Xie, J.; Zhong, F.; Zhang, X.; Su, Q.; Yang, Z.; Liu, Z.; Gao, H.; et al. Pyroptosis is involved in the inhibitory effect of FL118 on growth and metastasis in colorectal cancer. Life Sci. 2020, 257, 118065. [Google Scholar] [CrossRef]
- Qin, Y.; Yu, Y.; Yang, C.; Wang, Z.; Yang, Y.; Wang, C.; Zheng, Q.; Li, D.; Xu, W. Atractylenolide I Inhibits NLRP3 Inflammasome Activation in Colitis-Associated Colorectal Cancer via Suppressing Drp1-Mediated Mitochondrial Fission. Front. Pharmacol. 2021, 12, 674340. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Pi, D.; Zhou, S.; Yi, Z.; Dong, Y.; Wang, W.; Ye, H.; Chen, Y.; Zuo, Q.; Ouyang, M. Ginsenoside Rh3 induces pyroptosis and ferroptosis through the Stat3/p53/NRF2 axis in colorectal cancer cells. Acta Biochim. Biophys. Sin. 2023, 55, 587–600. [Google Scholar] [CrossRef]
- Dong, M.; Liu, H.; Cao, T.; Li, L.; Sun, Z.; Qiu, Y.; Wang, D. Huoxiang Zhengqi alleviates azoxymethane/dextran sulfate sodium-induced colitis-associated cancer by regulating Nrf2/NF-κB/NLRP3 signaling. Front. Pharmacol. 2022, 13, 1002269. [Google Scholar] [CrossRef]
- Liang, L.; Sun, W.; Wei, X.; Wang, L.; Ruan, H.; Zhang, J.; Li, S.; Zhao, B.; Li, M.; Cai, Z.; et al. Oxymatrine suppresses colorectal cancer progression by inhibiting NLRP3 inflammasome activation through mitophagy induction in vitro and in vivo. Phytother. Res. 2023, 37, 3342–3362. [Google Scholar] [CrossRef]
- Pan, H.; Liu, F.; Wang, J.; Zhao, M.; Wang, D.; Jia, C.; Wang, T.; Chen, Z.; Fan, Y.; Liang, D.; et al. Dihydromethysticin, a natural molecule from Kava, suppresses the growth of colorectal cancer via the NLRC3/PI3K pathway. Mol. Carcinog. 2020, 59, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; An, N.; Pang, D.; Cheng, Y.; Chen, Y.; Feng, X.; Lei, H.; He, W.; Yang, B.; Zhang, Y.; et al. The Green Walnut Husks Induces Apoptosis of Colorectal Cancer through Regulating NLRC3/PI3K Pathway. Curr. Pharm. Des. 2023, 29, 940–946. [Google Scholar] [CrossRef]
- Liu, R.; Truax, A.D.; Chen, L.; Hu, P.; Li, Z.; Chen, J.; Song, C.; Chen, L.; Ting, J.P. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: Correlation with cancer stages and inflammasome components. Oncotarget 2015, 6, 33456–33469. [Google Scholar] [CrossRef]
| NLR Signaling Pathways | Disease | Authors | Findings | Reference |
|---|---|---|---|---|
| NOD2 | UC 1 | Jamontt et al. | NOD2 signaling in IL-10-deficient mice enhanced ulcerative colitis | [76] |
| UC | Chen et al. | NOD2 activated NF-κB and IF-17F pathways via CARD3 after Fusobacterium nucleatum infection associated with ulcerative colitis | [77] | |
| UC | Natividad et al. | NOD2 was associated with paracellular permeability and susceptibility in DSS-induced colitis | [78] | |
| UC | Couturier-Maillard et al. | NOD2 changed change the microbial microenvironment to promote the risk of ulcerative colitis | [79] | |
| CD 2 | Jostins et al. | NOD2 gene polymorphism has been implicated in the etiology of Crohn’s disease | [93] | |
| CD | Franke et al. | The risk of developing Crohn’s disease has been linked to genes regulating components in the NOD2 signaling pathway | [94] | |
| CD | Ashton et al. | Polymorphism of NOD2 gene variants in Crohn’s disease led to decreased levels of NF-κB activation | [96] | |
| NLRP3 | UC | Ranson et al. | The expression of NLRP3 was upregulated in ulcerative colitis | [80] |
| UC | Chen et al. | NEK7 interacted with NLRP3 to impact DSS-induced colitis via pyroptosis | [81] | |
| UC | Hirota et al. | Anti-inflammatory cytokines IL-10 and TGF-β decreased in the colon of Nlrp3−/− mice | [83] | |
| UC | Yao et al. | Anomalously activated NLRP3 inflammasome remodeled the gut microbiota to resist the colitis | [84] | |
| CD | Zhen et al. | Loss-of-function in NLRP3 was closely associated with the development of Crohn’s disease | [97] | |
| CD | Mao et al. | Mutated CARD8 gene activated NLRP3 inflammasome to affect Crohn’s disease | [98] | |
| NLRC4 | UC | Steiner et al. | A gene variant of NLRC4, NLRC4 (A160T), showed a higher risk of developing ulcerative colitis | [85] |
| NLRP1 | UC | Tye et al. | NLRP1 upregulated IL-18 and IFN-γ in ulcerative colitis | [86] |
| NLRP12 | UC | Chen et al. | NLRP12 gene expression diminished in active ulcerative colitis | [87] |
| NLRP7 | UC | Onoufriadis et al. | A gene variant of NLRP7, NLRP7 (p.S361L), showed a higher risk of developing ulcerative colitis | [88] |
| NLRP6 | CD | Ranson et al. | The expression of NLRP6 increased with Crohn’s disease activity | [100] |
| NLRX1 | UC | Leber et al. | Loss of NLRX1 led to higher sensitivity to DSS-induced colitis | [90] |
| UC | Leber et al. | A greater number of Th17 and Th1 cells with higher proliferative capacity appeared in the colon of DSS-treated Nlrx1−/− mice | [91] |
| NLR Signaling Pathways | Authors | Findings | Reference |
|---|---|---|---|
| NLRC3 | Karki et al. | NLRC3 acted as an inhibitory sensor of PI3K-mTOR, mediating protection against colorectal cancer | [36] |
| Karki et al. | NLRC3 attenuated the development of colorectal cancer by suppressing c-Myc expression and activation of PI3K-AKT targets FoxO3a and FoxO1, which regulate cellular proliferation | [127] | |
| NLRC4 | Hu et al. | CASP1, mediated by the NLRC4 inflammasome, prevented colon inflammation-induced tumors by regulating the proliferation and apoptosis of colon epithelial cells | [128] |
| NLRP3 | Liu et al. | Deoxycholic acid disrupted the intestinal mucosa barrier and induced intestinal low-grade inflammation, which promoted intestinal tumorigenesis | [129] |
| Cambui et al. | rs2072443 variant in TMEM176B improved the prognosis of colorectal cancer | [130] | |
| Tezcan et al. | Rab5 enhanced the activation of NLRP3, while Rab7 and Rab11 played a role in enhancing the expression of NLRP3 gene | [131] | |
| NLRP6 | Frühbeck et al. | Downregulation of NLRP6 and IL-18 in the colon of colon cancer patients in the context of obesity led to reduced intestinal barrier integrity, triggering a vicious cycle of inflammatory cascades under the action of adipose tissue | [132] |
| NLRP7 | Li et al. | NLRP7 deubiquitination by USP10 promoted tumor progression and tumor-associated macrophage polarization in colorectal cancer | [133] |
| NLRP12 | Zaki et al. | NLRP12 negatively regulated NF-κB and ERK signaling in macrophages to inhibit pro-inflammatory cytokines and chemokines, thereby inhibiting colorectal tumorigenesis | [134] |
| Allen et al. | NLPR12 acted as a negative regulator of non-canonical NF-κB pathway, accompanied by activation of ERK and induction of NIK-dependent gene | [46] | |
| Kanneganti et al. | NLRP12 involved in the regulation of the Wnt/β-catenin pathway, inhibiting colorectal tumors by promoting β-catenin degradation | [135] | |
| NLRX1 | Tattoli et al. | NLRX1 played a role in colorectal cancer prevention by inhibiting TNF-induced intestinal epithelial cell proliferation | [136] |
| Koblansky et al. | NLRX1 acted as a tumor suppressor by attenuating Apcmin/+ colon tumorigenesis, cellular proliferation, NF-κB, MAPK, STAT3 activation and IL-6 levels | [137] | |
| NOD1 | Zhan et al. | NOD1 limited colitis-associated tumorigenesis by regulating IFN-γ production | [138] |
| Jiang et al. | NOD1 increased colon cancer cell adhesion, migration and metastasis through the p38 MAPK pathway | [139] | |
| Maisonneuve et al. | Myeloid-intrinsic NOD1 expression maintained intra-tumor arginase-1 levels to foster an immunosuppressor and tumor permissive microenvironment that promotes tumor development | [140] | |
| Wei et al. | CDC42 promoted liver metastasis of colorectal cancer cells by activating NOD1 in macrophages | [141] | |
| NOD2 | Udden et al. | NOD2 inhibited colorectal cancer by downregulating TLR-mediated NF-κB and MAPK pathways | [142] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, Y.; Yu, S.; Zhang, W. NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers. Int. J. Mol. Sci. 2023, 24, 14511. https://doi.org/10.3390/ijms241914511
Zhou Y, Yu S, Zhang W. NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers. International Journal of Molecular Sciences. 2023; 24(19):14511. https://doi.org/10.3390/ijms241914511
Chicago/Turabian StyleZhou, Yujie, Songyan Yu, and Wenyong Zhang. 2023. "NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers" International Journal of Molecular Sciences 24, no. 19: 14511. https://doi.org/10.3390/ijms241914511
APA StyleZhou, Y., Yu, S., & Zhang, W. (2023). NOD-like Receptor Signaling Pathway in Gastrointestinal Inflammatory Diseases and Cancers. International Journal of Molecular Sciences, 24(19), 14511. https://doi.org/10.3390/ijms241914511