
CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
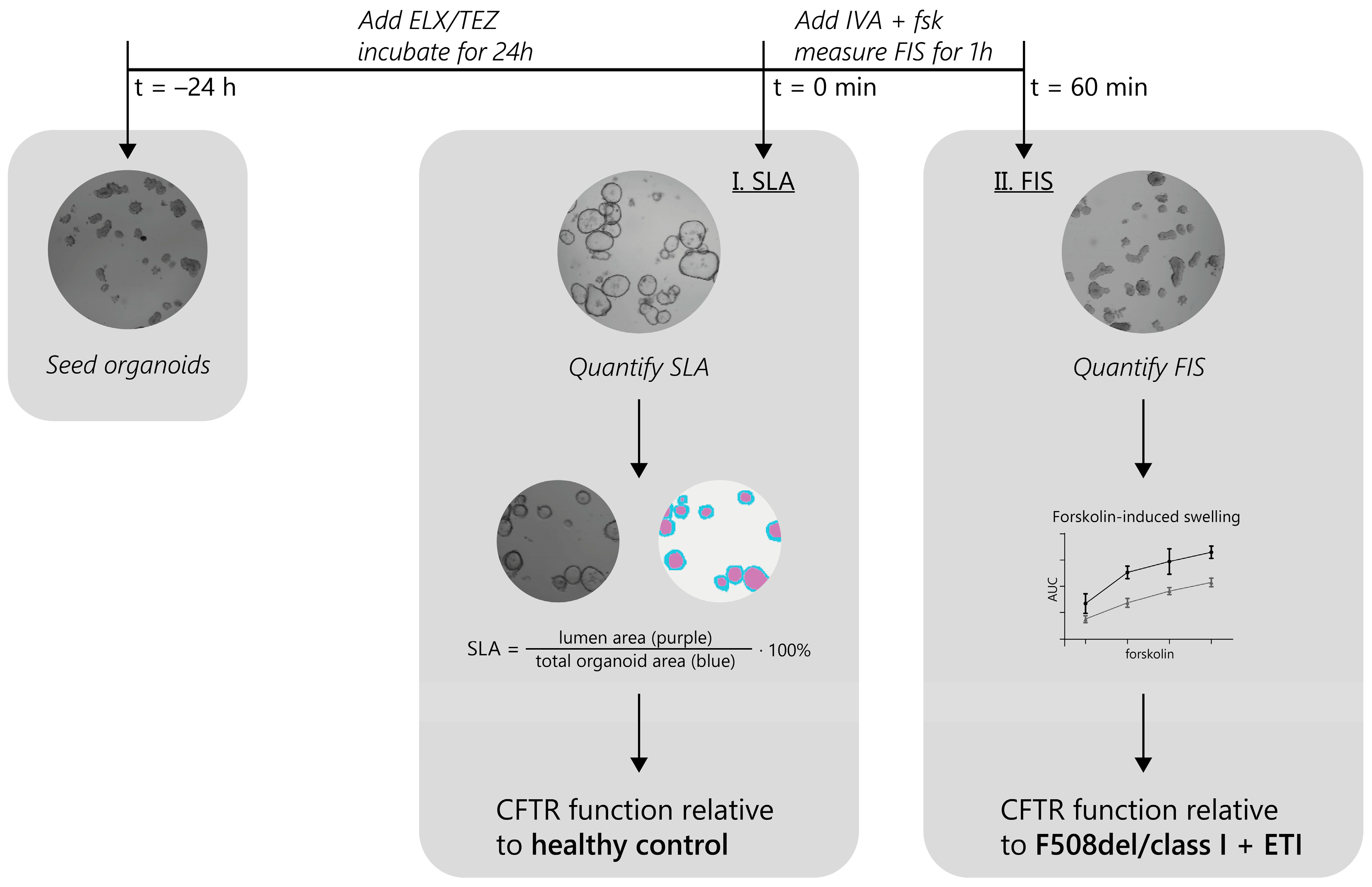
2.1. Experimental Approach for the Identification of ETI-Responsive CFTR Genotypes in PDIOs
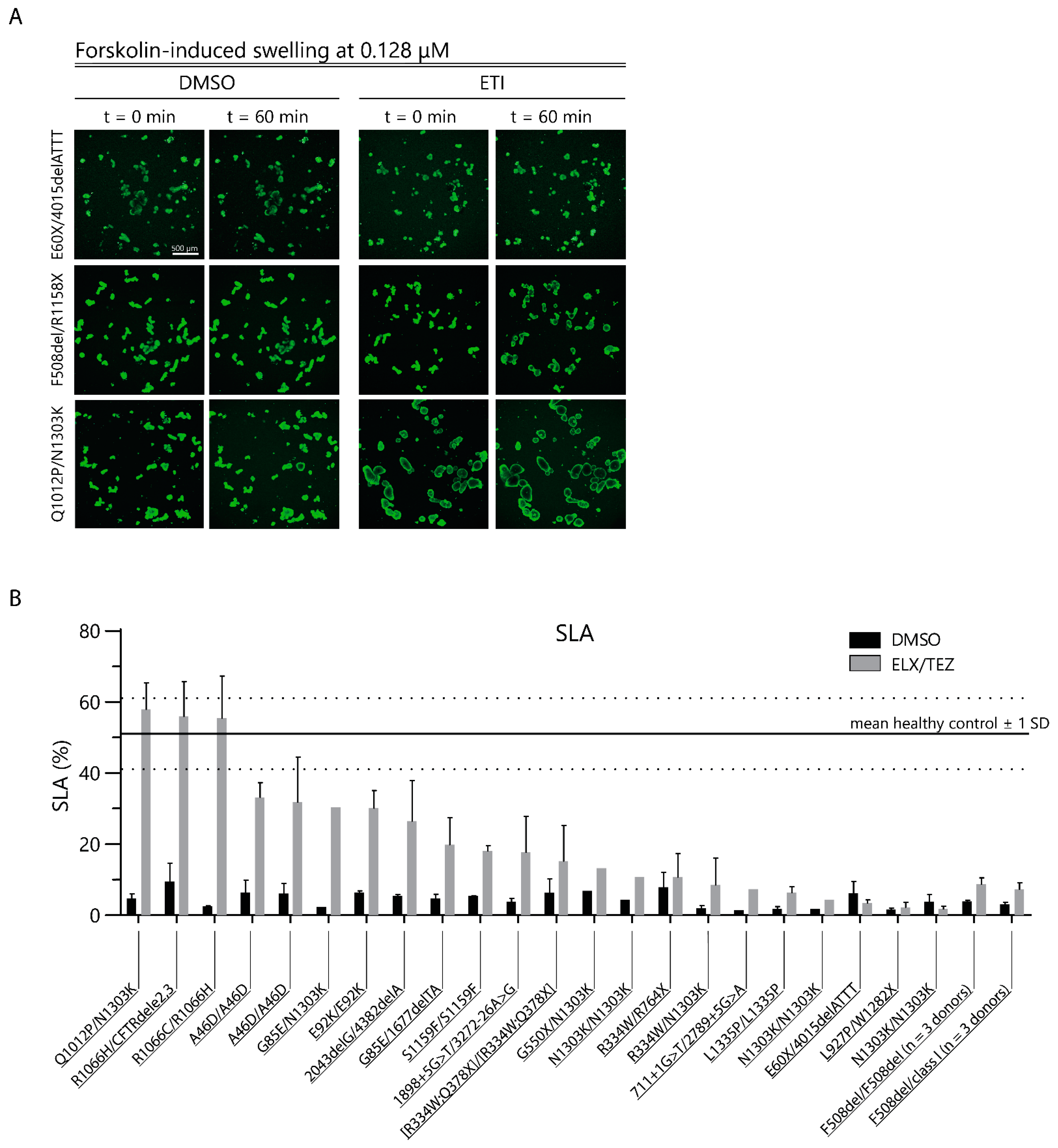
2.2. CFTR Modulator Effects in Rare Variant CF PDIOs in Relation to Wild-Type CFTR Function
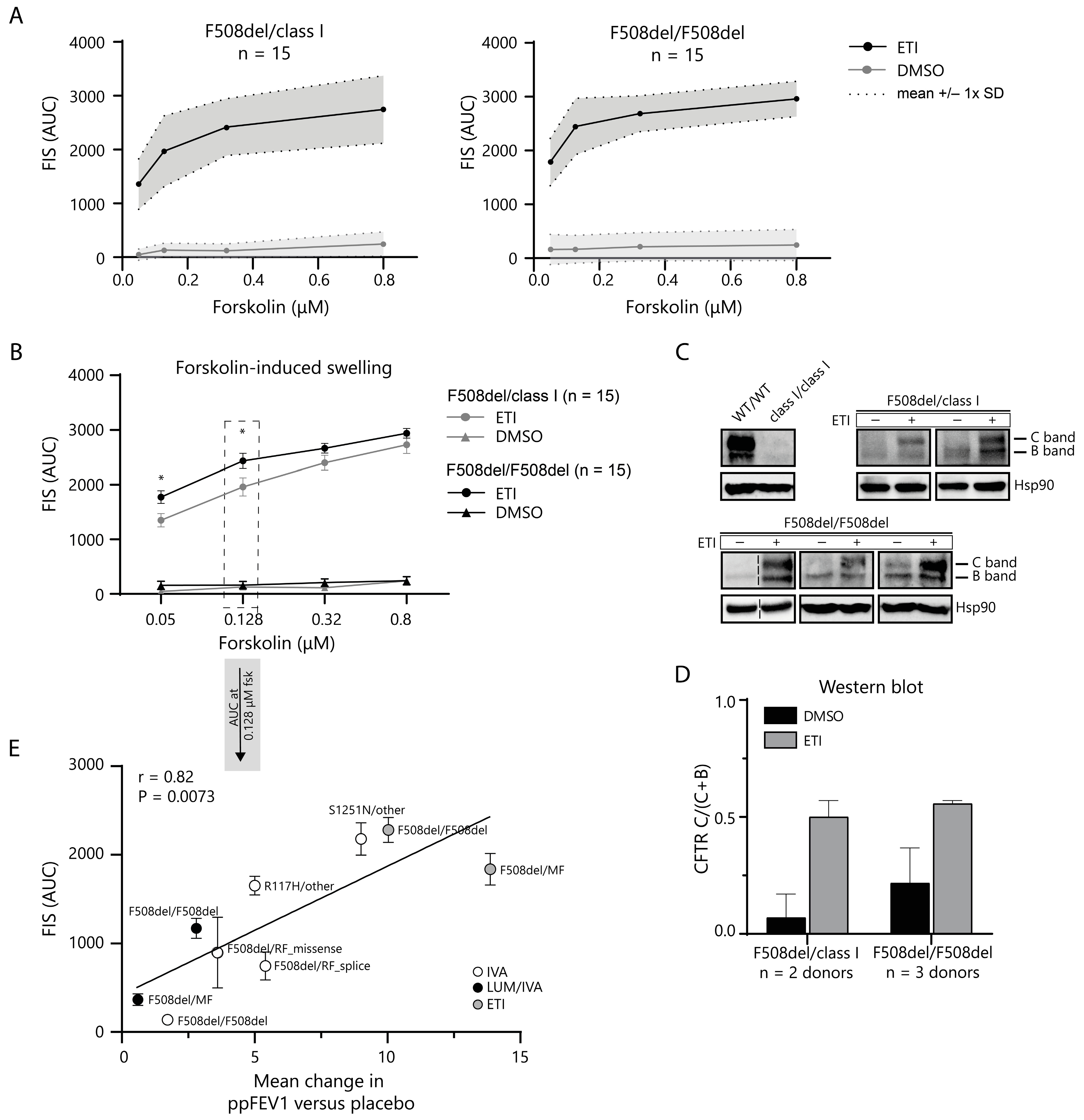
2.3. In Vitro CFTR Rescue by ETI Modulator Therapy Correlates with In Vivo Change in ppFEV1
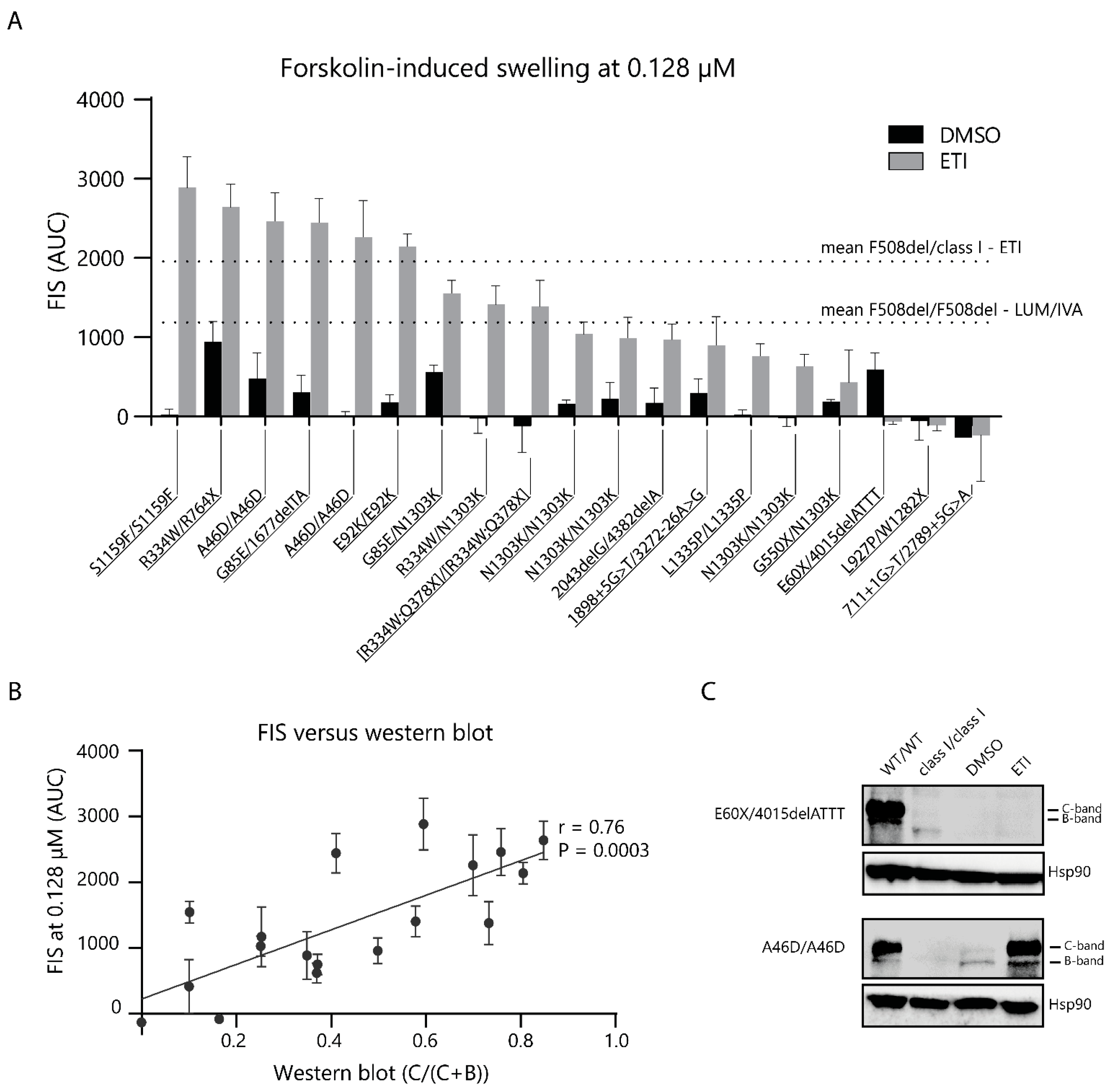
2.4. CFTR Modulator Effects in Rare Variant CF PDIOs in Relation to F508del ETI Induced Function
3. Discussion
4. Material and Methods
4.1. Ethical Approval for Organoid Use
4.2. PDIO Selection Rare Genotypes
4.3. PDIO Culture
4.4. Steady-State Lumen Area Quantification
4.5. Forskolin-Induced Swelling
4.6. Western Blot
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L. Identification of the Cystic Fibrosis Gene: Cloning and Characterization of Complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Rommens, J.M.; Iannuzzi, M.C.; Kerem, B.; Drumm, M.L.; Melmer, G.; Dean, M.; Rozmahel, R.; Cole, J.L.; Kennedy, D.; Hidaka, N. Identification of the Cystic Fibrosis Gene: Chromosome Walking and Jumping. Science 1989, 245, 1059–1065. [Google Scholar] [CrossRef] [PubMed]
- Sosnay, P.R.; Siklosi, K.R.; Van Goor, F.; Kaniecki, K.; Yu, H.; Sharma, N.; Ramalho, A.S.; Amaral, M.D.; Dorfman, R.; Zielenski, J.; et al. Defining the Disease Liability of Variants in the Cystic Fibrosis Transmembrane Conductance Regulator Gene. Nat. Genet. 2013, 45, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Clancy, J.P.; Cotton, C.U.; Donaldson, S.H.; Solomon, G.M.; VanDevanter, D.R.; Boyle, M.P.; Gentzsch, M.; Nick, J.A.; Illek, B.; Wallenburg, J.C.; et al. CFTR Modulator Theratyping: Current Status, Gaps and Future Directions. J. Cyst. Fibros. 2019, 18, 22–34. [Google Scholar] [CrossRef]
- Hudock, K.M.; Clancy, J.P. An Update on New and Emerging Therapies for Cystic Fibrosis. Expert Opin. Emerg. Drugs 2017, 22, 331–346. [Google Scholar] [CrossRef]
- Dittrich, A.-M.; Chuang, S.Y. Dual CFTR Modulator Therapy Efficacy in the Real World: Lessons for the Future. ERJ Open Res. 2022, 8, 00464–2022. [Google Scholar] [CrossRef]
- Rowe, S.M.; Verkman, A.S. Cystic Fibrosis Transmembrane Regulator Correctors and Potentiators. Cold Spring Harb. Perspect. Med. 2013, 3, a009761. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Chen, J. Molecular Structures Reveal Synergistic Rescue of D508 CFTR by Trikafta Modulators. Science 2022, 378, 284–290. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Correspondence, J.C. Mechanism of CFTR Correction by Type I Folding Correctors. Cell 2022, 185, 158–168.e11. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Z.; Levit, A.; Levring, J.; Touhara, K.K.; Shoichet, B.K.; Chen, J. Structural Identification of a Hotspot on CFTR for Potentiation. Science 2019, 364, 1184–1188. [Google Scholar] [CrossRef]
- Yu, H.; Burton, B.; Huang, C.J.; Worley, J.; Cao, D.; Johnson, J.P.; Urrutia, A.; Joubran, J.; Seepersaud, S.; Sussky, K.; et al. Ivacaftor Potentiation of Multiple CFTR Channels with Gating Mutations. J. Cyst. Fibros. 2012, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Moss, R.B.; Flume, P.A.; Elborn, J.S.; Cooke, J.; Rowe, S.M.; McColley, S.A.; Rubenstein, R.C.; Higgins, M. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis Who Have an Arg117His-CFTR Mutation: A Double-Blind, Randomised Controlled Trial. Lancet Respir. Med. 2015, 3, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, C.E.; Elborn, J.S.; Ramsey, B.W.; Marigowda, G.; Huang, X.; Cipolli, M.; Colombo, C.; Davies, J.C.; De Boeck, K.; Flume, P.A.; et al. Lumacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del CFTR. N. Engl. J. Med. 2015, 373, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Cousar, J.L.; Munck, A.; McKone, E.F.; van der Ent, C.K.; Moeller, A.; Simard, C.; Wang, L.T.; Ingenito, E.P.; McKee, C.; Lu, Y.; et al. Tezacaftor–Ivacaftor in Patients with Cystic Fibrosis Homozygous for Phe508del. N. Engl. J. Med. 2017, 377, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Kaftrio|European Medicines Agency. Available online: https://www.ema.europa.eu/en/medicines/human/EPAR/kaftrio (accessed on 11 May 2023).
- Trikafta: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=212273 (accessed on 11 May 2023).
- Durmowicz, A.G.; Lim, R.; Rogers, H.; Rosebraugh, C.J.; Chowdhury, B.A. The U.S. Food and Drug Administration’s Experience with Ivacaftor in Cystic Fibrosis: Establishing Efficacy Using in Vitro Data in Lieu of a Clinical Trial. Ann. Am. Thorac. Soc. 2018, 15, 1–2. [Google Scholar] [CrossRef]
- Vertex Announces FDA Approvals of TRIKAFTA® (Elexacaftor/Tezacaftor/Ivacaftor and Ivacaftor), SYMDEKO® (Tezacaftor/Ivacaftor and Ivacaftor) and KALYDECO® (Ivacaftor) for Use in People with CF with Certain Rare Mutations|Vertex Pharmaceuticals. Available online: https://investors.vrtx.com/news-releases/news-release-details/vertex-announces-fda-approvals-trikaftar (accessed on 1 August 2023).
- Cutting, G. Cystic Fibrosis Genetics: From Molecular Understanding to Clinical Application. Nat. Rev. Genet. 2015, 16, 45–56. [Google Scholar] [CrossRef]
- Amaral, M.D.; de Boeck, K.; Amaral, M.; Davies, J.C.; Drevinek, P.; Elborn, S.; Kerem, E.; Lee, T. Theranostics by Testing CFTR Modulators in Patient-Derived Materials: The Current Status and a Proposal for Subjects with Rare CFTR Mutations. J. Cyst. Fibros. 2019, 18, 685–692. [Google Scholar] [CrossRef]
- Allen, L.; Allen, L.; Carr, S.B.; Davies, G.; Downey, D.; Egan, M.; Forton, J.T.; Gray, R.; Haworth, C.; Horsley, A.; et al. Future Therapies for Cystic Fibrosis. Nat. Commun. 2023, 14, 693. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesisin a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Dekkers, J.F.; Wiegerinck, C.L.; De Jonge, H.R.; Bronsveld, I.; Janssens, H.M.; De Winter-De Groot, K.M.; Brandsma, A.M.; De Jong, N.W.M.; Bijvelds, M.J.C.; Scholte, B.J.; et al. A Functional CFTR Assay Using Primary Cystic Fibrosis Intestinal Organoids. Nat. Med. 2013, 19, 939–945. [Google Scholar] [CrossRef]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 Stem Cells Build Crypt-Villus Structures in Vitro without a Mesenchymal Niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, J.F.; Berkers, G.; Kruisselbrink, E.; Vonk, A.; De Jonge, H.R.; Janssens, H.M.; Bronsveld, I.; Van De Graaf, E.A.; Nieuwenhuis, E.E.S.; Houwen, R.H.J.; et al. Characterizing Responses to CFTR-Modulating Drugs Using Rectal Organoids Derived from Subjects with Cystic Fibrosis. Sci. Transl. Med. 2016, 8, 344ra84. [Google Scholar] [CrossRef] [PubMed]
- Cuyx, S.; Ramalho, A.S.; Corthout, N.; Fieuws, S.; Fürstová, E.; Arnauts, K.; Ferrante, M.; Verfaillie, C.; Munck, S.; Boon, M.; et al. Rectal Organoid Morphology Analysis (ROMA) as a Promising Diagnostic Tool in Cystic Fibrosis. Thorax 2021, 76, 1146–1149. [Google Scholar] [CrossRef] [PubMed]
- Muilwijk, D.; de Poel, E.; van Mourik, P.; Suen, S.W.F.; Vonk, A.M.; Brunsveld, J.E.; Kruisselbrink, E.; Oppelaar, H.; Hagemeijer, M.C.; Berkers, G.; et al. Forskolin-Induced Organoid Swelling Is Associated with Long-Term Cystic Fibrosis Disease Progression. Eur. Respir. J. 2022, 60, 2100508. [Google Scholar] [CrossRef]
- Berkers, G.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Dekkers, J.F.; de Winter-de Groot, K.M.; Arets, H.G.M.; Marck-van der Wilt, R.E.P.; Dijkema, J.S.; Vanderschuren, M.M.; et al. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep. 2019, 26, 1701–1708.e3. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Förstová, E.; Vonk, A.M.; Ferrante, M.; Verfailli, C.; Dupont, L.; Boon, M.; Proesmans, M.; Beekma, J.M.; Sarouk, I.; et al. Correction of CFTR Function in Intestinal Organoids to Guide Treatment of Cystic Fibrosis. Eur. Respir. J. 2020, 57, 1902426. [Google Scholar] [CrossRef]
- Graeber, S.Y.; van Mourik, P.; Vonk, A.M.; Kruisselbrink, E.; Hirtz, S.; van der Ent, C.K.; Mall, M.A.; Beekman, J.M. Comparison of Organoid Swelling and in Vivo Biomarkers of Cftr Function to Determine Effects of Lumacaftor-Ivacaftor in Patients with Cystic Fibrosis Homozygous for the F508del Mutation. Am. J. Respir. Crit. Care Med. 2020, 202, 1589–1592. [Google Scholar] [CrossRef]
- Cholon, D.M.; Quinney, N.L.; Fulcher, M.L.; Esther, C.R.; Das, J.; Dokholyan, N.V.; Randell, S.H.; Boucher, R.C.; Gentzsch, M. Potentiator Ivacaftor Abrogates Pharmacological Correction of DF508 CFTR in Cystic Fibrosis. Sci. Transl. Med. 2014, 6, 246ra96. [Google Scholar] [CrossRef]
- Middleton, P.G.; Mall, M.A.; Dřevínek, P.; Lands, L.C.; McKone, E.F.; Polineni, D.; Ramsey, B.W.; Taylor-Cousar, J.L.; Tullis, E.; Vermeulen, F.; et al. Elexacaftor-Tezacaftor-Ivacaftor for Cystic Fibrosis with a Single Phe508del Allele. N. Engl. J. Med. 2019, 381, 1809–1819. [Google Scholar] [CrossRef]
- De Boeck, K.; Munck, A.; Walker, S.; Faro, A.; Hiatt, P.; Gilmartin, G.; Higgins, M. Efficacy and Safety of Ivacaftor in Patients with Cystic Fibrosis and a Non-G551D Gating Mutation. J. Cyst. Fibros. 2014, 13, 674–680. [Google Scholar] [CrossRef]
- Flume, P.A.; Liou, T.G.; Borowitz, D.S.; Li, H.; Yen, K.; Ordoñez, C.L.; Geller, D.E. Ivacaftor in Subjects with Cystic Fibrosis Who Are Homozygous for the F508del-CFTR Mutation. Chest 2012, 142, 718–724. [Google Scholar] [CrossRef] [PubMed]
- Heijerman, H.G.M.; McKone, E.F.; Downey, D.G.; Van Braeckel, E.; Rowe, S.M.; Tullis, E.; Mall, M.A.; Welter, J.J.; Ramsey, B.W.; McKee, C.M.; et al. Efficacy and Safety of the Elexacaftor plus Tezacaftor plus Ivacaftor Combination Regimen in People with Cystic Fibrosis Homozygous for the F508del Mutation: A Double-Blind, Randomised, Phase 3 Trial. Lancet 2019, 394, 1940–1948. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; McColley, S.A.; Rietschel, E.; Li, X.; Bell, S.C.; Konstan, M.W.; Marigowda, G.; Waltz, D.; Boyle, M.P. Lumacaftor/Ivacaftor Treatment of Patients with Cystic Fibrosis Heterozygous for F508del-CFTR. Ann. Am. Thorac. Soc. 2017, 14, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Rowe, S.M.; Daines, C.; Ringshausen, F.C.; Kerem, E.; Wilson, J.; Tullis, E.; Nair, N.; Simard, C.; Han, L.; Ingenito, E.P.; et al. Tezacaftor–Ivacaftor in Residual-Function Heterozygotes with Cystic Fibrosis. N. Engl. J. Med. 2017, 377, 2024–2035. [Google Scholar] [CrossRef]
- Bihler, H.; Sivachenko, A.; Millen, L.; Bhatt, P.; Patel, A.T.; Chin, J.; Bailey, V.; Musisi, I.; LaPan, A.; Allaire, N.E.; et al. In Vitro Modulator Responsiveness of 655 CFTR Variants Found in People With CF. bioRxiv 2023. [Google Scholar] [CrossRef]
- Avramescu, R.G.; Kai, Y.; Xu, H.; Bidaud-Meynard, A.; Schnúr, A.; Frenkiel, S.; Matouk, E.; Veit, G.; Lukacs, G.L. Mutation-Specific Downregulation of CFTR2 Variants by Gating Potentiators. Hum. Mol. Genet. 2017, 26, 4873–4885. [Google Scholar] [CrossRef] [PubMed]
- Zomer-van Ommen, D.D.; de Poel, E.; Kruisselbrink, E.; Oppelaar, H.; Vonk, A.M.; Janssens, H.M.; van der Ent, C.K.; Hagemeijer, M.C.; Beekman, J.M. Comparison of ex vivo and in vitro intestinal cystic fibrosis models to measure CFTR-dependent ion channel activity. J. Cyst. Fibros. 2018, 17, 316–324. [Google Scholar] [CrossRef]
- Birimberg-Schwartz, L.; Ip, W.; Bartlett, C.; Avolio, J.; Vonk, A.M.; Gunawardena, T.; Du, K.; Esmaeili, M.; Beekman, J.M.; Rommens, J.; et al. Validating organoid-derived human intestinal monolayers for personalized therapy in cystic fibrosis. Life Sci. Alliance 2023, 6. [Google Scholar] [CrossRef] [PubMed]
- Railean, V.; Rodrigues, C.S.; Ramalho, S.S.; Silva, I.A.L.; Bartosch, J.; Farinha, C.M.; Pankonien, I.; Amaral, M.D. Personalized Medicine: Function of CFTR Variant p.Arg334Trp Is Rescued by Currently Available CFTR Modulators. Front. Mol. Biosci. 2023, 10, 1155705. [Google Scholar] [CrossRef]
- Rectal Organoid-Guided CFTR Modulator Therapy Restores Lung Function in a Cystic Fibrosis Patient with the Rare 1677delTA/R334W Genotype. Eur. Respir. J. 2022, 60, 2201341. [CrossRef]
- Ramalho, A.; Cuyx, S.; Boon, M.; Proesmans, M.; Dupont, L.; De Boeck, K.; Verhulst, S.; De Wachter, E.; Van Biervliet, S.; Vermeulen, F. The Importance of CFTR MRNA Testing to Uncover Other Variants in CF Genotype That Affect CFTR Expression. In Proceedings of the NACFC Philadelphia 2022, Philadelphia, PA, USA, 3–5 November 2022. [Google Scholar]
- Sadras, I.; Kerem, E.; Livnat, G.; Sarouk, I.; Breuer, O.; Reiter, J.; Gileles-Hillel, A.; Inbar, O.; Cohen, M.; Gamliel, A.; et al. Clinical and Functional Efficacy of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis Carrying the N1303K Mutation. J. Cyst. Fibros. 2023. online ahead of print. [Google Scholar] [CrossRef]
- Burgel, P.R.; Sermet-Gaudelus, I.; Durieu, I.; Kanaan, R.; MacEy, J.; Grenet, D.; Porzio, M.; Coolen-Allou, N.; Chiron, R.; Marguet, C.; et al. The French Compassionate Programme of Elexacaftor/Tezacaftor/Ivacaftor in People with Cystic Fibrosis with Advanced Lung Disease and No F508del CFTR Variant. Eur. Respir. J. 2023, 61, 2202437. [Google Scholar] [CrossRef]
- Vonk, A.M.; van Mourik, P.; Ramalho, A.S.; Silva, I.A.L.; Statia, M.; Kruisselbrink, E.; Suen, S.W.F.; Dekkers, J.F.; Vleggaar, F.P.; Houwen, R.H.J.; et al. Protocol for Application, Standardization and Validation of the Forskolin-Induced Swelling Assay in Cystic Fibrosis Human Colon Organoids. STAR Protoc. 2020, 1, 100019. [Google Scholar] [CrossRef]
- Labelbox Inc. Labelbox: The Leading Training Data Platform for Data Labeling. Available online: https://labelbox.com/ (accessed on 13 September 2023).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele 1/Allele 2 | PDIO Response | FRT ≥ 10% Allele 1 | FRT ≥ 10% Allele 2 | FDA-Approved Allele 1 | FDA-Approved Allele 2 |
|---|---|---|---|---|---|
| R1066C/R1066H | Healthy control | Non-responsive | Responsive | X | |
| R1066H/CFTRdele2,3 | Healthy control | Responsive | Not tested | X | |
| Q1012P/N1303K | Healthy control | Not tested | Borderline | ||
| S1159F/S1159F | ETI range | Responsive | Responsive | X | X |
| R334W/R764X | ETI range | Non-responsive | Not tested | ||
| A46D/A46D | ETI range | Responsive | Responsive | X | X |
| G85E/1677delTA | ETI range | Non-responsive | Not tested | X | |
| A46D/A46D | ETI range | Responsive | Responsive | X | X |
| E92K/E92K | ETI range | Responsive | Responsive | X | X |
| G85E/N1303K | ETI range | Non-responsive | Borderline | X | |
| R334W/N1303K | ETI range | Non-responsive | Borderline | ||
| [R334W;Q378X]/ [R334W;Q378X] | ETI range | Not tested | Not tested | ||
| N1303K/N1303K | LUM/IVA range | Borderline | Borderline | ||
| N1303K/N1303K | LUM/IVA range | Borderline | Borderline | ||
| 2043delG/4382delA | LUM/IVA range | Not tested | Not tested | ||
| 1898 + 5G > T/3272-26A > G | LUM/IVA range | Not tested | Not tested | ||
| L1335P/L1335P | Below LUM/IVA | Non-responsive | Non-responsive | X | X |
| N1303K/N1303K | Below LUM/IVA | Borderline | Borderline | ||
| G550X/N1303K | No swelling | Not tested | Borderline | ||
| E60X/4015delATTT | No swelling | Not tested | Not tested | ||
| L927P/W1282X | No swelling | Responsive | Not tested | ||
| 711 + 1G > T/2789 + 5G > A | No swelling | Not tested | Not tested |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefferts, J.W.; Bierlaagh, M.C.; Kroes, S.; Nieuwenhuijze, N.D.A.; Sonneveld van Kooten, H.N.; Niemöller, P.J.; Verburg, T.F.; Janssens, H.M.; Muilwijk, D.; van Beuningen, S.F.B.; et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. Int. J. Mol. Sci. 2023, 24, 14539. https://doi.org/10.3390/ijms241914539
Lefferts JW, Bierlaagh MC, Kroes S, Nieuwenhuijze NDA, Sonneveld van Kooten HN, Niemöller PJ, Verburg TF, Janssens HM, Muilwijk D, van Beuningen SFB, et al. CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes. International Journal of Molecular Sciences. 2023; 24(19):14539. https://doi.org/10.3390/ijms241914539
Chicago/Turabian StyleLefferts, Juliet W., Marlou C. Bierlaagh, Suzanne Kroes, Natascha D. A. Nieuwenhuijze, Heleen N. Sonneveld van Kooten, Paul J. Niemöller, Tibo F. Verburg, Hettie M. Janssens, Danya Muilwijk, Sam F. B. van Beuningen, and et al. 2023. "CFTR Function Restoration upon Elexacaftor/Tezacaftor/Ivacaftor Treatment in Patient-Derived Intestinal Organoids with Rare CFTR Genotypes" International Journal of Molecular Sciences 24, no. 19: 14539. https://doi.org/10.3390/ijms241914539