

Selective Adsorption of Organic Micro-Pollutants by Smectite Clays Revealed from Atomistic Simulations



Abstract
:1. Introduction
2. Results and Discussion
2.1. DIA and OXA Geometrical Characteristics
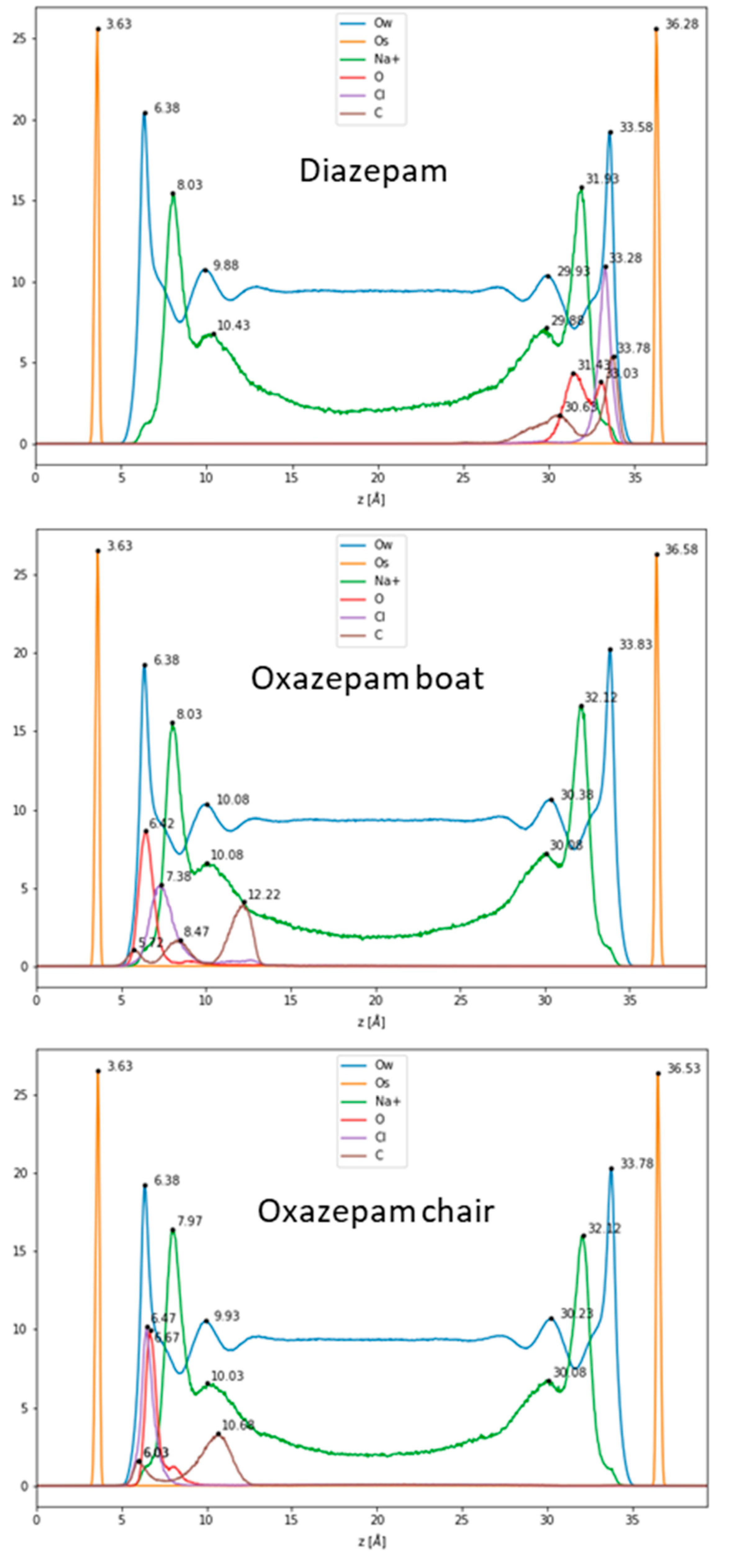
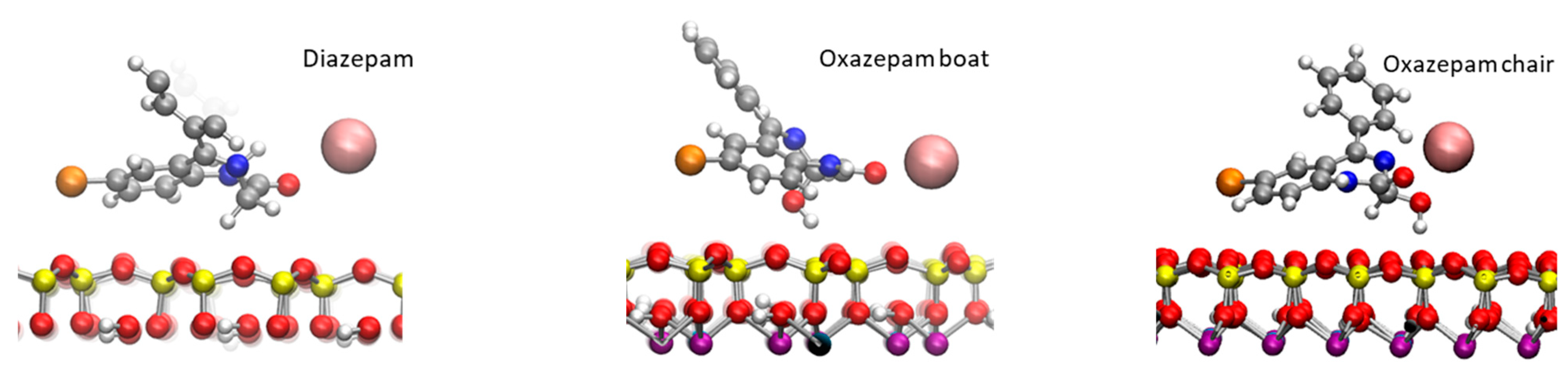
2.2. Adsorption of DIA and OXA on the External Surfaces of Na-Mt
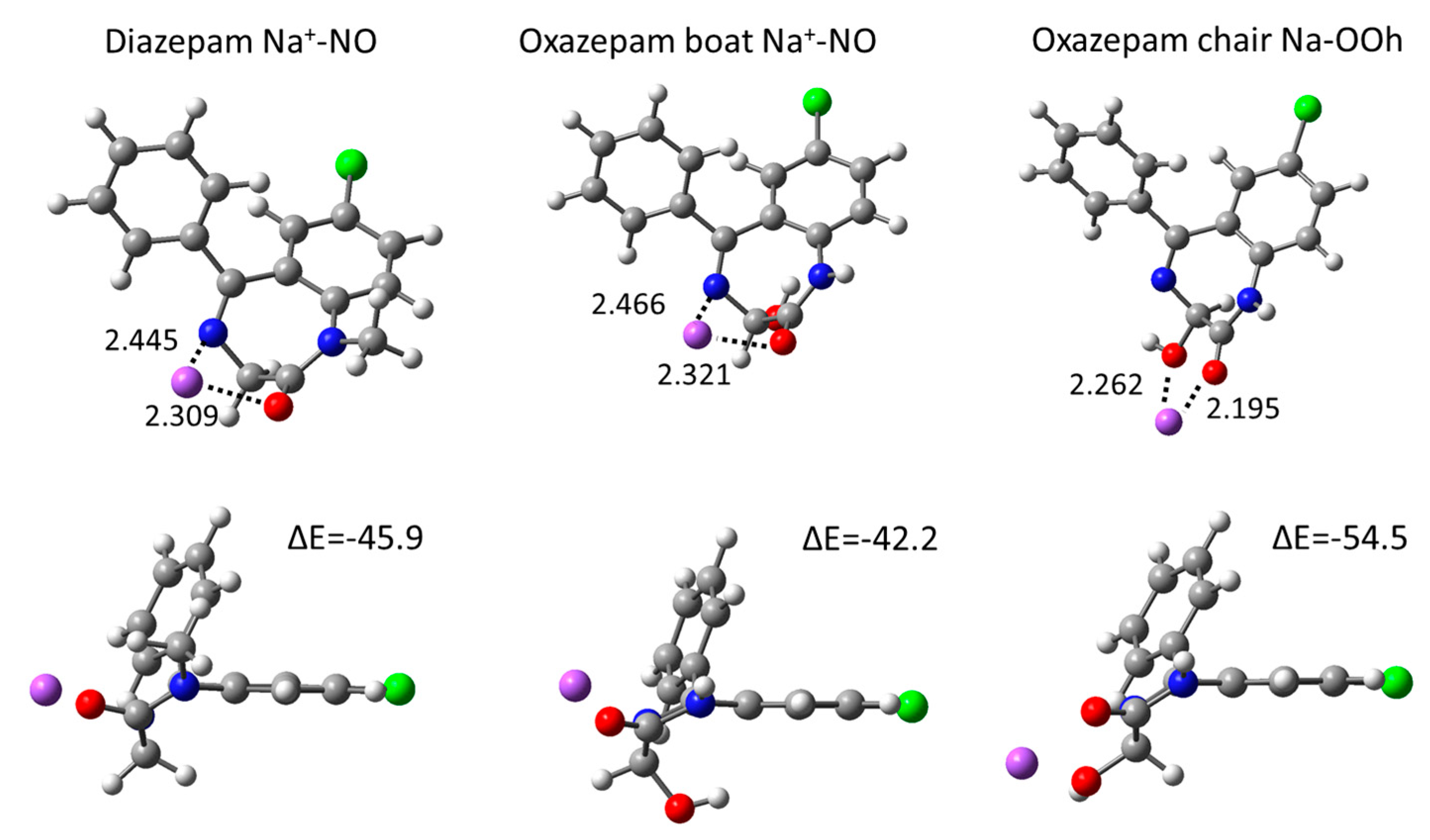
2.3. Gas Phase Na+ Coordination Interactions Derived from DFT Calculations
2.4. Intercalation of OXA Chair
3. Conclusions
4. Computational Methods
4.1. Classical MD Simulations on the Basal Surface
4.2. Gas Phase DFT Calculations
4.3. Classical MD Simulations of the Interlayer Space
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Clarke, B.O.; Smith, S.R. Review of ‘Emerging’ Organic Contaminants in Biosolids and Assessment of International Research Priorities for the Agricultural Use of Biosolids. Environ. Int. 2011, 37, 226–247. [Google Scholar] [CrossRef] [PubMed]
- Lapworth, D.J.; Baran, N.; Stuart, M.E.; Ward, R.S. Emerging Organic Contaminants in Groundwater: A Review of Sources, Fate and Occurrence. Environ. Pollut. 2012, 163, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Vodyanitskii, Y.N.; Yakovlev, A.S. Contamination of Soils and Groundwater with New Organic Micropollutants: A Review. Eurasian Soil Sci. 2016, 49, 560–569. [Google Scholar] [CrossRef]
- Freeling, F.; Alygizakis, N.A.; von der Ohe, P.C.; Slobodnik, J.; Oswald, P.; Aalizadeh, R.; Cirka, L.; Thomaidis, N.S.; Scheurer, M. Occurrence and Potential Environmental Risk of Surfactants and Their Transformation Products Discharged by Wastewater Treatment Plants. Sci. Total Environ. 2019, 681, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Gago-Ferrero, P.; Bletsou, A.A.; Damalas, D.E.; Aalizadeh, R.; Alygizakis, N.A.; Singer, H.P.; Hollender, J.; Thomaidis, N.S. Wide-Scope Target Screening of >2000 Emerging Contaminants in Wastewater Samples with UPLC-Q-ToF-HRMS/MS and Smart Evaluation of Its Performance through the Validation of 195 Selected Representative Analytes. J. Hazard. Mater. 2020, 387, 121712–121726. [Google Scholar] [CrossRef]
- Simon, E.; Duffek, A.; Stahl, C.; Frey, M.; Scheurer, M.; Tuerk, J.; Gehrmann, L.; Könemann, S.; Swart, K.; Behnisch, P.; et al. Biological Effect and Chemical Monitoring of Watch List Substances in European Surface Waters: Steroidal Estrogens and Diclofenac—Effect-Based Methods for Monitoring Frameworks. Environ. Int. 2022, 159, 107033–107044. [Google Scholar] [CrossRef]
- Loraine, G.A.; Pettigrove, M.E. Seasonal Variations in Concentrations of Pharmaceuticals and Personal Care Products in Drinking Water and Reclaimed Wastewater in Southern California. Environ. Sci. Technol. 2006, 40, 687–695. [Google Scholar] [CrossRef]
- Reemtsma, T.; Weiss, S.; Mueller, J.; Petrovic, M.; González, S.; Barcelo, D.; Ventura, F.; Knepper, T.P. Polar Pollutants Entry into the Water Cycle by Municipal Wastewater: A European Perspective. Environ. Sci. Technol. 2006, 40, 5451–5458. [Google Scholar] [CrossRef]
- Al-Rifai, J.H.; Gabelish, C.L.; Schäfer, A.I. Occurrence of Pharmaceutically Active and Non-Steroidal Estrogenic Compounds in Three Different Wastewater Recycling Schemes in Australia. Chemosphere 2007, 69, 803–815. [Google Scholar] [CrossRef]
- Ellis, J.B. Pharmaceutical and Personal Care Products (PPCPs) in Urban Receiving Waters. Environ. Pollut. 2006, 144, 184–189. [Google Scholar] [CrossRef]
- Yu, J.T.; Bouwer, E.J.; Coelhan, M. Occurrence and Biodegradability Studies of Selected Pharmaceuticals and Personal Care Products in Sewage Effluent. Agric. Water Manag. 2006, 86, 72–80. [Google Scholar] [CrossRef]
- Chang, P.-H.; Li, Z.; Jiang, W.-T.; Sarkar, B. Clay Minerals for Pharmaceutical Wastewater Treatment. In Modified Clay and Zeolite Nanocomposite Materials; Elsevier: Amsterdam, The Netherlands, 2019; pp. 167–196. ISBN 978-0-12-814617-0. [Google Scholar]
- Thiebault, T. Raw and Modified Clays and Clay Minerals for the Removal of Pharmaceutical Products from Aqueous Solutions: State of the Art and Future Perspectives. Crit. Rev. Environ. Sci. Technol. 2020, 50, 1451–1514. [Google Scholar] [CrossRef]
- Gao, J.; Pedersen, J.A. Adsorption of Sulfonamide Antimicrobial Agents to Clay Minerals. Environ. Sci. Technol. 2005, 39, 9509–9516. [Google Scholar] [CrossRef]
- Thiebault, T.; Boussafir, M.; Le Forestier, L.; Le Milbeau, C.; Monnin, L.; Guégan, R. Competitive Adsorption of a Pool of Pharmaceuticals onto a Raw Clay Mineral. RSC Adv. 2016, 6, 65257–65265. [Google Scholar] [CrossRef]
- Thiebault, T.; Boussafir, M. Adsorption Mechanisms of Psychoactive Drugs onto Montmorillonite. Colloid. Interface Sci. Commun. 2019, 30, 100183–100187. [Google Scholar] [CrossRef]
- De Oliveira, T.; Guégan, R.; Thiebault, T.; Milbeau, C.L.; Muller, F.; Teixeira, V.; Giovanela, M.; Boussafir, M. Adsorption of Diclofenac onto Organoclays: Effects of Surfactant and Environmental (PH and Temperature) Conditions. J. Hazard. Mater. 2017, 323, 558–566. [Google Scholar] [CrossRef]
- Aristilde, L.; Marichal, C.; Miéhé-Brendlé, J.; Lanson, B.; Charlet, L. Interactions of Oxytetracycline with a Smectite Clay: A Spectroscopic Study with Molecular Simulations. Environ. Sci. Technol. 2010, 44, 7839–7845. [Google Scholar] [CrossRef]
- Aristilde, L.; Lanson, B.; Charlet, L. Interstratification Patterns from the PH-Dependent Intercalation of a Tetracycline Antibiotic within Montmorillonite Layers. Langmuir 2013, 29, 4492–4501. [Google Scholar] [CrossRef]
- Belzunces, B.; Hoyau, S.; Benoit, M.; Tarrat, N.; Bessac, F. Theoretical Study of the Atrazine Pesticide Interaction with Pyrophyllite and Ca 2+-Montmorillonite Clay Surfaces. J. Comput. Chem. 2017, 38, 133–143. [Google Scholar] [CrossRef]
- Belzunces, B.; Hoyau, S.; Bessac, F. Interaction of Metamitron and Fenhexamid with Ca 2+-Montmorillonite Clay Surfaces: A Density Functional Theory Molecular Dynamics Study. J. Comput. Chem. 2019, 40, 1449–1462. [Google Scholar] [CrossRef]
- Böhm, L.; Grančič, P.; Scholtzová, E.; Heyde, B.J.; Düring, R.-A.; Siemens, J.; Gerzabek, M.H.; Tunega, D. Adsorption of the Hydrophobic Organic Pollutant Hexachlorobenzene to Phyllosilicate Minerals. Environ. Sci. Pollut. Res. 2022, 30, 36824–36837. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Rodríguez, D.; Jankovič, Ľ.; Scholtzová, E.; Tunega, D. Stability of Atrazine–Smectite Intercalates: Density Functional Theory and Experimental Study. Minerals 2021, 11, 554. [Google Scholar] [CrossRef]
- Galicia-Andrés, E.; Oostenbrink, C.; Gerzabek, M.H.; Tunega, D. On the Adsorption Mechanism of Humic Substances on Kaolinite and Their Microscopic Structure. Minerals 2021, 11, 1138. [Google Scholar] [CrossRef]
- Galicia-Andrés, E.; Tunega, D.; Gerzabek, M.H.; Oostenbrink, C. On Glyphosate-Kaolinite Surface Interactions. A Molecular Dynamic Study. Eur. J. Soil. Sci. 2020, 72, 1231–1242. [Google Scholar] [CrossRef] [PubMed]
- Corbin, G.; Vulliet, E.; Lanson, B.; Rimola, A.; Mignon, P. Adsorption of Pharmaceuticals onto Smectite Clay Minerals: A Combined Experimental and Theoretical Study. Minerals 2021, 11, 62. [Google Scholar] [CrossRef]
- Calisto, V.; Esteves, V.I. Psychiatric Pharmaceuticals in the Environment. Chemosphere 2009, 77, 1257–1274. [Google Scholar] [CrossRef]
- Thiebault, T.; Chassiot, L.; Fougère, L.; Destandau, E.; Simonneau, A.; Van Beek, P.; Souhaut, M.; Chapron, E. Record of Pharmaceutical Products in River Sediments: A Powerful Tool to Assess the Environmental Impact of Urban Management? Anthropocene 2017, 18, 47–56. [Google Scholar] [CrossRef]
- Thiebault, T.; Boussafir, M.; Fougère, L.; Destandau, E.; Monnin, L.; Le Milbeau, C. Clay Minerals for the Removal of Pharmaceuticals: Initial Investigations of Their Adsorption Properties in Real Wastewater Effluents. Environ. Nanotechnol. Monit. Manag. 2019, 12, 100266. [Google Scholar] [CrossRef]
- De Oliveira, T.; Boussafir, M.; Fougère, L.; Destandau, E.; Sugahara, Y.; Guégan, R. Use of a Clay Mineral and Its Nonionic and Cationic Organoclay Derivatives for the Removal of Pharmaceuticals from Rural Wastewater Effluents. Chemosphere 2020, 259, 127480. [Google Scholar] [CrossRef]
- Hounfodji, J.W.; Kanhounnon, W.G.; Kpotin, G.; Atohoun, G.S.; Lainé, J.; Foucaud, Y.; Badawi, M. Molecular Insights on the Adsorption of Some Pharmaceutical Residues from Wastewater on Kaolinite Surfaces. Chem. Eng. J. 2021, 407, 127176. [Google Scholar] [CrossRef]
- Carvalho, A.J.P.; Dordio, A.V.; Ramalho, J.P.P. A DFT Study on the Adsorption of Benzodiazepines to Vermiculite Surfaces. J. Mol. Model. 2014, 20, 2336. [Google Scholar] [CrossRef] [PubMed]
- Mignon, P.; Corbin, G.; Le Crom, S.; Marry, V.; Hao, J.; Daniel, I. Adsorption of Nucleotides on Clay Surfaces: Effects of Mineral Composition, PH and Solution Salts. Appl. Clay Sci. 2020, 190, 105544. [Google Scholar] [CrossRef]
- Myshakin, E.M.; Makaremi, M.; Romanov, V.N.; Jordan, K.D.; Guthrie, G.D. Molecular Dynamics Simulations of Turbostratic Dry and Hydrated Montmorillonite with Intercalated Carbon Dioxide. J. Phys. Chem. A 2014, 118, 7454–7468. [Google Scholar] [CrossRef] [PubMed]
- Voora, V.K.; Al-Saidi, W.A.; Jordan, K.D. Density Functional Theory Study of Pyrophyllite and M-Montmorillonites (M = Li, Na, K, Mg, and Ca): Role of Dispersion Interactions. J. Phys. Chem. A 2011, 115, 9695–9703. [Google Scholar] [CrossRef]
- Ferrage, E. Investigation of Smectite Hydration Properties by Modeling Experimental X-Ray Diffraction Patterns: Part I. Montmorillonite Hydration Properties. Am. Mineral. 2005, 90, 1358–1374. [Google Scholar] [CrossRef]
- Abramova, E.; Lapides, I.; Yariv, S. Thermo-XRD Investigation of Monoionic Montmorillonites Mechanochemically Treated with Urea. J. Therm. Anal. Calorim. 2007, 90, 99–106. [Google Scholar] [CrossRef]
- Lee, J.H.; Guggenheim, S. Single Crystal X-Ray Refinement of Pyrophyllite-1Tc. Am. Mineral. 1981, 66, 350–357. [Google Scholar]
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Open-Shell Transition Metals. Phys. Rev. B Condens. Matter Mater. Phys. 1993, 48, 13115–13118. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Cygan, R.T.; Liang, J.-J.; Kalinichev, A.G. Molecular Models of Hydroxide, Oxyhydroxide, and Clay Phases and the Development of a General Force Field. J. Phys. Chem. B 2004, 108, 1255–1266. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Hermans, J. Interaction Models for Water in Relation to Protein Hydration. Jerus. Symp. Quantum Chem. Biochem. 1981, 14, 331–342. [Google Scholar]
- Dang, L.X. Mechanism and Thermodynamics of Ion Selectivity in Aqueous Solutions of 18-Crown-6 Ether: A Molecular Dynamics Study. J. Am. Chem. Soc. 1995, 117, 6954–6960. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 09, Revision E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Woods, R.J.; Chappelle, R. Restrained Electrostatic Potential Atomic Partial Charges for Condensed-Phase Simulations of Carbohydrates. J. Mol. Struct. Theochem. 2000, 527, 149–156. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.; Caldwell, J.; Kollman, P.; Case, D. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Thyveetil, M.A.; Coveney, P.V.; Greenwell, H.C.; Suter, J.L. Computer Simulation Study of the Structural Stability and Materials Properties of DNA-Intercalated Layered Double Hydroxides. J. Am. Chem. Soc. 2008, 130, 4742–4756. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Coveney, P.V.; Greenwell, H.C. Clay Minerals Mediate Folding and Regioselective Interactions of RNA: A Large-Scale Atomistic Simulation Study. J. Am. Chem. Soc. 2010, 132, 13750–13764. [Google Scholar] [CrossRef] [PubMed]
- Swadling, J.B.; Coveney, P.V.; Christopher Greenwell, H. Stability of Free and Mineral-Protected Nucleic Acids: Implications for the RNA World. Geochim. Cosmochim. Acta 2012, 83, 360–378. [Google Scholar] [CrossRef]
- Swadling, J.B.; Suter, J.L.; Greenwell, H.C.; Coveney, P.V. Influence of Surface Chemistry and Charge on Mineral–RNA Interactions. Langmuir 2013, 29, 1573–1583. [Google Scholar] [CrossRef]
- Cheatham III, T.E.; Young, M.A. Molecular Dynamics Simulation of Nucleic Acids: Successes, Limitations, and Promise*. Biopolymers 2000, 56, 232–256. [Google Scholar] [CrossRef]
- Plimpton, S. Fast Parallel Algorithms for Short-Range Molecular Dynamics. J. Comput. Phys. 1995, 117, 1–19. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N—Na+ | O—Na+ | Oh—Na+ | Hh—Os | |||||
|---|---|---|---|---|---|---|---|---|
| 1st Peak | CN | 1st Peak | CN | 1st Peak | CN | 1st Peak | CN | |
| Diazepam | 4.42 | 0.64 | 2.41 | 0.54 | ||||
| Oxazepam boat | 4.46 | 0.44 | 2.43 | 0.32 | 2.52 | 0.03 | 1.41 | 0.68 |
| Oxazepam chair | 2.81 | 0.35 | 2.44 | 1.25 | 2.46 | 1.07 | 1.39 | 0.98 |
| Coordinating Atoms | ΔE | d Na—O | d Na—N | d Na—Oh | |
|---|---|---|---|---|---|
| Diazepam | O | −42.2 | 2.100 | ||
| N | −41.4 | 2.311 | |||
| O N | −45.9 | 2.309 | 2.445 | ||
| Oxazepam boat | O | −41.3 | 2.102 | ||
| N | −39.6 | 2.317 | |||
| O N | −42.2 | 2.321 | 2.466 | ||
| N Oh | −34.0 | 2.477 | 2.311 | ||
| Oh π | −36.7 | 2.240 | |||
| Oxazepam chair | O Oh | −54.5 | 2.195 | 2.262 | |
| N Oh | −52.0 | 2.331 | 2.279 | ||
| N O Oh | −51.3 | 2.379 | 2.647 | 2.282 |
| Dry | OXA 0 w | OXA 5 w | OXA 10 w | OXA 15 w | OXA 20 w | OXA 25 w | OXA 30 w | OXA 35 w | |
|---|---|---|---|---|---|---|---|---|---|
| MD | 9.5 | 13.1 | 12.8 | 12.4 | 12.7 | 13.2 | 12.9 | 13.3 | 13.6 |
| Exp. | 10.4 | 13.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cancade, M.; Thiebault, T.; Mignon, P. Selective Adsorption of Organic Micro-Pollutants by Smectite Clays Revealed from Atomistic Simulations. Int. J. Mol. Sci. 2023, 24, 14781. https://doi.org/10.3390/ijms241914781
Cancade M, Thiebault T, Mignon P. Selective Adsorption of Organic Micro-Pollutants by Smectite Clays Revealed from Atomistic Simulations. International Journal of Molecular Sciences. 2023; 24(19):14781. https://doi.org/10.3390/ijms241914781
Chicago/Turabian StyleCancade, Mathieu, Thomas Thiebault, and Pierre Mignon. 2023. "Selective Adsorption of Organic Micro-Pollutants by Smectite Clays Revealed from Atomistic Simulations" International Journal of Molecular Sciences 24, no. 19: 14781. https://doi.org/10.3390/ijms241914781
APA StyleCancade, M., Thiebault, T., & Mignon, P. (2023). Selective Adsorption of Organic Micro-Pollutants by Smectite Clays Revealed from Atomistic Simulations. International Journal of Molecular Sciences, 24(19), 14781. https://doi.org/10.3390/ijms241914781