Abstract
Advances in molecular biology have revolutionized the use of messenger RNA (mRNA) as a therapeutic. The concept of nucleic acid therapy with mRNA originated in 1990 when Wolff et al. reported successful expression of proteins in target organs by direct injection of either plasmid DNA or mRNA. It took decades to bring the transfection efficiency of mRNA closer to that of DNA. The next few decades were dedicated to turning in vitro-transcribed (IVT) mRNA from a promising delivery tool for gene therapy into a full-blown therapeutic modality, which changed the biotech market rapidly. Hundreds of clinical trials are currently underway using mRNA for prophylaxis and therapy of infectious diseases and cancers, in regenerative medicine, and genome editing. The potential of IVT mRNA to induce an innate immune response favors its use for vaccination and immunotherapy. Nonetheless, in non-immunotherapy applications, the intrinsic immunostimulatory activity of mRNA directly hinders the desired therapeutic effect since it can seriously impair the target protein expression. Targeting the same innate immune factors can increase the effectiveness of mRNA therapeutics for some indications and decrease it for others, and vice versa. The review aims to present the innate immunity-related ‘barriers’ or ‘springboards’ that may affect the development of immunotherapies and non-immunotherapy applications of mRNA medicines.
1. Introduction
With the broad clinical application of mRNA-based therapeutics, mRNA technology has made a real breakthrough in medical biotechnology, regenerative medicine, replacement therapy, and genome editing [1,2]. Although the principle of using in vitro-transcribed (IVT) mRNA in gene therapy was described many years ago, its actual clinical application became possible only in recent years, as non-viral delivery of nucleic acids was extensively developed [2,3]. To date, mRNA is regarded largely as a promising tool for preventive and therapeutic immunization.
The fundamental mechanism underlying the mRNA vaccine technology is based on a vector that delivers a nucleic acid molecule encoding a gene of interest into a target cell, thus allowing the cell to produce the target protein and to express the antigen to elicit an immune response [4]. mRNA vaccines have potential benefits over other nucleic-acid-based therapies, such as viral vectors or plasmid DNA. mRNA does not need to penetrate nucleus barriers and thus it does not lead to irreversible changes to the cell or its genome. It provides almost immediate initiation of protein translation as soon as it appears in the cytoplasm. In contrast with viral vaccines, mRNA-based therapeutics do not lead to the production of infectious particles and have no potential to cause adverse effects such as immune-mediated hepatitis [1,5,6].
When applying mRNA technology, one should consider molecular and biological consequences of the cellular response to foreign RNA. If a eukaryotic cell detects foreign RNA, it responds to the viral invasion. The innate immune system has nucleic-acid sensors, RNA sensors in particular, which constantly scan for and detect foreign RNA and endogenous RNA of cells under stress. Numerous RNA-binding proteins in mammalian cells control immune reactions by modulating gene expression, splicing, nuclear export, mRNA modification, translation, and degradation [7].
Multiple molecular mechanisms evolved to distinguish self and non-self RNAs to secure the initiation of an antiviral response, including the expression of type I interferon (IFN) and IFN-stimulated genes. The main RNA response mechanism is related to the recognition of pathogen-associated molecular patterns (PAMPs) by pattern recognition receptors (PRRs). PRRs can be categorized into several families based on structure, localization, and ligands [8]. They include toll-like receptors (TLRs), C-type lectin receptors (CLRs), retinoic acid-inducible gene I (RIG-I), melanoma differentiation-associated protein 5 (MDA5), the cyclic GMP-AMP synthase (cGAS)/stimulator of interferon genes (STING) second messenger system, and NOD-like receptors (NLRs). Usually, mRNA enters the cell by endocytosis. There are specific receptors in endosomes that can recognize RNA, such as TLR3 for double-stranded RNA (dsRNA) and TLR7/8 for single-stranded RNA (ssRNA). Other receptors in the cytosol—e.g., RIG-I, MDA5, NLRP3, and NOD2—recognize exogenic RNA [9].
The recognition of RNA activates transcription factors called IRF3 and NF-κB. NF-κB induces inflammation, whereas IRF3 inhibits the translation of exogenous RNA via interferon synthesis [9,10]. IFNs regulate various cell functions by inducing expression of IFN-stimulated genes, e.g., signal transduction pathways of Janus kinases and transcription activators [10]. The nuclear factor NF-κB plays a major role in a wide range of cell functions, including launching the innate immune response mechanism that targets viral nucleic acids [10]. NF-κB is found in various mammalian cell types, including immune cells such as macrophages and dendritic cells.
NF-κB activation directly impacts the function of immune cells [11], such as the differentiation of dendritic cells into antigen-presenting cells, T-cell differentiation, recruitment of neutrophils to an inflammation site, and macrophage polarization. At the supramolecular level, NF-κB plays a pivotal part as a transcription factor. When activated, it induces transcription of pro-inflammatory genes [11,12], leading to the synthesis of cytokines, chemokines, cell cycle regulators, anti-apoptotic factors, and adhesion molecules [10,11,12] (see Table 1).

Table 1.
Biomolecules that are synthesized in response to NF-κB activation.
The ability of RNA to induce an immune response through activation can be regarded both as an advantage and a limitation of RNA-based therapeutics. On the one hand, an mRNA vaccine is an adjuvant itself that facilitates the survival and proliferation of immune cells presenting the target antigen. On the other hand, the inappropriate activation of the innate immune response (mostly due to IFN) can lead to insufficient expression of the target gene and weaken the mRNA’s effect in non-immunotherapy settings [20]. In the text below, we will focus in detail on the specific pathways of innate immune response activation in the context of the development and clinical application of mRNA-based therapeutics (Figure 1).
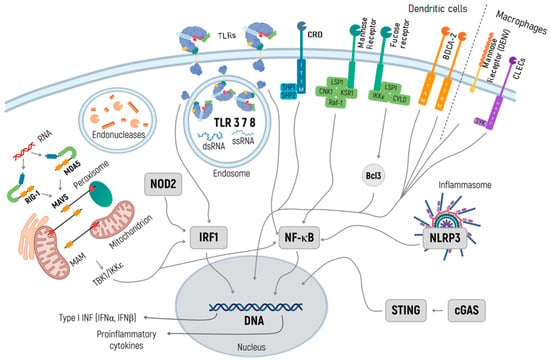
Figure 1.
Specific pathways of the innate immune response activation in the context of development and clinical application of mRNA-based therapeutics. TLRs in endosomes recognize both dsRNA and ssRNA and activate transcription factors NF-κB and IRF 1. NF-κB induces inflammation, while IRF3 launches the transcription of IFN genes, which inhibit the translation of exogenous RNA. Through binding to dsRNA and triggering IRF1 and NF-κB, RIG-I and MDA5 launch signaling cascades initiating the transcription of a type I IFN gene. NOD2 recognizes the exogenic RNA and activates IFN gene transcription via IRF1. The inflammasome appears in response to any exogenic particles, particularly delivery systems, and, by activating NF-κB, launches the transcription of proinflammatory cytokines. The cGAS/STING pathway recognizes the DNA and rare RNA and initiates the transcription of proinflammatory cytokines. RIG-I and MDA5 are activated by immunostimulatory RNA, for example, viral RNAs. They then undergo conformational changes that expose and multimerize their CARDs (not shown for simplicity), which allows homotypic CARD–CARD interactions with MAVS. MAVS is anchored with its TM into mitochondria, mitochondrial-associated membranes (MAMs), and peroxisomes, and relays the signal to TBK1 and IκB kinase-ε (IKKε). This signal activates IRF3 and IRF7, which together with the transcription factor NF-κB, induce the expression of type I INF and other genes. When designing RNA-based therapeutics, one should be aware of the endonuclease activity in endosomes, and further in lysosomes, and consider using RNAse inhibitors.
2. Innate Immune Response Pathways Recognizing mRNA
2.1. RIG-I and MDA5 Signaling
PRRs such as helicase proteins RIG-I (retinoic acid-inducible gene I) and MDA5 (melanoma differentiation-associated protein 5) are key participants in the early phase of the immune response to viral infections. RIG-I and MDA5 (together called RIG-I-like receptors or RLRs) are localized to the cytoplasm in the phosphorylated state and inactive conformation [21,22]. Binding to PAMP (usually with dsRNA or other types of RNA [22]) sequentially leads to conformational transformation, dephosphorylation of the C-terminal domain (CTD) and the N-terminal caspase activation and recruitment domains (CARD1 or 2), and ubiquitination [23,24]. The activation of RIG-I and MDA5 is accompanied by their oligomerization on dsRNA, with ubiquitin marks playing a pivotal role in this process [25,26].
Next, the oligomerized and ubiquitinated complexes of RIG-I or MDA5 with dsRNA interact through CARD domains with mitochondrial antiviral signaling (MAVS) proteins [27], which consequently activate transcription factors NF-κB and IFN-regulatory factors IRF3/7 through the assembly of kinase complexes IKKα/β/γ and TBK1/IKKΣ (TANK-binding kinase 11/inhibitor of nuclear factor kappa-B kinase subunit epsilon), respectively [28,29]. NF-κB triggers the expression of IL-1β and IL-6, IRF3 triggers the expression of IFNβ and INFγ, while IRF7 initiates the expression of IFNα [30]. IFNs are secreted out of the cell and bind to their receptors, resulting in a positive feedback signaling loop and increasing local secretion of IFNs and other immunostimulatory factors, leading to the antiviral response, RNA degradation, and suppressed translation [31].
Laboratory of genetics and physiology 2 (LGP2) is the third and least well-understood member of RLRs. It modulates the function of RIG-I and MDA5 during viral infection in two opposite ways. On the one hand, it works with MDA5 to increase the sensing of RNA viruses. LGP2 primarily binds the ends of dsRNA or even coats the molecule and then incorporates itself into MDA5 filaments. Thus, it enhances the interaction between MDA5 and RNA, which leads to an increase in downstream signaling and a type I IFN response [32,33]. On the other hand, LGP2 suppresses RIG-I signaling by applying different mechanisms, such as immunostimulatory RNA sequestration, prevention of RIG-I binding to MAVS, and inhibition of RIG-I ubiquitination [32,34].
While developing mRNA vaccines, one has to consider the activation of RIG-I and MDA5 because their main ligand—dsRNA—is a typical side product of IVT. In the past few years, IVT has been used as a dominant technology in mRNA production for therapeutic applications. The phage RNA polymerase, the pivotal enzyme used in IVT, is known to have RNA-dependent RNA polymerase activity and takes part in the assembly of ssRNA higher-order structures [31]. Additionally, phage RNA polymerase generates 5′-phosphorylated RNA, which is also typical for viruses and whose double-stranded version is a powerful RIG-I agonist [22,35].
Recent studies showed that modulating the activity of RLRs and other PRRs can significantly boost the yield of the target protein, ensuring a better efficacy of both non-immunotherapeutic modalities. For example, the reduced activation of RIG-I and other PRRs due to the chemical modification of mRNA leads to a 4-fold rise in erythropoietin expression and a significant increase of hematocrit and prevents the production of surfactant protein B (SP-B), which is lethal for mice [36].
The stimulation of RIG-I 5′ppp-RNA enhances the protective antiviral response against influenza (IAV-influenza virus) and chikungunya virus [37]. The stimulation of MDA5 leads to a 40% tumor growth delay in mouse models [38]. The available data about the mechanisms of RIG-I and MDA5 action and about their similarities and differences open up the opportunity for regulating their activity in order to improve the efficiency and safety of non-immunotherapeutic applications as well as vaccines against infectious diseases and cancer.
As a starting point for the RIG-I and MDA5 activity control strategy, one can employ information about the size of the therapeutic mRNA in question. It has been established that dsRNAs activate RIG-I and MDA5 in different ways: RIG-I preferentially recognizes short sequences (no longer than 2 kb), and MDA5 recognizes long dsRNA sequences [39].
At the same time, RIG-I initiates IFN synthesis in response to at least 20 bp of dsRNA in length [40], while MDA5 requires at least 100 bp [41]. For the RIG-I stimulation, one can use miRNA and siRNA that are 20–24 bp in length. Hence, the signaling pathway in question can be determined based on the size of the RNA to be delivered.
Another factor in controlling RLR activation is the desired duration of the therapeutic effect from delivered mRNA. In the case of vaccines against communicable diseases, where mRNA plays the role of adjuvant and thus should not stay in the cell for long, modulation of the RIG-I is required. It is RIG-I that is responsible for the initial immune response to exogenous RNA [42]. In contrast, the stimulation of MDA5 could come in handy when we need a prolonged therapeutic effect, as in the production of self-amplifying RNA for non-immunotherapeutic applications. MDA5 maintains anti-inflammatory signaling for up to 5 days [43].
A precise understanding of RIG-I and MDA5 signaling pathways can be useful for designing therapeutic vaccines against specific communicable diseases. Consequently, NF-κB activation via an IκBα-independent pathway, which is typical for MDA5-signaling [22], can be used to overcome the NF-κB pathway suppression, related to a decline of IκBα phosphorylation associated with Mycobacterium tuberculosis infection [44]. The activation of RIG-I in turn initiates caspase-1–dependent inflammasome activation and processing of mature IL-1® [45], which can also compensate for some processes, as observed in tuberculosis [46].
The activation of RIG-I and MDA5 in non-immunotherapeutic approaches can be diminished with standard methods for decreasing the immunogenicity of RNA, in particular by capping, reducing the uridine (U) content via nucleoside modifications, polyadenylation, reducing the number of double-stranded products of in vitro transcription, and by inhibition of PRRs [47]. As a specific way to decrease RLR signaling, one can use the cap-1 structure, which prevents RNA from binding to both RIG-I and MDA5 [48,49]. Another possible way to reduce the activity or even inhibit RIG-I completely is to introduce at least a single unpaired 5′ triphosphorylated nucleotide or a 3′ overhang (next to a 5′ triphosphate end at a dsRNA terminus) and to reduce the number of phosphate groups at the 5′ end [22].
Viral proteins, for example, NS-1 influenza protein and protein V in paramyxoviruses, are capable of inhibiting RIG-I and MDA5, respectively [50,51], and such viral proteins can reduce immunogenicity if delivered together with RNA. Viral RNA, its derivatives, and imitation of viral RNA structure can be regarded as possible stimulators of RLR activity and could be used as antivirals, cancer therapeutics, and vaccine adjuvants [52]. It has been demonstrated that an optimal protective effect can be achieved with the simultaneous activation of RIG-I and MDA5 [53,54].
At the same time, the immune response can be modulated. For instance, the administration of polyinosinic:polycytidylic acid [poly(I:C)] results in a response with the prevalence of IFN III [55] and can help to minimize the adverse effects of IFN I activation [56].
2.2. TLR Signaling
Toll-like receptors (TLRs) are a major group of receptors that belong to the PRR family [57,58,59]. TLRs are the transmembrane proteins capable of recognizing molecules frequently found in pathogens (PAMPs) or molecules released by damaged cells (damage-associated molecular patterns: DAMPs) [59]. TLRs can be engaged by various viral, bacterial, and mucosal structures as well as components of damaged or dying cells [58,59,60,61,62]. They trigger the signaling cascades that amplify the transcription of immune-defense genes [62,63]. Considering the constitutive membrane expression on various cell structures, TLRs can be viewed as innate-immunity guards against invading microbial pathogens at the frontier of host defense [64].
TLRs consist of two domains: an extracellular transmembrane domain and an intracellular C-terminal domain (Figure 2) [65]. The N-terminal domain, also known as an ectodomain, is located outside the plasma membrane and has the shape of a horseshoe. It acts as a receptor and contains 19–25 highly conserved leucine-rich repeats (LRRs), each consisting of 24–29 amino acid residues [66,67]. A similar structure can be found in variable receptors of lymphocytes in early vertebrates [68]. Tandem LRRs have also been discovered in certain bacteria [69] and most plants, where they are a part of the protein structures responsible for infection resistance [70,71]. This observation highlights evolutionary conservatism and structural universality in terms of recognition of PAMPs and DAMPs. Notably, glycosylation of the TLR ectodomain has an impact on ligand–receptor interaction [72].
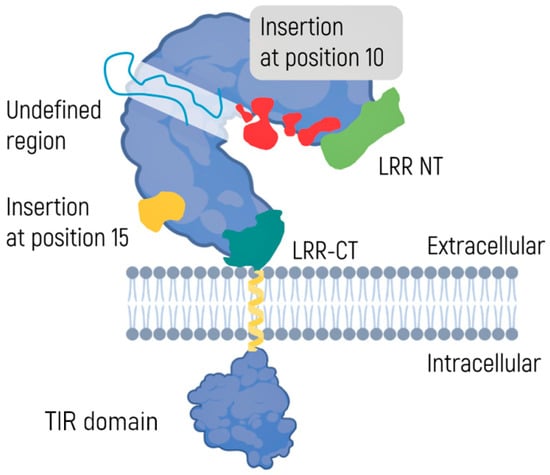
Figure 2.
A toll-like receptor: All TLRs are integral membrane glycoproteins with an N-terminal ectodomain and a signal transmembrane domain. The ectodomain of TLR7, TLR8, and TLR9 is depicted, with the LRR solenoid shown with a gray molecular surface, and the N- and C-terminal flanking regions shown in green and purple. Insertions at position 10, indicated in red, might contribute to the information on the PAMP binding site. The insert at position 15, indicated in yellow, is expected to originate on the convex face of the TLR. Also shown is a scheme of the transmembrane domain (presumed to be a single a-helix) followed by a molecular representation of the Toll-IL-1 and IL-18R (TIR) domain [66].
The intracellular C-terminal domain consists of ~150 amino acid residues and is called Toll/interleukin 1 receptor (IL-1R) homology or TIR. The domain recruits adapter proteins and launches intracellular signals [73,74]. To date, 10 human subtypes of TLRs have been identified [75]. Although the functions of different TLRs may overlap, each is characterized by a specific set of recognized PAMP or DAMP molecules. Some TLRs, such as TLR1, TLR6, and TLR10, can form homo- or heterodimers, but the biological role of these complexes is yet to be elucidated [76].
TLRs are located on the external cell membrane and the surface of intracellular vesicles, endosomes, lysosomes, endolysosomes, and other structures. On the surface of the cell, there are TLR1, TLR2, TLR4, TLR5, TLR6, and TLR10, and on the inside of the cell, there are TLR3, TLR7, TLR8, TLR9, TLR11, TLR12, and TLR13 [77]. TLR2 and TLR4 can also be found in dendritic and epithelial cells [78]. The location of TLRs is determined by their functions and depends on the types of pathogens recognized.
The binding of TLRs to a ligand leads to the internalization of the complex and proteolytic cleavage of the receptor ectodomain; this process is required to initiate TLR signaling [62,79]. The engagement of TLRs initiates a complicated cascade of biochemical reactions that starts with the C-terminal TIR domain [80]. This domain interacts with similar TIR domains of adaptor proteins, which determine the specificity of TLR signaling by their type [77,81].
A collection of PAMPs, which characterizes a given pathogen, stimulates a specific set of TLRs. In this way, the resultant PAMP signal affects the speed and intensity of the immune response and determines the balance among major populations of T helper cells (Th1/Th2/Th17/Treg) [82]. In other words, due to the small diversity of TLRs, the innate immune system is capable of recognizing different groups of pathogenic and symbiotic microorganisms. In most cases, this arrangement enables the immune system to choose the optimal type of secondary immune response required for homeostasis [83,84].
The activation of interferon-regulatory factors (IRFs) and NF-κB are the key steps of the TLRs’ signaling pathway [85]. The activation of NF-κB induces the expression of proinflammatory cytokines, while the activation of IRFs provides optimal production of type I IFNs [86].
The production of IFNs works simultaneously in two directions by hindering and facilitating mRNA vaccines. On the one hand, it induces activation of protein kinase R (PKR) and 2′-5′-oligoadenylate synthetase (OAS), and these systems inhibit translation and cause degradation of cellular and ribosomal RNA, respectively, consequently decreasing the efficiency of mRNA-based products [87,88]. On the other hand, the elevated production of IFNs stimulates the immune response against the target antigen and this effect may be useful in the case of mRNA vaccines [89].
Discussing the development of mRNA-based therapeutics, TLRs are of particular interest because they are located on different components of cells and are responsible for different ways of antiviral immunity. TLRs can significantly affect the efficacy of mRNA-containing products [90,91]. Except for TLR1 and TLR5 (which recognize bacterial lipoproteins and flagellin), almost all subtypes of TLRs are involved in the recognition of viral particles [92,93]. Namely, TLR2, TLR4, TLR6, and TLR10 recognize glycoproteins of the virus envelope and some viral enzymes. At the same time, TLR3 recognizes dsRNA, whereas TLR7 and TLR8 recognize ssRNA, and TLR9 recognizes ssDNA [93]. Synthetized mRNA preparations contain dsRNA impurities in addition to ssRNA and thus can be recognized by TLR3, causing sustained production of type I IFN [94]. Exogenous ssRNA, entering the cell within a therapeutic formulation, can also cause the production of IFN-I by engaging TLR7 and TLR8 [95,96].
During evolution, many viruses have developed ways to resist the host immune surveillance mechanisms [97]. Such mechanisms include inhibition of TLR synthesis in the host’s cells [98], a reduction in TLRs’ activity through disruption of their plasma membrane localization [99], and various ways to impair adapter proteins of TLRs [100,101]. Some viruses have developed proteins that, by binding to TLRs, can suppress their function directly, reducing IFN production and disrupting the NF-κB pathway [101,102,103]. One can use these mechanisms to create therapeutic mRNA products, which control the amplitude of an immune response or shift the balance of T helper populations (Th1/Th2/Th17/Treg). Thus, TLR agonists are well tolerated and significantly increase the efficacy of mRNA vaccines.
Moreover, the FDA has recently approved TLR2, TLR4, and TLR9 agonists as adjuvants for the BCG vaccine [104,105,106]. The latest SARS-CoV-2 pandemic has spurred the development and use of mRNA vaccines that have shown high safety and efficacy [107]. Such promising results are based on almost four decades of research, having led to the development of reliable methods for protecting artificial mRNA from degradation and enhancing its translation in the host cell [108,109,110]. Early methods included 5′-capping, 3′-polyadenylation, various base modifications such as 1-methyl pseudouridine (m1Ψ), and the use of an optimized Kozak consensus sequence in the 5′UTR region, etc. [111,112,113,114].
Over the last two decades, a number of agonists have been designed for almost all types of TLRs, and some of them have reached clinical use as vaccine adjuvants [115,116,117]. This, together with emerging methods for protecting mRNA against degradation, opens bright prospects for modulation of the primary immune response through selective action on various members of the TLR family [118] as well as extensive possibilities regarding manipulation of various components of innate immunity.
Artificial mRNA is characterized by its own adjuvant activity because it can stimulate TLR7/8. This effect can be enhanced by additional agonists, such as protamine, a cationic TLR7 activator protein that will enhance the production of type I IFNs and potentially increase the amplitude of a secondary immune response [118].
Karikó et al. showed that removing dsRNA from the mRNA mixture using high-performance liquid chromatography enhances translation of the target mRNA up to 1000-fold. Moreover, the purified mRNA causes less induction of IFNs and inflammatory cytokines [94]. This study unveils two significant points. Firstly, chromatography application in the production of mRNA vaccines helps to improve significantly the quality of final vaccine preparation. The technique aims to purify samples from non-target nucleic acids that are, among other things, ligands for TLRs. In light of such advanced purification methods, there are ongoing discussions regarding whether base modifications are necessary, as the level of translation enhancement is the greatest when mRNA is unmodified. The second point refers to the role of dsRNA in an immune response. Compared with other types of exogenous RNAs, it is the main trigger of cellular response. Hence, one should keep in mind both points when creating mRNA vaccines.
Alternatively, a reduction of immunogenicity can be attained by using modified bases such as pseudouridine or m1Ψ, which change mRNA secondary structures (hairpins) and thus prevent receptor TLR3 or RIG-I from recognizing mRNA [119]. As a result, TLR3 continues downregulating the production of type I IFNs [120,121]. Of note, this approach shows better performance than chromatographic purification for the removal of dsRNA from the original product. Because of the large amount of RNA in the cell, RNA polymerase can synthesize a small number of antisense oligonucleotides and trigger TLR3. Thus, the incorporation of modified bases into mRNA decreases to a certain extent its immunogenicity, avoiding its degradation and eliminating the need for its thorough purification [94,122,123,124].
Furthermore, modified RNA bases can change mRNA’s secondary structure and, therefore, prevent its recognition by TLR3 and RIG-I receptors [119]. There is also indirect evidence that the presence of m1Ψ in the mRNA structure alters the steric organization of hydrogen bonds and prevents them from binding to TLR7/8 [119,123]. In this way, the incorporation of the m1Ψ nucleoside allows, in a sense, to hide RNA from immune surveillance and extend its lifetime in the cell, thereby potentially increasing the protein yield.
Nevertheless, it would be naive to believe that the steric changes associated with the introduction of m1Ψ will not alter the dynamics of the translation process [125]. Although most research has documented a significant enhancement of protein expression [126], free energy calculations have shown that the presence of m1Ψ in the mRNA strand may negatively affect protein synthesis by reducing the thermodynamic efficiency of interaction with the ribosome [127,128,129]. This assumption has later been experimentally confirmed only for some artificial mRNAs [130]. Thus, the incorporation of m1Ψ into the mRNA strand allows it to extend its lifetime significantly and prevents the activation of TLRs and RIG-I, thereby significantly reducing immunogenicity.
The development of various delivery systems opens up opportunities for the simultaneous use of several TLR agonists and non-TLR adjuvants in one mRNA product [109,115,131,132]. This combined approach helps to shape a desired immune response [133,134]. For example, simultaneous stimulation of TLR7/8 and TLR/9 can elicit a strong Th1 response and a high level of IgG2a antibodies [133,134]. In the meantime, mRNA administration, together with simultaneous stimulation of TLR4 and TLR9, elicits a strong Th2 response and a high level of IgG1 [133,134]. Hence, this evidence confirms the possibility of using combinations of TLR agonists with various mRNA constructs in one therapeutic product to induce an immune response with specified parameters.
2.3. NOD-like Receptors
Another equally important group of PRRs is NOD-like receptors (NLRs). This is a conserved family of intracellular receptors that are thought to be sensors capable of recognizing DAMPs and PAMPs in the cytoplasm [135]. To date, 22 NLR genes have been identified in humans [136].
The proteins of the NLR family share many structural features, whereas the structure, in general, is similar to that of TLRs [137,138]. All NLRs consist of three domains, two of which (C-terminal and central domains) are common for all members of the family [8] (Figure 3). The extracellular C-terminal leucine-rich repeat (LRR) domain has the form of a horseshoe. The central nucleoside-binding domain is a component of the larger NACHT domain. The NACHT domain oligomerizes during NLR activation using ATP energy and interacts with an N-terminal effector domain.
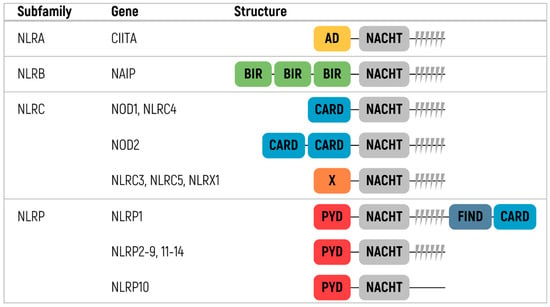
Figure 3.
Classification and protein structure of human NOD-like receptor family [8,135]. NLR—NOD-Like Receptors, CIITA—Class II Major Histocompatibility Complex Transactivator, NAIP—NLR family apoptosis inhibitory protein, NOD1—Nucleotide-binding Oligomerization Domain-containing protein 1, NOD2—Nucleotide-binding Oligomerization Domain containing protein 2, NLRP—Nucleotide-binding oligomerization domain Leucine-rich Repeat and Pyrin domain, CARD—Caspase Activation and Recruitment Domain, BIR—Baculovirus Inhibitor of apoptosis protein Repeat, PYD—Pyrin Domain, AD—Activation Domain.
The N-terminal domain differs across NLRs and divides the family into subfamilies [135,136,139,140]. There are four different subfamilies: NLRA, NLRBs (also known as NAIPs), NLRCs, and NLRPs. The NLRA subfamily contains an acidic transactivating domain. CIITA (MHC class II transcription activator) is the only member of the NLRA subfamily. The NLRB subfamily contains an N-terminal BIR-like domain. NAIP is the only protein of the subfamily detected in humans.
The two most prominent of these subfamilies are NLRC and NLRP. The NLRC proteins, namely NOD1, NLRX1, NOD2, and NLRC3–5, have one or more caspase activation and recruitment domains (CARD). NLRP proteins have pyrin domains and are the best characterized among other subfamilies. This subfamily consists of 14 members named NLRP1–14 [8,135,136].
NLRs are broadly categorized into two groups according to their ability to form inflammasomes. The first group is called canonical and signals through the formation of caspase-1–activation inflammasomes. Another group is called non-canonical because NLR proteins act through either an alternate caspase or an inflammasome-independent mechanism. Of particular interest are NLRPs, whose function is well-studied [8]. Some proteins, such as NLRP3, have both canonical and non-canonical functions.
NLRs take part in antigen presentation (NLRC5 and CIITA), pathogen/damage sensing (NOD1-2, NLRC4, NAIP, and NLRP3), inflammasome activation (NLRP3, NLRP1, NLRP2, NLRP6, and NLRP7), and inflammatory signaling inhibition (NLRC3, NLRC5, NLRX1, and NLRP6) [141]. Furthermore, Lupfer and Kanneganti showed that NLRP2, NLRP5, and NLRP7 are involved in non-immune pathways such as embryonic development [142]. The study highlighted alternative functions for some NLRs in different cell types and a multitude of activation mechanisms that provide independent downstream effects for other NLRs.
In the next section, we are going to discuss inflammasome activation in detail on the example of NLRP3 inflammasome activation. Here, it is worth discussing other NLR functions that could be relevant to the development of mRNA vaccines.
The CIITA expression is activated by the expression of IFNγ and is regulated mostly at a transcriptional level [143]. It acts as a transcriptional coactivator implicated in the regulation of MHC class II expression. However, different viruses and other pathogens, such as HIV, VZV, human cytomegalovirus, Epstein–Barr virus, and mycobacteria, can inhibit CIIPA and thereby suppress MHC II gene expression [143,144,145,146,147,148].
The NLRC5 expression is also regulated by IFNγ and can be additionally induced (although less effectively) by type I IFNs, LPS, polyinosinic:polycytidylic acid [poly(I:C)], and some viruses [149,150]. It was first identified as a transcriptional regulator of MHC class I genes and referred to as CITA or class I transactivator [151]. At the same time, there was quite contradictory data showing that NLRC5 may play both negative and positive roles in type I IFN production [150]. On the one hand, NLRC5 was shown to interact directly with NF-κB or RIG-I and repress TLR4 signaling and type-I IFN responses. Meanwhile, it could enhance type I IFN production [152].
NOD2 is known to control cytokine expression through its activation of NF-κB and p38 MAPK-dependent signaling in response to bacterial infection. However, it also takes part in the response to exogenous dsRNA and ssRNA [153]. dsRNA molecules induce OAS-1 to interact with NOD2. In turn, this leads to the production of 3′-5′ oligoadenylate synthetase type 2, which activates antiviral responses, particularly RNase L activation.
Furthermore, it was shown that NOD2 functions as a cytoplasmic viral PRR and recognizes viral ssRNA. It utilizes the adaptor protein MAVS to activate IRF3, leading to the production of IFNβ [154]. Other NLRs, such as NOD1, NLRC4, NAIP, or NLRC3, did not show the ability to activate IRF3 in response to ssRNA. Hence, NOD2 utilizes either RICK or MAVS depending on the stimulus (e.g., MDP or. ssRNA) to activate either NF-κB and MARK or IRF3, respectively. It is worth mentioning that NOD1 also activates NF-κB and MAPK signaling pathways, recognizing bacterial γ-D-glutamyl-meso-diaminopimelic acid [137,155]. NLRX1 is a negative regulator of MAVS signaling during viral dsRNA and ssRNA invasion [153,156]. This NLR protein is interesting as it is located on the mitochondria and contains mitochondrial targeting sequences. NLRX1 interacts with MAVS and prevents its binding to RIG-I. This interaction leads to disruption of the activation of NF-κB and IRF3 and the inhibition of type I IFN and proinflammatory cytokine production [153].
One could consider modified muramyl dipeptide molecules as potential adjuvants for mRNA vaccines [157]. These molecules are produced during bacterial peptidoglycans degradation, and they induce NALP3-mediated activation of caspase-1 and maturation of proIL-1β [158]. The muramyl dipeptide molecules also require NOD2 to boost humoral and cellular immune responses to their optimum level.
Several studies show that the cooperation of NLRs and TLRs is worth considering when developing mRNA vaccines. NLR stimulation in various combinations with TLRs causes stable polarization of an immune response and shifts the balance between Th1/Th2 populations. The synthetic peptide FK-156 alone can stimulate NOD1, launching a Th2 response [159,160]. Furthermore, the bi-functional ligand CL-429 can activate both NOD2 and TLR2 and, therefore, suppress virus-induced inflammation [161].
NLR agonists lead to persistent changes in the innate immune system affecting immunity to various infections [157,162]. Such “memory” of innate immunity is called trained immunity [163,164], in which PRRs play a substantial part. It was shown that bacillus Calmette-Guérin (BCG) interacts with TLR2 or TLR4 and signals via NOD2 pathways, leading to the production of IL-1β, TNF, IL-6, and IL-32 cytokines [165]. The combined effect on various types of PRRs in the context of the application of mRNA opens vast opportunities for managing immune responses.
2.4. NLRP3 Inflammasome Activation
As a representative of the nucleotide-binding leucine-rich repeat proteins (NLRPs) family, the NLRP3 protein mainly contains three components: an LRR domain, the adenosine triphosphatase (ATPase) domain known as NACHT, and the N-terminal pyrin domain (PYD) [166,167].
NLRP3 responds to numerous stimuli, including viral/bacterial DNA and RNA, pore-forming toxins, silica, asbestos, uric acid, ion flux, and ATP [168]. Owing to such a broad range of NLRP3 activators, direct binding of all these activators to NLRP3 seems unlikely. Recruitment of different cofactors might help to determine ligand specificity.
Once the LRR domain detects a pathogen or an endogenous danger signal, the oligomeric NLRP3 inflammasome binds its subunits through NACHT domains. Then, the complex recruits ASC through PYD–PYD interactions thereby nucleating PYD filaments of ASC. The adaptor protein ASC attracts pro-caspase 1 via CARD–CARD interactions and induces the self-cleavage of caspase 1 [169].
The activation of the NLRP3 inflammasome has been described as a two-step signal model. The first signal (priming) is provided by TLR and cytokine receptors, such as the interleukin 1 receptor (IL-1R) and tumor necrosis factor receptor (TNFR) [170]. After the recognition of microbes or inflammatory cytokines by NF-κB-activating receptors, NF-κB is immediately translocated to the nucleus with the assistance of FADD and caspase 8. They raise the amount of NLRP3 and pro-IL-1β by boosting transcription of relevant genes and translation of the resultant mRNAs [170,171].
The second signal (activation) is triggered by diverse stimuli, including pore-forming toxins, ATP, and different particulates [172,173]. Owing to the second step, the NLRP3 inflammasome completes the assembly and activates caspase 1. The latter processes pro-IL-1β and pro-IL-18 into mature proteins at the microtubule-organizing center, distributed in the perinuclear and punctate regions [166,174].
It has been suggested that NLRP3 inflammasome activation is mediated by intricate cellular signaling events, including potassium efflux, calcium overload, reactive oxygen species (ROS) production, mitochondrial dysfunction, and lysosomal rupture [175,176,177,178,179]. Yet, the specific mechanism underlying ion flux changes or organelle dysfunction during the activation of the NLRP3 inflammasome remains controversial.
Available experimental data suggests that human macrophages sense all bacterial RNA components (mRNA, tRNA, and rRNAs) and synthetic ssRNA, activating the NLRP3 inflammasome, whereas murine cells preferentially recognize bacterial mRNA [180]. Other studies have shown that DHX33 launches the assembly of NLRP3–ASC–caspase 1 by binding directly to polyI:C dsRNA or to E. coli total RNA [181].
A number of recent articles have linked NLRP3 inflammasome activation to mitochondrial dysfunction. In particular, the production of reactive oxygen species by phagocytosis or mitochondria is thought to be critical for the activation of the NLRP3 inflammasome in response to various stimuli, including microbial RNAs and endogenous noncoding RNA transcripts of the Alu retrotransposon [168,182].
The dysregulated NLRP3 inflammasome has been implicated in the pathogenesis of several autoimmune and autoinflammatory conditions, infections, and tumorigenesis [183,184]. Elucidating the role of the NLRP3 inflammasome in the pathogenesis of various disorders is a matter of ongoing research. Undoubtedly, a further clarified and precise understanding of the molecular mechanism behind the NLRP3 inflammasome regulation will guide the development of new effective therapeutics.
Being a key component of inflammatory response and pyroptosis pathways, NLRP3 activation has a detrimental effect on mRNA transfection efficiency [185]. According to clinical data on the administration of the mRNA-1273 COVID-19 vaccine [186], this activation also may contribute to some adverse events of mRNA therapeutics.
As activated TLRs prime an NLRP3-mediated response, potential contaminants, such as LPS and bacterial RNA components (mRNA, tRNA, and rRNAs), should be removed. Furthermore, the formulation of mRNA vaccines should be carefully examined to avoid NLRP3 activation.
Forster Iii J et al. have tested a panel of six mRNA lipid nanoparticle (LNP) formulations while varying concentrations of different lipid components. Of particular interest were nanoparticles containing a high concentration of the ionizable DLin-MC3-DMA lipid in tandem, highly cationic lipid DOTAP, and a low cholesterol concentration. Such nanoparticles dramatically activate the NLRP3 inflammasome, with associated cytotoxicity and decreased GFP transfection efficiency [185]. These data indicate that optimization of key lipid concentrations in mRNA LNPs partially affects the degree of NLRP3 inflammasome activation. Hence, one should examine the lipid composition of an mRNA transfection tool in order to balance endosomal escape, inflammasome activation for disease-specific expression, and the development of self-adjuvant mRNA delivery systems.
Owing to the extensive participation of the NLRP3 pathway in several inflammatory diseases and cancers, a variety of chemical NLRP3 inhibitors has been proposed, with some of them tested in a clinical setting [173]. Nevertheless, because the usefulness of this pathway for the field of mRNA therapeutics only recently received well-deserved attention, our literature search did not reveal comprehensive data on the influence of known NLRP3 inhibitors (including Glyburide, 16673-34-0, FC11A-2, VX-765, VX-740, BHB, MCC950, CY-09, tranilast, OLT1177, oridonin, parthenolide, and Furanochalcone Velutone) on mRNA transfection efficiency [187]. A notable exception is BAY 11-7082, which results in a reproducible increase in the expression of OCT4 upon synthetic mRNA transfection into human skin cells [188]. Therefore, the utility of NLRP3 inhibitors for mRNA gene therapies is a promising subject for further research.
The central role of NLRP3 in inflammatory responses to various external stimuli has made this complex a common target for the development of vaccine adjuvants. Among clinically relevant NLRP3 adjuvants, there are aluminum salts [189], saponins [190], and emulsions that commonly include water-in-oil (W/O) and oil-in-water (O/W) such as prototypes Montanide 720 and MF59 [164], TLR activators such as TLR4 agonists MPL-A or GLA [191], TLR3 ligands [dsRNA analogs such as poly(I:C)], TLR5 agonists (flagellin), TLR7/TLR8 agonists (imidazoquinolines such as imiquimod), TLR9 activators (CpG oligodeoxynucleotides), chitosan [192], trehalose-6,6′-dibehenate [193], calcium phosphate nanoparticles [194], and PLGA [195].
To sum up, the importance of NLRP3 as the target of conventional vaccine adjuvants is generally recognized, and this complex is studied extensively. Nonetheless, its activation by mRNA therapeutics should be wisely balanced via tuning of transgene structure and carrier composition.
2.5. C-Type Lectin Receptors
C-type lectin receptors (CLRs) are a superfamily of proteins, which, in the broader sense, contain a C-type lectin-like domain [196]. These receptors can be divided into two groups based on their ability to recognize carbohydrate or non-carbohydrate ligands. Those that bind carbohydrates function in either a Ca2+ dependent or independent manner. CLRs are transmembrane and soluble pattern recognition receptors. They are expressed mostly by myeloid cells, such as dendritic cells (DC) and macrophages, and recognize both PAMPs and DAMPs [197,198]. Binding CLRs to PAMPs induces signaling, internalization of a pathogenic organism, its lysosomal degradation, and presentation of the epitopes to lymphocytes.
We are going to discuss a particular group of CLRs, capable of recognizing specific carbohydrate sequences. In terms of therapeutic mRNAs, special attention should be given to dendritic cells immunoreceptor (DCIR), blood dendritic cells antigen 2, CD303 (BDCA-2), and dendritic cell-specific ICAM-3 grabbing non-integrin (DC-SIGN) (Figure 4).
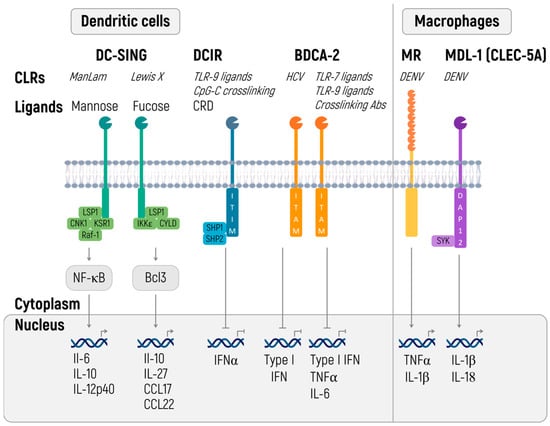
Figure 4.
C-type lectin receptors (CLRs) shape innate and adaptive immune responses. CLRs induce innate and adaptive immune responses. Certain CLRs contain ITIM domains and signal via SHP1 and SHP2 phosphatases, whereas other CLRs signal via their ITAM motif. DC-SIGN signaling is carbohydrate specific (CRD) and either signals via Raf-1 signalosome or IKKε and deubiquitinase CYLD, with distinct outcomes [199].
Apart from carbohydrate patterns of a pathogen, DCIR and BDCA-1 are capable of binding ssDNA [199]. Signaling from both receptors leads to the inhibition of the inflammatory response. DCIR, containing the immunoreceptor tyrosine-based inhibitory motif (ITIM), inhibits TLR8-mediated production of IL-12 and TNF through effects on various signaling pathways [200], CpG-ODN-induced expression of IL-1® and IL-6 [201], and STAT1-dependent production of IFN I under the influence of M. tuberculosis during the infection [202].
BDCA-2 transmits the signal through the immunoreceptor tyrosine-based activation motif (ITAM) of the adaptor molecule FcRy [203]. In contrast to most cases, this process launches the anti-inflammatory signals that perhaps are related to the activation of phospholipase Cγ2 (PLCy2) and consequent mobilization of Ca2+, which inhibits activation of a protein called myeloid differentiation primary response 88 or MyD88, responsible for TLR-mediated production of cytokines [204]. BDCA-2 ligation is associated with the suppression of IFN I production and other proinflammatory cytokines that are triggered by TLR7 and TLR9 signaling [203,205]. In this way, the commonality of recognizable patterns of pathogenicity (ssRNA) and the functional antagonism of known receptors of the CLR and TLR families enable investigators to fine-tune an immune response when administering therapeutic RNA [199,204].
In the manufacturing of vaccines against infectious diseases and cancer, it is worth considering the structure of the therapeutic RNAs because this biomolecule should engage TLR7/8 and 9 rather than suppress DCIR and BDCA-2. For non-immunological applications, the increased stimulation of DCIR and BDCA-2 helps to overcome activation signals from TLR and thus to increase the therapeutic effect. Unfortunately, little is known about the specific RNA that would allow for separate stimulation of TLR and CLR. It is a matter of future research.
When producing vaccines against specific infectious diseases, one should keep in mind that CLRs contribute to the polarization of an immune response because of ongoing modulation of the cytokine response. For instance, eliminating DCIR activity restores the production of IL-12 and shifts the immune response toward Th1, which favorably affects the course of M. tuberculosis infection [202].
Strikingly, it was shown that CLRs such as DC-SIGN are involved in virus transmission [202]. For instance, DC-SIGN can mediate the release of HIV-1 virions associated with exosomes and help their dissemination to CD4+ T cells. While binding to DC-SIGN, the HCV particles are kept from lysosomal degradation in immature DC and transmitted to hepatocytes [206].
2.6. The cGAS/STING Pathway
The cyclic GMP–AMP synthase (cGAS)/stimulator of IFN genes (STING) pathway emerged as a critical mechanism linking DNA sensing to induce robust innate immune defense programs [207]. Its activation lacks any pathogen-specific attributes, which sets it apart from several other innate immune signaling mechanisms [208]. Within this pathway, the binding of cGAS to double-stranded DNA (dsDNA) allosterically activates its catalytic activity. Activated cGAS leads to the production of 2′-3′-cyclic GMP–AMP (cGAMP), a secondary messenger molecule and potent agonist of STING [209]. Aside from cGAMP, STING also directly senses other cyclic dinucleotides secreted by some bacteria [210].
cGAMP binds to a stimulator of IFN genes (STING) dimers localized at the endoplasmic reticulum (ER) membrane, thereby causing profound conformational changes that trigger STING oligomerization, its liberation from anchoring factors (such as STIM1), interaction with trafficking factors (for example, SEC24/23 and STEEP), and, finally, incorporation into coatomer protein complex II (COPII) vesicles. While passing through the ER–Golgi intermediate compartment (ERGIC) and Golgi, STING recruits TANK-binding kinase 1 (TBK1), promoting TBK1 autophosphorylation, STING phosphorylation at Ser366, and recruitment of IRF3. The phosphorylation of IRF3 by TBK1 enables IRF3 dimerization and translocation to the nucleus thus inducing expression of type I IFN, IFN-stimulated genes, and several other inflammatory mediators, pro-apoptotic genes, and chemokines.
The activation of STING also leads to NF-κB activation and the formation of LC3+ vesicles (autophagosomes) via a non-canonical mechanism of autophagy. In the end, both STING within autophagosomes and from the Golgi apparatus are trafficked to lysosomes, where STING degradation occurs. Steady-state STING translocation through the secretory pathway in the absence of robust cGAMP stimulation is counteracted by continuous retrograde transport to the ER, mediated by COPI vesicles and facilitated by the interaction between STING and SURF4.
Emerging evidence indicates that, in addition to its well-established role in the sensing of cytosolic DNA, the cGAS–STING pathway is involved in restricting RNA virus infection, possibly indicating crosstalk between the innate sensing of cytosolic DNA and innate sensing of cytosolic RNA [211]. Although cGAS is reported to bind dsRNA, this interaction does not lead to cGAMP production [212]. STING interacts with RIG-I and MAVS [213,214], which are key components of the RNA sensing pathway, indicating that it might participate in the RNA virus-induced cytokine production. Although a concerted action of RIG-I and STING in the RNA virus-induced defense responses has been reported [213,215], the underlying mechanism remains unclear.
A recent study suggests that rather than inducing IFN expression, STING initiates global translation inhibition to restrict the production of both viral and host proteins in a RIG-I/MDA5-dependent but MAVS-independent manner [216]. Thus, the recognition of an RNA virus infection by RIG-I/MDA5 probably results in two distinct responses—one is mediated by MAVS to induce IFNs and cytokines, and the other is mediated by STING to inhibit translation.
Growing evidence also indicates that RNA viruses have developed strategies to antagonize cGAS and STING activity. The nonstructural protein NS4B of yellow fever virus (YFV) was the first reported viral protein that inhibits STING through its interaction, although the mechanism is unclear [209]. Additional studies have revealed that hepatitis C virus (HCV) NS4B, a homolog of YFV NS4B, disturbs STING signaling by attenuating the STING–TBK1 interaction [217]. The papain-like proteases from several coronaviruses such as SARS–CoV–1, human coronavirus NL63 (HCoV-NL63), and porcine epidemic diarrhea virus (PEDV) have been shown to associate with STING and block its dimerization and K63-linked ubiquitination, thereby inhibiting the production of IFNβ [218].
To sum up, the utility of STING for mRNA vaccine treatment may lie in its involvement in the signaling initiated by other immune sensors, such as RIG-I. One should probably avoid the activation of cGAS/STING pathway members because it may lower the efficiency of mRNA translation and mediate cytotoxicity. Nonetheless, at present, there are no reports describing the exact involvement of this pathway in the latest mRNA therapies.
Several reports have detailed the structure-based design of cGAS inhibitors because of cGAS’s amenability to crystallography [219,220,221,222,223]. Most of the cGAS antagonists have been discovered by their manner of binding to the active site and thus competing with either ATP/GTP substrates or the cGAMP product. Another major class of cGAS antagonists competes with DNA for coupling with cGAS and interferes with the initial cGAS activation step [224,225,226,227,228].
Currently, there are two main approaches to identifying STING inhibitors. The first one is to design molecules that occupy the cyclic-dinucleotide–binding site and, therefore, serve as competitive antagonists of STING activators. The second approach is to identify inhibitors that bind to either Cys88 or Cys91 near the transmembrane domain of STING, each subject to palmitoylation [229]. In the present literature review, we failed to find reports of experimental assessment of well-known STING inhibitors such as nitrofurans (C-176 and C-178), indole urease (H-151), nitro fatty acids (NO2-FA and NO2-OA), acrylamides (BPK-21 and BPK-25), and brefeldin A and could not determine whether they influence the efficiency of therapeutic mRNA expression. Given that STING acts in a coordinated manner with RIG-I, strategies, addressing the latter may prevent the activation of an immune response involving STING (described above).
cGAS-STING pathway components and their agonists represent promising vaccine adjuvants [230], and their efficacy is currently being tested. Tse et al. have used a constitutively active mutant (V155M) of STING as a genetic adjuvant for encapsulated in lipid nanoparticles (LNP) mRNA vaccines [231]. The mRNA-encoded constitutively active STINGV155M was most effective at maximizing CD8+ T cell responses at the antigen/adjuvant mass ratio of 5:1. The mRNA-encoded STING V155M increased the efficacy of the mRNA vaccines that encode oncoproteins E6 and E7 of human papillomavirus (HPV). The vaccines led to a reduction in HPV + TC-1 tumor growth and thus prolonged the survival of vaccinated mice. The study offers proof of concept of STING application as an mRNA-encoded genetic adjuvant, and this approach requires further evaluation.
cGAS-STING pathway is known to take part in restricting RNA virus infection. Recently, it has also been recognized as a crucial mechanism that regulates tumor immunity [232]. Recently, Miao et al. created STING-activating mRNA-loaded LNPs and elicited vigorous antitumor immune responses in multiple tumor models, including B16-OVA, B16F10, and TC-1 tumors [233]. The ionizable lipids with a cyclic tertiary amine group in the LNPs not only provide adjuvanticity by stimulating the STING pathway but also facilitate cytosolic delivery of mRNA encoding the antigens. Single-dose vaccination with an optimized STING-activating LNPs loaded with OVA-encoded mRNA (A18 mOVA LNPs) cured 3 out of 11 mice of established B16-OVA tumors. STING has proven to be an efficient genetic adjuvant for LNP-encapsulated mRNA vaccines [231].
2.7. Endonuclease Activity
It has been widely believed that mRNA decay in higher eukaryotes proceeds mainly at the ends of the molecule along two exonucleolytic pathways [234]. Nowadays, endonuclease activity is proven to be important for mRNA degradation [235]. Endonucleases cut mRNA thus forming short products, which are further degraded by exonucleases.
Exosomes are complexes of ribonucleases, which are present in both the nucleus and the cytoplasm and are involved in mRNA breakdown [236]. Exosomes are thought to destroy mRNA solely due to 3′-5′ exonuclease activity, but it has been demonstrated that one of their components has endoribonuclease activity (e.g., in Rrp44). In addition, SMG6 also engages endoribonuclease activity during mRNA destruction [237]. The discovery of other specific enzymes, such as PMR-1, inositol-requiring enzyme-1 (IRE1), RNase L, aldolase C, activator of RNA decay 1 (ARD-1), Ras GTPase-activating protein SH3 domain-binding protein (G3BP), and apurinic/apyrimidinic endonuclease 1 (APE1), has uncovered their participation in the regulation of individual mRNAs [238].
Nonetheless, this is a hindrance to the development of mRNA vaccines. Their in vivo use requires special modifications to IVT mRNA [239]. For instance, mRNAs should contain recognition sequences such as AU-rich elements (ARE) found in the 3′ untranslated regions of many mRNA transcripts and the coding region determinant of c-Myc mRNA [240,241].
3. Applied Aspects: mRNA “Adjuvants” and “Tolerogens”
As mentioned above, mRNA itself can possess some adjuvant properties, when being a part of a vaccine [122,242,243,244]. First and foremost, in vitro synthesized dsRNA and ssRNA can be immunogenic. The former is recognized by TLR-3-related cytoplasmic dsRNA receptors: RIG-I and MDA5 [122]. The latter, if containing unmodified uridine, is recognized by TLR7 [245] and TLR8 [246], and by RIG-I [247] and PKR [248]. Signaling through these receptors causes activation of IRF3, IRF7, and NF-κB, their consequent translocation into the nucleus, and transcription of type I IFN genes (IFN-α and IFN-β) accompanied by the expression of proinflammatory cytokines [249].
There are many ways to reduce the reactogenicity of mRNA in the vaccine. These approaches include efficient purification of IVT mRNA by chromatographic methods [250], the improvement of transcription methods [31,251], polyadenylation of mRNA, and the replacement of uridine with modified nucleotides, which are mostly pseudouridine (Ψ) and m1Ψ [120,123].
Naked RNA is unstable and is quickly degraded by extracellular RNases once it enters the human body [134]. Apart from that, naked RNA cannot be internalized efficiently by the cell. Hence, a variety of transfection reagents have been created to deliver RNA-based therapeutics into the cell, facilitate cellular mRNA uptake, and protect it from degradation [252]. Most RNA transfection agents are characterized by similar adjuvant activity, e.g., protamine (a cationic peptide), O/W cationic nanoemulsion, polyethylene glycol (PEG)-LNPs, a chemically modified dendrimer, polyethyleneimine (PEI), chitosan (polysaccharide), cationic LNPs (in particular containing ionizable cationic lipids), and some combinations thereof [3,253,254]. Taken together, the above observations indicate that mRNA combined with various delivery agents has excellent adjuvant properties.
LNPs used in mRNA-based vaccines activate multiple inflammatory pathways; induce IL-1, IL-1β, and IL-6 [251,255]; and exacerbate inflammation [256]. The FDA has approved LNPs (used in ongoing COVID-19 clinical trials) including a charge-ionizable lipid, a neutral ionizable lipid, a PEG–lipid complex, cholesterol, and DSPC [255]. Parhiz et al. have demonstrated that the major impact of LNPs on inflammation could be attributed to various ionizable lipids. Apparently, they are recognized by TLR4 because LNP-treated TLR4 knockout mice do not mount a significant inflammatory response [256]. This finding is consistent with earlier studies showing the ability of TLR2 and TLR4 to recognize immunostimulatory lipids on the surface of macrophages and other cells [257]. Notably, LNPs with removed ionizable lipids do not initiate inflammation. However, their stability and kinetics of in vivo delivery remain the subject of future research [256]. The immunogenicity of modern LNP-mRNA vaccines can be tailored by changing the lipid composition of LNPs [258,259]. That is probably the reason why the number of studies that addressed the usage of additional adjuvants in RNA-based therapeutics is limited [3,260].
Another promising area of research is the combining of mRNA-LNPs with classic adjuvants to improve the immunogenicity and the persistence of vaccine components at the injection area [260]. The classic adjuvants used in live, attenuated, recombinant, and DNA-based vaccines are listed in Table 2 [260,261,262,263].
Table 2.
The components of classical vaccine adjuvants.
The traditional adjuvant MF59 consisting of squalene droplets stabilized with small amounts of surfactants Tween 80 and Span 85 [252] has been employed in different RNA-based therapeutics tested in animal models [272]. Combinations of various classic adjuvants and immune stimulators gave rise to so-called adjuvant systems, such as AS01, AS02, AS03, AS04, and AS15. They are frequently utilized in clinical studies to improve immunogenicity [260,261].
A special place among adjuvants is occupied by genetic adjuvants, which are widely used in DNA-based vaccines (see Table 3) [273,274]. Such adjuvants can be categorized into several groups. The first group comprises cytokines and chemokines. For the purpose in question, the most popular coding sequences are those encoding cytokines IL-2, IL-6, IL-12, IL-15, and GM-CSF, which specifically induce the proliferation of NK cells, B cells, and T cells and the stimulation of dendritic-cell maturation and IFNγ production [275,276,277]. Chemokines IP-10 and CCR7 promote the accumulation and activation of dendritic cells and T cells [278]. The second group contains costimulatory biomolecules, such as CD28, CD40, CD80, and CD86. They specifically induce T-cell proliferation and the maturation of dendritic cells and B cells [279,280]. The third group are genetic adjuvants based on immune-signaling molecules, such as MDA5, RIG-I, MyD88, TRIF, HSP70, NF-κB, IRF1-3-7, T-bet, PD-1, HMGB1, ZBP1/DAI, and TBK1. The last group includes bacterial proteins, namely flagellin [281].
TriMix, another genetic adjuvant, is a mixture of three mRNAs (encoding costimulatory ligand CD70, CD40L, and a constitutively active TLR4) that creates a T-cell–attracting and stimulatory environment, including recruitment of antigen-specific CD4+ and CD8+ T cells. The adjuvant has been used with naked mRNA and mRNA in LNPs and manifested good efficacy [258,282,283,284].
Table 3.
Examples of genetic adjuvants and their signaling pathways.
Table 3.
Examples of genetic adjuvants and their signaling pathways.
| Name | Molecular Function | Main Signaling Pathway | Ref. |
|---|---|---|---|
| MyD88 | Adaptor protein | MyD88/TRAF6/NFkB | [285,286] |
| TRIF | Adaptor protein | TRIF/TRAF3/IRF3 | [285,287] |
| IRF1 | Transcriptional factor | IRF1 targets | [288,289] |
| IRF3 | Transcriptional factor | IRF3 targets | [288,290] |
| IRF7 | Transcriptional factor | IRF7 targets | [288,290] |
| HSP70 | Chaperone | HSP70-TLR4-NLRP3 | [291] |
| PD-1 | Membrane receptor | PD-1/SHP2/STAT1/T-bet | [292] |
| MDA5 | Intracellular receptor | MDA5-TBK1-IRF3 | [293] |
| NF-κB | Transcriptional factor | NF-κB targets | [294] |
| T-bet | Transcriptional factor | NF-κB targets | [294,295] |
| RIG-I | Intracellular receptor | RIG-I/TBK1/IRF3 | [293] |
| TBK1 | Kinase | TBK1-IRF3 | [296] |
| HMGB1 | Signaling protein | HMGB1/TLR4/NF-κB | [297,298] |
| DAI/ZBP1 | Transcriptional factor | DAI/ZBP1 targets | [299] |
| GM-CSF | Cytokine | GM-CSF/GM-CSFR | [300,301,302,303] |
| IL-4 | Cytokine | IL4/IL4R | [300] |
| IL-12 | Cytokine | IL-12/IL-12R | [304,305,306,307,308,309,310] |
| IL-2 | Cytokine | IL-2/IL-2R | [304,311,312,313] |
| IL-22 | Cytokine | IL-22/IL-22R | [314] |
| IL-21 | Cytokine | IL-21/IL-21R | [315,316] |
| IL-15 | Cytokine | IL-15/IL-15R | [315,316,317,318,319] |
| IL-6 | Cytokine | IL-6/IL-6R | [319] |
| IL-7 | Cytokine | IL-7/IL-7R | [319] |
| IL-23 | Cytokine | IL-23/IL-23R | [320] |
| CCL3 (MIP-1α) | Chemokine | MIP-1α/CCR1 | [321] |
| CCL20 (MIP-3α) | Chemokine | CCL20/CCR6 | [321] |
| CCL19 (MIP-3β) | Chemokine | CCL19/CCR7 | [321,322] |
| CCL5 (RANTES) | Chemokine | CCL5/CCR5 | [319] |
| CXCL10 (IP-10) | Chemokine | CXCL10/CXCR3 | [323] |
| CCR7 | Membrane receptor | CCL19/CCR7 | [324,325] |
| CCL21 | Chemokine | CCL19/CCR7 | [326] |
| CD80 | Membrane receptor | CD28-CD80 | [327,328] |
| CD86 | Membrane receptor | CD28-CD80 | [327,328,329] |
| CD40L | Cytokine | CD40L/CD40 | [330,331] |
| Flagellin | Bacterial protein | TLR5/MyD88/NF-κB | [281] |
Genetic adjuvants have been used in several ongoing studies of RNA-based therapeutics. Recent publications describe the use of a fusion of the IgG-Fc domain to the RBD of SARS-CoV-2 as the target antigen within one RNA molecule [258,282,283,284,332,333]. Fc-fusion proteins are commonly used in the clinic and promote immune responses [334]. In another study, researchers used mRNA as a target antigen in combination with mRNA of TriMix adjuvants (CD40L, CD70, and TLR4) within the same LNPs; this approach significantly improved the immunogenic properties of the vaccine [258]. Altogether, these examples represent different approaches to the use of genetic adjuvants in mRNA-LNPs.
Finally, mRNA-LNP vaccines can be utilized for coadministration with antigens and other adjuvants. Different classes of adjuvants (i.e., proteins, peptides, antibodies, antigens, DNA, and small-molecule compounds) can be either encapsulated in LNPs or attached through electrostatic binding or via conjugation with surface molecules [260]. Examples of uses of such combinatorial adjuvants in mRNA-LNP vaccines are limited to a study by Goswami and coauthors [335]. They evaluated mannosylated LNP-mRNA obtained by the incorporation of a stable mannose-cholesterol amine conjugate into the LNPs for ensuring a more rapid onset of an antibody response against an antigen in comparison with unglycosylated LNP-mRNA.
The application of combinatorial adjuvants to LNP-mRNA therapeutics creates a lot of opportunities; however, in vitro and in vivo studies are not sufficient for exploring all the possible options, and supplementary bioinformatic algorithms are recommended for predicting the efficiency of combinatorial adjuvants.
When mRNA is used for non-immunological indications, diminished activation of innate-immunity sensors can improve the expression of the target protein and the efficacy of gene therapy. Therefore, one should strike a balance between high immunogenicity and protein translation efficiency. There are various techniques to achieve this goal (see Table 4).
Table 4.
Examples of methods for reducing the immunogenicity of mRNA, tolerogenic agents, and their signaling pathways.
It should be noted that the described mechanisms of innate immunity in a population can be heterogeneous [336]. Therefore, vaccine reactivity is individual [337]. This fact should be taken into account when formulating new adjuvants/tolerogens that activate different pathways. The option of “balancing” adjuvant and tolerogenic mechanisms should certainly be taken into account in the development of RNA therapeutics including the personalized medicine context.
4. Final Remarks
We discussed a multitude of cellular immune responses to RNA invasion. Nonetheless, there are antiviral mechanisms—protein kinase RNA-activated (PKR) and the OAS/RNase L system—which are initiated by the production of interferons. These systems are regarded as one of the most critical aspects in the regulation of gene expression in the presence of dsRNA and should be considered during the development of RNA-based therapeutics.
PKR, also known as eukaryotic initiation factor 2-alpha kinase 2 (eIF2αK2), is a ubiquitously expressed enzyme that is synthesized in response to IFNs [338]. In infected cells, PKR recognizes viral dsRNAs and undergoes dimerization and autophosphorylation. Activated PKR, also called phosphorylated or pPKR, suppresses global translation by phosphorylating eIF2α on Ser51. By phosphorylating other substrates, pPKR also triggers multiple signaling pathways, such as MAPK and NF-κB [339].
Similarly, the presence of interferon triggers the OAS/RNase L system, which is one of the actors in the IFN effector pathways [340]. During viral infection, IFN induces the expression of oligoadenylate synthetase (OAS). It synthesizes 2–5A, which act as second messengers to trigger dimerization and activation of latent RNase L. Activated RNase L cleaves viral and cellular ssRNA and leads to the limitation of virus replication and furthers the apoptosis of infected cells. In addition, many RNA helicases play a critical role during the conformational changes of RNA and ribonucleoprotein (RNP) complexes, and may also have a significant impact on the efficacy of mRNA therapeutics [341,342].
Researchers have already had a wide range of compounds in their arsenal that allow them to manipulate the efficacy of mRNA therapeutics. At academic and industrial research centers, basic research in translation biology and chemistry of nucleic acids makes it possible to create fundamentally new therapeutics and get closer to addressing many socially significant diseases.
Two mRNA vaccines against SARS-CoV-2 have been approved by the FDA recently, but they still pose a risk of side effects, although they are minimal. [343,344]. They may be related to genetic characteristics of immune system reactivity leading to individual adverse reactions to the vaccine [343]. Individual differences in human immunity include polymorphisms in genes encoding TLRs, HLA molecules, cytokines, and cytokine receptors [345]. One of the most troubling adverse effects of SARS-CoV 2 mRNA vaccines that is associated with genetic characteristics is myocarditis [346,347]. The prevalence of this complication is around 0.3–5.0 per 100,000 doses of COVID-19 mRNA vaccines, and it can result in fatality in very rare cases. Recently, the contribution of genetic variants was confirmed in monochorionic diamniotic twins and may be related to HLA alleles [345]. Also, it is important to consider adverse effects like intense homeostatic proliferation and immune imprinting when designing mRNA vaccines [341,348].
Extensive evidence from the Pfizer-BioNTech Vaccine Adverse Effects Analysis suggests that carriers of the HLA-A∗03:01 genotype are likely to experience chills, fever, fatigue, and generally feeling unwell after the vaccination [349]. Taken together, an understanding of individual genetics and the creation of genetic passports can reduce the number of adverse effects of RNA-based therapeutics [350,351].
In conclusion, it can be assumed that the determination of the pattern of immunoreactivity can be adapted for individual selection of adjuvant combinations for the rapid effectiveness of personalized mRNA vaccines for cancer immunotherapy.
Author Contributions
Conceptualization, A.M.; methodology, A.M. and V.R; data curation, A.M.; writing—original draft preparation, V.R., D.S., V.T., K.L. and A.R.; writing—review and editing, A.M.; visualization, A.R.; supervision, R.I.; project administration, R.I.; funding acquisition, R.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Ministry of Science and Higher Education of the Russian Federation (agreement No. 075-10-2021-113, unique project ID RF----193021X0001). The work of Albert Muslimov was supported by the Russian Science Foundation under grant # 22-25-00516.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. MRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [PubMed]
- Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Dewitte, H. Three Decades of Messenger RNA Vaccine Development. Nano Today 2019, 28, 100766. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. Mrna Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.; Liu, X.; Li, M.; Zhang, Z.; Song, L.; Zhu, B.; Wu, X.; Liu, J.; Zhao, D.; Li, Y. Advances in COVID-19 MRNA Vaccine Development. Signal Transduct. Target. Ther. 2022, 7, 94. [Google Scholar] [CrossRef]
- Prieve, M.G.; Harvie, P.; Monahan, S.D.; Roy, D.; Li, A.G.; Blevins, T.L.; Paschal, A.E.; Waldheim, M.; Bell, E.C.; Galperin, A.; et al. Targeted MRNA Therapy for Ornithine Transcarbamylase Deficiency. Mol. Ther. 2018, 26, 801–813. [Google Scholar] [CrossRef]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral Vectors: A Look Back and Ahead on Gene Transfer Technology. New Microbiol. 2013, 36, 1–22. [Google Scholar]
- Kafasla, P.; Skliris, A.; Kontoyiannis, D.L. Post-Transcriptional Coordination of Immunological Responses by RNA-Binding Proteins. Nat. Immunol. 2014, 15, 492–502. [Google Scholar] [CrossRef]
- Platnich, J.M.; Muruve, D.A. NOD-like Receptors and Inflammasomes: A Review of Their Canonical and Non-Canonical Signaling Pathways. Arch. Biochem. Biophys. 2019, 670, 4–14. [Google Scholar] [CrossRef]
- Chen, N.; Xia, P.; Li, S.; Zhang, T.; Wang, T.T.; Zhu, J. RNA Sensors of the Innate Immune System and Their Detection of Pathogens. IUBMB Life 2017, 69, 297–304. [Google Scholar] [CrossRef]
- Pfeffer, L.M. The Role of Nuclear Factor Κb in the Interferon Response. J. Interf. Cytokine Res. 2011, 31, 553–559. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-ΚB Signaling in Inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [PubMed]
- Tak, P.P.; Firestein, G.S. NF-ΚB: A Key Role in Inflammatory Diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-ΚB Pathway for the Therapy of Diseases: Mechanism and Clinical Study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Ma, C.; Zhang, Z.; Zhang, H.; Hu, H. NF-ΚB Signaling in Inflammation and Cancer. MedComm 2021, 2, 618–653. [Google Scholar] [CrossRef] [PubMed]
- Ledoux, A.C.; Perkins, N.D. NF-ΚB and the Cell Cycle. Biochem. Soc. Trans. 2014, 42, 76–81. [Google Scholar] [CrossRef] [PubMed]
- Timucin, A.C.; Basaga, H. Pro-Apoptotic Effects of Lipid Oxidation Products: HNE at the Crossroads of NF-ΚB Pathway and Anti-Apoptotic Bcl-2. Free Radic. Biol. Med. 2017, 111, 209–218. [Google Scholar] [CrossRef]
- Liang, C.; Oh, B.H.; Jung, J.U. Novel Functions of Viral Anti-Apoptotic Factors. Nat. Rev. Microbiol. 2015, 13, 7–12. [Google Scholar] [CrossRef]
- Lawrence, T. The Nuclear Factor NF-KappaB Pathway in Inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Zhong, L.; Simard, M.J.; Huot, J. Endothelial MicroRNAs Regulating the NF-KB Pathway and Cell Adhesion Molecules during Inflammation. FASEB J. 2018, 32, 4070–4084. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic MRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]
- Feng, M.; Ding, Z.; Xu, L.; Kong, L.; Wang, W.; Jiao, S.; Shi, Z.; Greene, M.I.; Cong, Y.; Zhou, Z. Structural and Biochemical Studies of RIG-I Antiviral Signaling. Protein Cell 2013, 4, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Brisse, M.; Ly, H. Comparative Structure and Function Analysis of the RIG-I-like Receptors: RIG-I and MDA5. Front. Immunol. 2019, 10, 1586. [Google Scholar] [CrossRef] [PubMed]
- Rawling, D.C.; Pyle, A.M. Parts, Assembly and Operation of the RIG-I Family of Motors. Curr. Opin. Struct. Biol. 2014, 25, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Matsumoto, M.; Hatakeyama, S.; Seya, T. Riplet/RNF135, a RING Finger Protein, Ubiquitinates RIG-I to Promote Interferon-β Induction during the Early Phase of Viral Infection. J. Biol. Chem. 2009, 284, 807–817. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Sun, L.; Jiang, X.; Chen, X.; Hou, F.; Adhikari, A.; Xu, M.; Chen, Z.J. Reconstitution of the RIG-I Pathway Reveals a Signaling Role of Unanchored Polyubiquitin Chains in Innate Immunity. Cell 2010, 141, 315–330. [Google Scholar] [CrossRef]
- Zheng, Y.; Gao, C. E3 Ubiquitin Ligases, the Powerful Modulator of Innate Antiviral Immunity. Cell. Immunol. 2019, 340, 103915. [Google Scholar] [CrossRef]
- Kawai, T.; Takahashi, K.; Sato, S.; Coban, C.; Kumar, H.; Kato, H.; Ishii, K.J.; Takeuchi, O.; Akira, S. IPS-1, an Adaptor Triggering RIG-I- and Mda5-Mediated Type I Interferon Induction. Nat. Immunol. 2005, 6, 981–988. [Google Scholar] [CrossRef]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory Syncytial Virus-Mediated NF-KB P65 Phosphorylation at Serine 536 Is Dependent on RIG-I, TRAF6, and IKKβ. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef]
- Paz, S.; Sun, Q.; Nakhaei, P.; Romieu-Mourez, R.; Goubau, D.; Julkunen, I.; Lin, R.; Hiscott, J. Induction of IRF-3 and IRF-7 Phosphorylation Following Activation of the RIG-I Pathway. Cell. Mol. Biol. 2006, 52, 17–28. [Google Scholar]
- Chow, K.T.; Gale, M.; Loo, Y.M. RIG-I and other RNA Sensors in Antiviral Immunity. Annu. Rev. Immunol. 2018, 36, 667–694. [Google Scholar] [CrossRef]
- Wu, M.Z.; Asahara, H.; Tzertzinis, G.; Roy, B. Synthesis of Low Immunogenicity RNA with High-Temperature in vitro Transcription. RNA 2020, 26, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, K.R.; Bruns, A.M.; Horvath, C.M. MDA5 and LGP2: Accomplices and Antagonists of Antiviral Signal Transduction. J. Virol. 2014, 88, 8194–8200. [Google Scholar] [CrossRef] [PubMed]
- Stok, J.E.; Oosenbrug, T.; Haar, L.R.; Gravekamp, D.; Bromley, C.P.; Zelenay, S.; Reis e Sousa, C.; Veen, A.G. RNA Sensing via the RIG-I-like Receptor LGP2 Is Essential for the Induction of a Type I IFN Response in ADAR1 Deficiency. EMBO J. 2022, 41, e109760. [Google Scholar] [CrossRef] [PubMed]
- Rehwinkel, J.; Gack, M.U. RIG-I-like Receptors: Their Regulation and Roles in RNA Sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef]
- Kim, D.H.; Longo, M.; Han, Y.; Lundberg, P.; Cantin, E.; Rossi, J.J. Interferon Induction by SiRNAs and SsRNAs Synthesized by Phage Polymerase. Nat. Biotechnol. 2004, 22, 321–325. [Google Scholar] [CrossRef]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of Therapeutic Proteins after Delivery of Chemically Modified MRNA in Mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef]
- Chiang, C.; Beljanski, V.; Yin, K.; Olagnier, D.; Ben Yebdri, F.; Steel, C.; Goulet, M.-L.; DeFilippis, V.R.; Streblow, D.N.; Haddad, E.K.; et al. Sequence-Specific Modifications Enhance the Broad-Spectrum Antiviral Response Activated by RIG-I Agonists. J. Virol. 2015, 89, 8011–8025. [Google Scholar] [CrossRef]
- Claudepierre, M.-C.; Hortelano, J.; Schaedler, E.; Kleinpeter, P.; Geist, M.; Remy-Ziller, C.; Brandely, R.; Tosch, C.; Laruelle, L.; Jawhari, A.; et al. Yeast Virus-Derived Stimulator of the Innate Immune System Augments the Efficacy of Virus Vector-Based Immunotherapy. J. Virol. 2014, 88, 5242–5255. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-Dependent Recognition of Double-Stranded Ribonucleic Acids by Retinoic Acid-Inducible Gene-I and Melanoma Differentiation-Associated Gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Anchisi, S.; Guerra, J.; Garcin, D. RIG-I Atpase Activity and Discrimination of Self-RNA versus Non-Self-RNA. MBio 2015, 6, e02349. [Google Scholar] [CrossRef]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J.M. MDA5 Detects the Double-Stranded RNA Replicative form in Picornavirus-Infected Cells. Cell Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Guan, X.; Yoboua, F.; Zucchini, N.; Fink, K.; Doyon, P.; Martin, L.; Servant, M.J.; Chartier, S. Sustained Activation of Interferon Regulatory Factor 3 during Infection by Paramyxoviruses Requires MDA5. J. Innate Immun. 2014, 6, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Gitlin, L.; Benoit, L.; Song, C.; Cella, M.; Gilfillan, S.; Holtzman, M.J.; Colonna, M. Melanoma Differentiation-Associated Gene 5 (MDA5) Is Involved in the Innate Immune Response to Paramyxoviridae Infection in vivo. PLoS Pathog. 2010, 6, e1000734. [Google Scholar] [CrossRef]
- Xia, A.; Li, X.; Quan, J.; Chen, X.; Xu, Z.; Jiao, X. Mycobacterium Tuberculosis Rv0927c Inhibits NF-ΚB Pathway by Downregulating the Phosphorylation Level of IκBα and Enhances Mycobacterial Survival. Front. Immunol. 2021, 12, 721370. [Google Scholar] [CrossRef]
- Loo, Y.M.; Gale, M. Immune Signaling by RIG-I-like Receptors. Immunity 2011, 34, 680–692. [Google Scholar] [CrossRef]
- Zhai, W.; Wu, F.; Zhang, Y.; Fu, Y.; Liu, Z. The Immune Escape Mechanisms of Mycobacterium Tuberculosis. Int. J. Mol. Sci. 2019, 20, 340. [Google Scholar] [CrossRef] [PubMed]
- Minnaert, A.K.; Vanluchene, H.; Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Raemdonck, K.; Sanders, N.N.; Remaut, K. Strategies for Controlling the Innate Immune Activity of Conventional and Self-Amplifying MRNA Therapeutics: Getting the Message Across. Adv. Drug Deliv. Rev. 2021, 176, 113900. [Google Scholar] [CrossRef]
- Devarkar, S.C.; Wang, C.; Miller, M.T.; Ramanathan, A.; Jiang, F.; Khan, A.G.; Patel, S.S.; Marcotrigiano, J. Structural Basis for M7G Recognition and 2′-O-Methyl Discrimination in Capped RNAs by the Innate Immune Receptor RIG-I. Proc. Natl. Acad. Sci. USA 2016, 113, 596–601. [Google Scholar] [CrossRef]
- Züst, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-Methylation Provides a Molecular Signature for the Distinction of Self and Non-Self MRNA Dependent on the RNA Sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef]
- Rückle, A.; Haasbach, E.; Julkunen, I.; Planz, O.; Ehrhardt, C.; Ludwig, S. The NS1 Protein of Influenza A Virus Blocks RIG-I-Mediated Activation of the Noncanonical NF-ΚB Pathway and P52/RelB-Dependent Gene Expression in Lung Epithelial Cells. J. Virol. 2012, 86, 10211–10217. [Google Scholar] [CrossRef]
- Motz, C.; Schuhmann, K.M.; Kirchhofer, A.; Moldt, M.; Witte, G.; Conzelmann, K.K.; Hopfner, K.P. Paramyxovirus V Proteins Disrupt the Fold of the RNA Sensor MDA5 to Inhibit Antiviral Signaling. Science 2013, 339, 690–693. [Google Scholar] [CrossRef]
- Kasumba, D.M.; Grandvaux, N. Therapeutic Targeting of RIG-I and MDA5 Might Not Lead to the Same Rome. Trends Pharmacol. Sci. 2019, 40, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Broquet, A.H.; Hirata, Y.; McAllister, C.S.; Kagnoff, M.F. RIG-I/MDA5/MAVS Are Required to Signal a Protective IFN Response in Rotavirus-Infected Intestinal Epithelium. J. Immunol. 2011, 186, 1618–1626. [Google Scholar] [CrossRef] [PubMed]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M. Establishment and Maintenance of the Innate Antiviral Response to West Nile Virus Involves Both RIG-I and MDA5 Signaling through IPS-1. J. Virol. 2008, 82, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Linehan, M.M.; Dickey, T.H.; Molinari, E.S.; Fitzgerald, M.E.; Potapova, O.; Iwasaki, A.; Pyle, A.M. A Minimal RNA Ligand for Potent RIG-I Activation in Living Mice. Sci. Adv. 2018, 4, e1701854. [Google Scholar] [CrossRef]
- Sleijfer, S.; Bannink, M.; Van Gool, A.R.; Kruit, W.H.J.; Stoter, G. Side Effects of Interferon-α Therapy. Pharm. World Sci. 2005, 27, 423–431. [Google Scholar] [CrossRef]
- Lemaitre, B.; Nicolas, E.; Michaut, L.; Reichhart, J.M.; Hoffmann, J.A. The Dorsoventral Regulatory Gene Cassette Spatzle/Toll/Cactus Controls the Potent Antifungal Response in Drosophila Adults. Cell 1996, 86, 973–983. [Google Scholar] [CrossRef]
- González-Crespo, S.; Levine, M. Related Target Enhancers for Dorsal and NF-ΚB Signaling Pathways. Science 1994, 264, 255–258. [Google Scholar] [CrossRef]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A Human Homologue of the Drosophila Toll Protein Signals Activation of Adaptive Immunity. Nature 1997, 388, 394–397. [Google Scholar] [CrossRef]
- Gay, N.J.; Keith, F.J. Drosophila Toll and IL-1 Receptor. Nature 1991, 351, 355–356. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, K.A.; Kagan, J.C. Toll-like Receptors and the Control of Immunity. Cell 2020, 180, 1044–1066. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Toll-like Receptors and Innate Immunity. Nat. Rev. Immunol. 2001, 1, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Medzhitov, R. Regulation of Adaptive Immunity by the Innate Immune System. Science 2010, 327, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.K.; Mullen, G.E.D.; Leifer, C.A.; Mazzoni, A.; Davies, D.R.; Segal, D.M. Leucine-Rich Repeats and Pathogen Recognition in Toll-like Receptors. Trends Immunol. 2003, 24, 528–533. [Google Scholar] [CrossRef]
- Chang, Z.L. Important Aspects of Toll-like Receptors, Ligands and Their Signaling Pathways. Inflamm. Res. 2010, 59, 791–808. [Google Scholar] [CrossRef]
- Matsushima, N.; Tachi, N.; Kuroki, Y.; Enkhbayar, P.; Osaki, M.; Kamiya, M.; Kretsinger, R.H. Structural Analysis of Leucine-Rich-Repeat Variants in Proteins Associated with Human Diseases. Cell. Mol. Life Sci. 2005, 62, 2771–2791. [Google Scholar] [CrossRef]
- Boehm, T.; McCurley, N.; Sutoh, Y.; Schorpp, M.; Kasahara, M.; Cooper, M.D. VLR-Based Adaptive Immunity. Annu. Rev. Immunol. 2012, 30, 203–220. [Google Scholar] [CrossRef]
- Szalkowski, A.M.; Anisimova, M. Graph-Based Modeling of Tandem Repeats Improves Global Multiple Sequence Alignment. Nucleic Acids Res. 2013, 41, e162. [Google Scholar] [CrossRef]
- Leister, D. Tandem and Segmental Gene Duplication and Recombination in the Evolution of Plant Disease Resistance Genes. Trends Genet. 2004, 20, 116–122. [Google Scholar] [CrossRef]
- Schaper, E.; Anisimova, M. The Evolution and Function of Protein Tandem Repeats in Plants. New Phytol. 2015, 206, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Sato, R.; Shibata, T.; Tanaka, Y.; Kato, C.; Yamaguchi, K.; Furukawa, Y.; Shimizu, E.; Yamaguchi, R.; Imoto, S.; Miyano, S.; et al. Requirement of Glycosylation Machinery in TLR Responses Revealed by CRISPR/Cas9 Screening. Int. Immunol. 2017, 29, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Li, W. Structures and Recognition Modes of Toll-like Receptors. Proteins Struct. Funct. Bioinform. 2017, 85, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Song, D.H.; Lee, J.O. Sensing of Microbial Molecular Patterns by Toll-like Receptors. Immunol. Rev. 2012, 250, 216–229. [Google Scholar] [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bull. Natl. Res. Cent. 2019, 43, 187. [Google Scholar] [CrossRef]
- Lee, S.M.Y.; Yip, T.F.; Yan, S.; Jin, D.Y.; Wei, H.L.; Guo, R.T.; Peiris, J.S.M. Recognition of Double-Stranded RNA and Regulation of Interferon Pathway by Toll-like Receptor 10. Front. Immunol. 2018, 9, 516. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef]
- Uronen-Hansson, H.; Allen, J.; Osman, M.; Squires, G.; Klein, N.; Callard, R.E. Toll-like Receptor 2 (TLR2) and TLR4 Are Present inside Human Dendritic Cells, Associated with Microtubules and the Golgi Apparatus but Are Not Detectable on the Cell Surface: Integrity of Microtubules Is Required for Interleukin-12 Production in Response to Internalized Bacteria. Immunology 2004, 111, 173–178. [Google Scholar] [CrossRef]
- De Zoete, M.R.; Bouwman, L.I.; Keestra, A.M.; Van Putten, J.P.M. Cleavage and Activation of a Toll-like Receptor by Microbial Proteases. Proc. Natl. Acad. Sci. USA 2011, 108, 4968–4973. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Bowie, A.G. The Family of Five: TIR-Domain-Containing Adaptors in Toll-like Receptor Signalling. Nat. Rev. Immunol. 2007, 7, 353–364. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takeda, K.; Akira, S. TIR Domain-Containing Adaptors Define the Specificity of TLR Signaling. Mol. Immunol. 2004, 40, 861–868. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological Decision-Making: How Does the Immune System Decide to Mount a Helper T-Cell Response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Zhao, Y. Toll-like Receptors and Immune Regulation: Their Direct and Indirect Modulation on Regulatory CD4+ CD25+ T Cells. Immunology 2007, 122, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Takeda, K. Regulatory Mechanisms of Immune Responses to Intestinal Bacteria. Mucosal Immunol. 2009, 2, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Taniguchi, T. Toll-like Receptor Signaling and IRF Transcription Factors. IUBMB Life 2006, 58, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Iwanaszko, M.; Kimmel, M. NF-ΚB and IRF Pathways: Cross-Regulation on Target Genes Promoter Level. BMC Genomics 2015, 16, 307. [Google Scholar] [CrossRef]
- De Haro, C.; Méndez, R.; Santoyo, J. The EIF-2α Kinases and the Control of Protein Synthesis 1. FASEB J. 1996, 10, 1378–1387. [Google Scholar] [CrossRef]
- Liang, S.L.; Quirk, D.; Zhou, A. RNase L: Its Biological Roles and Regulation. IUBMB Life 2006, 58, 508–514. [Google Scholar] [CrossRef]
- Isaacs, A.; Cox, R.A.; Rotem, Z. Foreign nucleic acids as the stimulus to make interferon. Lancet 1963, 282, 113–116. [Google Scholar] [CrossRef]
- Mosaheb, M.M.; Reiser, M.L.; Wetzler, L.M. Toll-like Receptor Ligand-Based Vaccine Adjuvants Require Intact MyD88 Signaling in Antigen-Presenting Cells for Germinal Center Formation and Antibody Production. Front. Immunol. 2017, 8, 225. [Google Scholar] [CrossRef]
- Shen, C.F.; Yen, C.L.; Fu, Y.C.; Cheng, C.M.; Shen, T.C.; De Chang, P.; Cheng, K.H.; Liu, C.C.; Chang, Y.T.; Chen, P.L.; et al. Innate Immune Responses of Vaccinees Determine early Neutralizing Antibody Production after ChAdOx1nCoV-19 Vaccination. Front. Immunol. 2022, 13, 807454. [Google Scholar] [CrossRef] [PubMed]
- Carty, M.; Guy, C.; Bowie, A.G. Detection of Viral Infections by Innate Immunity. Biochem. Pharmacol. 2021, 183, 114316. [Google Scholar] [CrossRef] [PubMed]
- Song, W.S.; Jeon, Y.J.; Namgung, B.; Hong, M.; Yoon, S. Il A Conserved TLR5 Binding and Activation Hot Spot on Flagellin. Sci. Rep. 2017, 7, srep40878. [Google Scholar] [CrossRef] [PubMed]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the Optimal MRNA for Therapy: HPLC Purification Eliminates Immune Activation and Improves Translation of Nucleoside-Modified, Protein-Encoding MRNA. Nucleic Acids Res. 2011, 39, e142. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ohto, U.; Shibata, T.; Krayukhina, E.; Taoka, M.; Yamauchi, Y.; Tanji, H.; Isobe, T.; Uchiyama, S.; Miyake, K.; et al. Structural Analysis Reveals that Toll-like Receptor 7 Is a Dual Receptor for Guanosine and Single-Stranded RNA. Immunity 2016, 45, 737–748. [Google Scholar] [CrossRef]
- Tanji, H.; Ohto, U.; Shibata, T.; Taoka, M.; Yamauchi, Y.; Isobe, T.; Miyake, K.; Shimizu, T. Toll-like Receptor 8 Senses Degradation Products of Single-Stranded RNA. Nat. Struct. Mol. Biol. 2015, 22, 109–115. [Google Scholar] [CrossRef]
- Bowie, A.G.; Unterholzner, L. Viral Evasion and Subversion of Pattern-Recognition Receptor Signalling. Nat. Rev. Immunol. 2008, 8, 911–922. [Google Scholar] [CrossRef]
- Datta, A.; Sinha-Datta, U.; Dhillon, N.K.; Buch, S.; Nicot, C. The HTLV-I P30 Interferes with TLR4 Signaling and Modulates the Release of pro- and anti-Inflammatory Cytokines from Human Macrophages. J. Biol. Chem. 2006, 281, 23414–23424. [Google Scholar] [CrossRef]
- Chase, A.J.; Sedaghat, A.R.; German, J.R.; Gama, L.; Zink, M.C.; Clements, J.E.; Siliciano, R.F. Severe Depletion of CD4 + CD25 + Regulatory T Cells from the Intestinal Lamina Propria but Not Peripheral Blood or Lymph Nodes during Acute Simian Immunodeficiency Virus Infection. J. Virol. 2007, 81, 12748–12757. [Google Scholar] [CrossRef]
- Gale, M.; Sen, G.C. Overview: Viral Evasion of the Interferon System. J. Interf. Cytokine Res. 2009, 29, 475–476. [Google Scholar] [CrossRef]
- Fekonja, O.; Avbelj, M.; Jerala, R. Suppression of TLR Signaling by Targeting TIR Domain-Containing Proteins. Curr. Protein Pept. Sci. 2013, 13, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Chen, J.; Wu, M.; Chen, H.; Kato, N.; Yuan, Z. Hepatitis B Virus Polymerase Inhibits RIG-I- and Toll-like Receptor 3-Mediated Beta Interferon Induction in Human Hepatocytes through Interference with Interferon Regulatory Factor 3 Activation and Dampening of the Interaction between TBK1/IKKε and DDX3. J. Gen. Virol. 2010, 91, 2080–2090. [Google Scholar] [CrossRef]
- Xu, Y.; Hu, Y.; Shi, B.; Zhang, X.; Wang, J.; Zhang, Z.; Shen, F.; Zhang, Q.; Sun, S.; Yuan, Z. HBsAg Inhibits TLR9-Mediated Activation and IFN-α Production in Plasmacytoid Dendritic Cells. Mol. Immunol. 2009, 46, 2640–2646. [Google Scholar] [CrossRef] [PubMed]
- Vacchelli, E.; Galluzzi, L.; Eggermont, A.; Fridman, W.H.; Galon, J.; Sautès-Fridman, C.; Tartour, E.; Zitvogel, L.; Kroemer, G. Trial Watch: FDA-Approved Toll-like Receptor Agonists for Cancer Therapy. Oncoimmunology 2012, 1, 894–907. [Google Scholar] [CrossRef] [PubMed]
- Andón, F.T.; Leon, S.; Ummarino, A.; Redin, E.; Allavena, P.; Serrano, D.; Anfray, C.; Calvo, A. Innate and Adaptive Responses of Intratumoral Immunotherapy with Endosomal Toll-Like Receptor Agonists. Biomedicines 2022, 10, 1590. [Google Scholar] [CrossRef]
- Lobo, N.; Brooks, N.A.; Zlotta, A.R.; Cirillo, J.D.; Boorjian, S.; Black, P.C.; Meeks, J.J.; Bivalacqua, T.J.; Gontero, P.; Steinberg, G.D.; et al. 100 Years of Bacillus Calmette–Guérin Immunotherapy: From Cattle to COVID-19. Nat. Rev. Urol. 2021, 18, 611–622. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 MRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Decker, C.J.; Parker, R. Mechanisms of MRNA Degradation in Eukaryotes. Trends Biochem. Sci. 1994, 19, 336–340. [Google Scholar] [CrossRef]
- Hou, X.; Zaks, T.; Langer, R.; Dong, Y. Lipid Nanoparticles for MRNA Delivery. Nat. Rev. Mater. 2021, 6, 1078–1094. [Google Scholar] [CrossRef]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct Gene Transfer into Mouse Muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Babendure, J.R.; Babendure, J.L.; Ding, J.H.; Tsien, R.Y. Control of Mammalian Translation by MRNA Structure near Caps. RNA 2006, 12, 851–861. [Google Scholar] [CrossRef]
- Furuichi, Y.; LaFiandra, A.; Shatkin, A.J. 5′-Terminal Structure and MRNA Stability. Nature 1977, 266, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Cooke, C.; Alwine, J.C. The Cap and the 3′ Splice Site Similarly Affect Polyadenylation Efficiency. Mol. Cell. Biol. 1996, 16, 2579–2584. [Google Scholar] [CrossRef]
- Lockard, R.E.; Lingrel, J.B. The Synthesis of Mouse Hemoglobin Chains in a Rabbit Reticulocyte Cell-Free System Programmed with Mouse Reticulocyte 9S RNA. Biochem. Biophys. Res. Commun. 1969, 37, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Sartorius, R.; Trovato, M.; Manco, R.; D’Apice, L.; De Berardinis, P. Exploiting Viral Sensing Mediated by Toll-like Receptors to Design Innovative Vaccines. Npj Vaccines 2021, 6, 127. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Kandimalla, E.R. Synthetic Agonists of Toll-like Receptors 7, 8 and 9. Biochem. Soc. Trans. 2007, 35, 1461–1467. [Google Scholar] [CrossRef]
- Owen, A.M.; Fults, J.B.; Patil, N.K.; Hernandez, A.; Bohannon, J.K. TLR Agonists as Mediators of Trained Immunity: Mechanistic Insight and Immunotherapeutic Potential to Combat Infection. Front. Immunol. 2021, 11, 622614. [Google Scholar] [CrossRef]
- Schlake, T.; Thess, A.; Fotin-Mleczek, M.; Kallen, K.J. Developing MRNA-Vaccine Technologies. RNA Biol. 2012, 9, 1319–1330. [Google Scholar] [CrossRef]
- Mauger, D.M.; Joseph Cabral, B.; Presnyak, V.; Su, S.V.; Reid, D.W.; Goodman, B.; Link, K.; Khatwani, N.; Reynders, J.; Moore, M.J.; et al. MRNA Structure Regulates Protein Expression through Changes in Functional Half-Life. Proc. Natl. Acad. Sci. USA 2019, 116, 24075–24083. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef]
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Greenwald, E.; Ahmad, S.; Hur, S. An Origin of the Immunogenicity of in vitro Transcribed RNA. Nucleic Acids Res. 2018, 46, 5239–5249. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.; Sorensen, E.W.; Mintri, S.; Rabideau, A.E.; Zheng, W.; Besin, G.; Khatwani, N.; Su, S.V.; Miracco, E.J.; Issa, W.J.; et al. Impact of MRNA Chemistry and Manufacturing Process on Innate Immune Activation. Sci. Adv. 2020, 6, eaaz6893. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.K.M.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.D.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 MRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther. Nucleic Acids 2018, 12, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-Methyl-Pseudouridine in MRNA Enhances Translation through EIF2α-Dependent and Independent Mechanisms by Increasing Ribosome Density. Nucleic Acids Res. 2017, 45, 6023–6036. [Google Scholar] [CrossRef] [PubMed]
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-Methylpseudouridine-Incorporated MRNA Outperforms Pseudouridine-Incorporated MRNA by Providing Enhanced Protein Expression and Reduced Immunogenicity in Mammalian Cell Lines and Mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of Translation via MRNA Structure in Prokaryotes and Eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef]
- Lind, C.; Esguerra, M.; Jespers, W.; Satpati, P.; Gutierrez-de-Terán, H.; Åqvist, J. Free Energy Calculations of RNA Interactions. Methods 2019, 162–163, 85–95. [Google Scholar] [CrossRef]
- Kierzek, E.; Malgowska, M.; Lisowiec, J.; Turner, D.H.; Gdaniec, Z.; Kierzek, R. The Contribution of Pseudouridine to Stabilities and Structure of RNAs. Nucleic Acids Res. 2014, 42, 3492–3501. [Google Scholar] [CrossRef]
- Thess, A.; Grund, S.; Mui, B.L.; Hope, M.J.; Baumhof, P.; Fotin-Mleczek, M.; Schlake, T. Sequence-Engineered MRNA Without Chemical Nucleoside Modifications Enables an Effective Protein Therapy in Large Animals. Mol. Ther. 2015, 23, 1456–1464. [Google Scholar] [CrossRef]
- Heit, A.; Schmitz, F.; Haas, T.; Busch, D.H.; Wagner, H. Antigen Co-Encapsulated with Adjuvants Efficiently Drive Protective T Cell Immunity. Eur. J. Immunol. 2007, 37, 2063–2074. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.N.; Mahon, K.P.; Chikh, G.; Kim, P.; Chung, H.; Vicari, A.P.; Love, K.T.; Goldberg, M.; Chen, S.; Krieg, A.M.; et al. Lipid-Derived Nanoparticles for Immunostimulatory RNA Adjuvant Delivery. Proc. Natl. Acad. Sci. USA 2012, 109, E797–E803. [Google Scholar] [CrossRef] [PubMed]
- Duthie, M.S.; Windish, H.P.; Fox, C.B.; Reed, S.G. Use of Defined TLR Ligands as Adjuvants within Human Vaccines. Immunol. Rev. 2011, 239, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimian, M.; Hashemi, M.; Maleki, M.; Hashemitabar, G.; Abnous, K.; Ramezani, M.; Haghparast, A. Co-Delivery of Dual Toll-like Receptor Agonists and Antigen in Poly(Lactic-Co-Glycolic) Acid/Polyethylenimine Cationic Hybrid Nanoparticles Promote Efficient in vivo Immune Responses. Front. Immunol. 2017, 8, 1077. [Google Scholar] [CrossRef]
- Proell, M.; Riedl, S.J.; Fritz, J.H.; Rojas, A.M.; Schwarzenbacher, R. The Nod-Like Receptor (NLR) Family: A Tale of Similarities and Differences. PLoS ONE 2008, 3, e2119. [Google Scholar] [CrossRef]
- Jin, J.; Zhou, T.-J.; Ren, G.-L.; Cai, L.; Meng, X.-M. Novel Insights into NOD-like Receptors in Renal Diseases. Acta Pharmacol. Sin. 2022, 43, 2789–2806. [Google Scholar] [CrossRef]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef]
- Askari, N.; Correa, R.G.; Zhai, D.; Reed, J.C. Expression, Purification, and Characterization of Recombinant NOD1 (NLRC1): A NLR Family Member. J. Biotechnol. 2012, 157, 75–81. [Google Scholar] [CrossRef]
- Faustin, B.; Reed, J.C. Sunburned Skin Activates Inflammasomes. Trends Cell Biol. 2008, 18, 4–8. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The Inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Chu, J.Q.; Gao, F.F.; Wu, W.; Li, C.; Pan, Z.; Sun, J.; Wang, H.; Huang, C.; Lee, S.H.; Quan, J.-H.; et al. Expression profiles of NOD-like receptors and regulation of NLRP3 inflammasome activation in Toxoplasma gondii-infected human small intestinal epithelial cells. Parasites Vectors 2021, 14, 153. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Kanneganti, T.D. Unsolved Mysteries in NLR Biology. Front. Immunol. 2013, 4, 285. [Google Scholar] [CrossRef] [PubMed]
- León Machado, J.A.; Steimle, V. The Mhc Class Ii Transactivator Ciita: Not (Quite) the Odd-One-out Anymore among Nlr Proteins. Int. J. Mol. Sci. 2021, 22, 1074. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Asamitsu, K.; Nishimura, H.; Kamatani, N.; Okamoto, T. Reciprocal Modulation of Transcriptional Activities between HIV-1 Tat and MHC Class II Transactivator CIITA. Biochem. Biophys. Res. Commun. 2000, 279, 494–499. [Google Scholar] [CrossRef] [PubMed]
- Abendroth, A.; Slobedman, B.; Lee, E.; Mellins, E.; Wallace, M.; Arvin, A.M. Modulation of Major Histocompatibility Class II Protein Expression by Varicella-Zoster Virus. J. Virol. 2000, 74, 1900–1907. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Lin, J.Y.; Chou, Y.C.; Chen, M.R.; Yeh, T.H.; Lin, C.W.; Lin, S.J.; Tsai, C.H. Epstein-Barr Virus LMP2A Suppresses MHC Class II Expression by Regulating the B-Cell Transcription Factors E47 and PU.1. Blood 2015, 125, 2228–2238. [Google Scholar] [CrossRef]
- Sandhu, P.K.; Buchkovich, N.J. Human Cytomegalovirus Decreases Major Histocompatibility Complex Class II by Regulating Class II Transactivator Transcript Levels in a Myeloid Cell Line. J. Virol. 2020, 94, 10–1128. [Google Scholar] [CrossRef]
- Pai, R.K.; Convery, M.; Hamilton, T.A.; Boom, W.H.; Harding, C.V. Inhibition of IFN-γ-Induced Class II Transactivator Expression by a 19-KDa Lipoprotein from Mycobacterium Tuberculosis: A Potential Mechanism for Immune Evasion. J. Immunol. 2003, 171, 175–184. [Google Scholar] [CrossRef]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 Limits the Activation of Inflammatory Pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef]
- Vijayan, S.; Sidiq, T.; Yousuf, S.; van den Elsen, P.J.; Kobayashi, K.S. Class I Transactivator, NLRC5: A Central Player in the MHC Class I Pathway and Cancer Immune Surveillance. Immunogenetics 2019, 71, 273–282. [Google Scholar] [CrossRef]
- Meissner, T.B.; Li, A.; Biswas, A.; Lee, K.H.; Liu, Y.J.; Bayir, E.; Iliopoulos, D.; Van Den Elsen, P.J.; Kobayashi, K.S. NLR Family Member NLRC5 Is a Transcriptional Regulator of MHC Class I Genes. Proc. Natl. Acad. Sci. USA 2010, 107, 13794–13799. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, S.; Till, A.; Winkler, M.; Häsler, R.; Lipinski, S.; Jung, S.; Grötzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The Nucleotide-Binding Oligomerization Domain-like Receptor NLRC5 Is Involved in IFN-Dependent Antiviral Immune Responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Ting, J.P.Y.; Duncan, J.A.; Lei, Y. How the Noninflammasome NLRs Function in the Innate Immune System. Science 2010, 327, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Sabbah, A.; Chang, T.H.; Harnack, R.; Frohlich, V.; Tominaga, K.; Dube, P.H.; Xiang, Y.; Bose, S. Activation of Innate Immune Antiviral Responses by Nod2. Nat. Immunol. 2009, 10, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD Proteins: Regulators of Inflammation in Health and Disease. Nat. Rev. Immunol. 2013, 14, 9–23, Erratum in 2014, 14, 131. [Google Scholar] [CrossRef]
- Moore, C.B.; Bergstralh, D.T.; Duncan, J.A.; Lei, Y.; Morrison, T.E.; Zimmermann, A.G.; Accavitti-Loper, M.A.; Madden, V.J.; Sun, L.; Ye, Z.; et al. NLRX1 Is a Regulator of Mitochondrial Antiviral Immunity. Nature 2008, 451, 573–577. [Google Scholar] [CrossRef]
- Maisonneuve, C.; Bertholet, S.; Philpott, D.J.; De Gregorio, E. Unleashing the Potential of NOD- and Toll-like Agonists as Vaccine Adjuvants. Proc. Natl. Acad. Sci. USA 2014, 111, 12294–12299. [Google Scholar] [CrossRef]
- Martinon, F.; Agostini, L.; Meylan, E.; Tschopp, J. Identification of Bacterial Muramyl Dipeptide as Activator of the NALP3/Cryopyrin Inflammasome. Curr. Biol. 2004, 14, 1929–1934. [Google Scholar] [CrossRef]
- Magalhaes, J.G.; Fritz, J.H.; Le Bourhis, L.; Sellge, G.; Travassos, L.H.; Selvanantham, T.; Girardin, S.E.; Gommerman, J.L.; Philpott, D.J. Nod2-Dependent Th2 Polarization of Antigen-Specific Immunity. J. Immunol. 2008, 181, 7925–7935. [Google Scholar] [CrossRef]
- Barazzone, G.C.; Teixeira, A.F.; Azevedo, B.O.P.; Damiano, D.K.; Oliveira, M.P.; Nascimento, A.L.T.O.; Lopes, A.P.Y. Revisiting the Development of Vaccines Against Pathogenic Leptospira: Innovative Approaches, Present Challenges, and Future Perspectives. Front. Immunol. 2022, 12, 760291. [Google Scholar] [CrossRef]
- Rice, T.A.; Brenner, T.A.; Percopo, C.M.; Ma, M.; Keicher, J.D.; Domachowske, J.B.; Rosenberg, H.F. Signaling via Pattern Recognition Receptors NOD2 and TLR2 Contributes to Immunomodulatory Control of Lethal Pneumovirus Infection. Antivir. Res. 2016, 132, 131–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lemesre, J.L.; Holzmuller, P.; Gonçalves, R.B.; Bourdoiseau, G.; Hugnet, C.; Cavaleyra, M.; Papierok, G. Long-Lasting Protection against Canine Visceral Leishmaniasis Using the LiESAp-MDP Vaccine in Endemic Areas of France: Double-Blind Randomised Efficacy Field Trial. Vaccine 2007, 25, 4223–4234. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Domínguez-Andrés, J.; Barreiro, L.B.; Chavakis, T.; Divangahi, M.; Fuchs, E.; Joosten, L.A.B.; van der Meer, J.W.M.; Mhlanga, M.M.; Mulder, W.J.M.; et al. Defining Trained Immunity and Its Role in Health and Disease. Nat. Rev. Immunol. 2020, 20, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Reinke, S.; Thakur, A.; Gartlan, C.; Bezbradica, J.S.; Milicic, A. Inflammasome-Mediated Immunogenicity of Clinical and Experimental Vaccine Adjuvants. Vaccines 2020, 8, 554. [Google Scholar] [CrossRef] [PubMed]
- Brueggeman, J.M.; Zhao, J.; Schank, M.; Yao, Z.Q.; Moorman, J.P. Trained Immunity: An Overview and the Impact on COVID-19. Front. Immunol. 2022, 13, 837524. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 Inflammasome: Molecular Activation and Regulation to Therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Dick, M.S.; Sborgi, L.; Rühl, S.; Hiller, S.; Broz, P. ASC Filament Formation Serves as a Signal Amplification Mechanism for Inflammasomes. Nat. Commun. 2016, 7, 11929. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Hornung, V. Of Inflammasomes and Pathogens—Sensing of Microbes by the Inflammasome. EMBO Mol. Med. 2013, 5, 814–826. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of Asc-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef]
- Sharma, B.R.; Kanneganti, T.D. NLRP3 Inflammasome in Cancer and Metabolic Diseases. Nat. Immunol. 2021, 22, 550–559. [Google Scholar] [CrossRef]
- Bauernfeind, F.G.; Horvath, G.; Stutz, A.; Alnemri, E.S.; MacDonald, K.; Speert, D.; Fernandes-Alnemri, T.; Wu, J.; Monks, B.G.; Fitzgerald, K.A.; et al. Cutting Edge: NF-ΚB Activating Pattern Recognition and Cytokine Receptors License NLRP3 Inflammasome Activation by Regulating NLRP3 Expression. J. Immunol. 2009, 183, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and Reacting to Microbes through the Inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the Nlrp3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed]
- Magupalli, V.G.; Negro, R.; Tian, Y.; Hauenstein, A.V.; Di Caprio, G.; Skillern, W.; Deng, Q.; Orning, P.; Alam, H.B.; Maliga, Z.; et al. HDAC6 Mediates an Aggresome-like Mechanism for NLRP3 and Pyrin Inflammasome Activation. Science 2020, 369, eaas8995. [Google Scholar] [CrossRef] [PubMed]
- Beckwith, K.S.; Beckwith, M.S.; Ullmann, S.; Sætra, R.S.; Kim, H.; Marstad, A.; Åsberg, S.E.; Strand, T.A.; Haug, M.; Niederweis, M.; et al. Plasma Membrane Damage Causes NLRP3 Activation and Pyroptosis during Mycobacterium Tuberculosis Infection. Nat. Commun. 2020, 11, 2270. [Google Scholar] [CrossRef]
- Di, A.; Xiong, S.; Ye, Z.; Malireddi, R.K.S.; Kometani, S.; Zhong, M.; Mittal, M.; Hong, Z.; Kanneganti, T.D.; Rehman, J.; et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity 2018, 49, 56–65.e4. [Google Scholar] [CrossRef]
- Lee, G.S.; Subramanian, N.; Kim, A.I.; Aksentijevich, I.; Goldbach-Mansky, R.; Sacks, D.B.; Germain, R.N.; Kastner, D.L.; Chae, J.J. The Calcium-Sensing Receptor Regulates the NLRP3 Inflammasome through Ca2+ and CAMP. Nature 2012, 492, 123–127. [Google Scholar] [CrossRef]
- Yu, X.; Lan, P.; Hou, X.; Han, Q.; Lu, N.; Li, T.; Jiao, C.; Zhang, J.; Zhang, C.; Tian, Z. HBV Inhibits LPS-Induced NLRP3 Inflammasome Activation and IL-1β Production via Suppressing the NF-ΚB Pathway and ROS Production. J. Hepatol. 2017, 66, 693–702. [Google Scholar] [CrossRef]
- Zhong, Z.; Liang, S.; Sanchez-Lopez, E.; He, F.; Shalapour, S.; Lin, X.J.; Wong, J.; Ding, S.; Seki, E.; Schnabl, B.; et al. New Mitochondrial DNA Synthesis Enables NLRP3 Inflammasome Activation. Nature 2018, 560, 198–203. [Google Scholar] [CrossRef]
- Sha, W.; Mitoma, H.; Hanabuchi, S.; Bao, M.; Weng, L.; Sugimoto, N.; Liu, Y.; Zhang, Z.; Zhong, J.; Sun, B.; et al. Human NLRP3 Inflammasome Senses Multiple Types of Bacterial RNAs. Proc. Natl. Acad. Sci. USA 2014, 111, 16059–16064. [Google Scholar] [CrossRef]
- Mitoma, H.; Hanabuchi, S.; Kim, T.; Bao, M.; Zhang, Z.; Sugimoto, N.; Liu, Y.J. The DHX33 RNA Helicase Senses Cytosolic RNA and Activates the NLRP3 Inflammasome. Immunity 2013, 39, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 Loss and Alu RNA Induce Age-Related Macular Degeneration via the NLRP3 Inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Milner, M.T.; Maddugoda, M.; Götz, J.; Burgener, S.S.; Schroder, K. The NLRP3 Inflammasome Triggers Sterile Neuroinflammation and Alzheimer’s Disease. Curr. Opin. Immunol. 2021, 68, 116–124. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, W.T.; Pham, L.; Symons, G.F.; Monif, M.; Shultz, S.R.; McDonald, S.J. The NLRP3 Inflammasome in Traumatic Brain Injury: Potential as a Biomarker and Therapeutic Target. J. Neuroinflamm. 2020, 17, 104. [Google Scholar] [CrossRef]
- Forster, J.; Nandi, D.; Kulkarni, A. MRNA-Carrying Lipid Nanoparticles That Induce Lysosomal Rupture Activate NLRP3 Inflammasome and Reduce MRNA Transfection Efficiency. Biomater. Sci. 2022, 10, 5566–5582. [Google Scholar] [CrossRef]
- Won, T.; Gilotra, N.A.; Wood, M.K.; Hughes, D.M.; Talor, M.V.; Lovell, J.; Milstone, A.M.; Steenbergen, C.; Čiháková, D. Increased Interleukin 18-Dependent Immune Responses Are Associated with Myopericarditis after COVID-19 MRNA Vaccination. Front. Immunol. 2022, 13, 851620. [Google Scholar] [CrossRef]
- Zhang, R.; Hong, F.; Zhao, M.; Cai, X.; Jiang, X.; Ye, N.; Su, K.; Li, N.; Tang, M.; Ma, X.; et al. New Highly Potent NLRP3 Inhibitors: Furanochalcone Velutone F Analogues. ACS Med. Chem. Lett. 2022, 13, 560–569. [Google Scholar] [CrossRef]
- Awe, J.P.; Crespo, A.V.; Li, Y.; Kiledjian, M.; Byrne, J.A. BAY11 Enhances OCT4 Synthetic MRNA Expression in Adult Human Skin Cells. Stem Cell Res. Ther. 2013, 4, 15. [Google Scholar] [CrossRef]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome: A Molecular Platform Triggering Activation of Inflammatory Caspases and Processing of ProIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Den Brok, M.H.; Büll, C.; Wassink, M.; De Graaf, A.M.; Wagenaars, J.A.; Minderman, M.; Thakur, M.; Amigorena, S.; Rijke, E.O.; Schrier, C.C.; et al. Saponin-Based Adjuvants Induce Cross-Presentation in Dendritic Cells by Intracellular Lipid Body Formation. Nat. Commun. 2016, 7, 13324. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Walle, L.V.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-Canonical Inflammasome Activation Targets Caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef]
- Muñoz-Wolf, N.; McCluskey, S.; Lavelle, E.C. The Role of Inflammasomes in Adjuvant-Driven Humoral and Cellular Immune Responses. In Immunopotentiators in Modern Vaccines, 2nd ed.; Academic Press: New York, NY, USA, 2017. [Google Scholar]
- Davidsen, J.; Rosenkrands, I.; Christensen, D.; Vangala, A.; Kirby, D.; Perrie, Y.; Agger, E.M.; Andersen, P. Characterization of Cationic Liposomes Based on Dimethyldioctadecylammonium and Synthetic Cord Factor from M. tuberculosis (Trehalose 6,6′-Dibehenate)—A Novel Adjuvant Inducing Both Strong CMI and Antibody Responses. Biochim. Biophys. Acta Biomembr. 2005, 1718, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, X.; Huang, X.; Zhang, J.; Xia, N.; Zhao, Q. Calcium Phosphate Nanoparticles as a New Generation Vaccine Adjuvant. Expert Rev. Vaccines 2017, 16, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.A.; Ruane, D.; Claass, B.; Creagh, E.; Harris, J.; Malyala, P.; Singh, M.; O’Hagan, D.T.; Pétrilli, V.; Tschopp, J.; et al. Uptake of Particulate Vaccine Adjuvants by Dendritic Cells Activates the NALP3 Inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Zelensky, A.N.; Gready, J.E. The C-Type Lectin-like Domain Superfamily. FEBS J. 2005, 272, 6179–6217. [Google Scholar] [CrossRef] [PubMed]
- Drouin, M.; Saenz, J.; Chiffoleau, E. C-Type Lectin-like Receptors: Head or Tail in Cell Death Immunity. Front. Immunol. 2020, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Sancho, D.; Reis e Sousa, C. Sensing of Cell Death by Myeloid C-Type Lectin Receptors. Curr. Opin. Immunol. 2013, 25, 46–52. [Google Scholar] [CrossRef]
- Bermejo-Jambrina, M.; Eder, J.; Helgers, L.C.; Hertoghs, N.; Nijmeijer, B.M.; Stunnenberg, M.; Geijtenbeek, T.B.H. C-Type Lectin Receptors in Antiviral Immunity and Viral Escape. Front. Immunol. 2018, 9, 590. [Google Scholar] [CrossRef]
- Meyer-Wentrup, F.; Cambi, A.; Joosten, B.; Looman, M.W.; de Vries, I.J.M.; Figdor, C.G.; Adema, G.J. DCIR Is Endocytosed into Human Dendritic Cells and Inhibits TLR8-Mediated Cytokine Production. J. Leukoc. Biol. 2009, 85, 518–525. [Google Scholar] [CrossRef]
- Zhao, X.; Shen, Y.; Hu, W.; Chen, J.; Wu, T.; Sun, X.; Yu, J.; Wu, T.; Chen, W. DCIR Negatively Regulates CpG-ODN-Induced IL-1β and IL-6 Production. Mol. Immunol. 2015, 68, 641–647. [Google Scholar] [CrossRef]
- Troegeler, A.; Mercier, I.; Cougoule, C.; Pietretti, D.; Colom, A.; Duval, C.; Vu Manh, T.P.; Capilla, F.; Poincloux, R.; Pingris, K.; et al. C-Type Lectin Receptor DCIR Modulates Immunity to Tuberculosis by Sustaining Type I Interferon Signaling in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2017, 114, E540–E549. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhang, L.; Rosen, D.B.; Bover, L.; Watanabe, G.; Bao, M.; Lanier, L.L.; Liu, Y.J. BDCA2/FcεRIγ Complex Signals through a Novel BCR-like Pathway in Human Plasmacytoid Dendritic Cells. PLoS Biol. 2007, 5, e248. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.H.; Gringhuis, S.I. Signalling through C-Type Lectin Receptors: Shaping Immune Responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Dzionek, A.; Sohma, Y.; Nagafune, J.; Cella, M.; Colonna, M.; Facchetti, F.; Günther, G.; Johnston, I.; Lanzavecchia, A.; Nagasaka, T.; et al. BDCA-2, a Novel Plasmacytoid Dendritic Cell-Specific Type II C-Type Lectin, Mediates Antigen Capture and Is a Potent Inhibitor of Interferon α/β Induction. J. Exp. Med. 2001, 194, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, I.S.; Lekkerkerker, A.N.; Depla, E.; Bosman, F.; Musters, R.J.P.; Depraetere, S.; van Kooyk, Y.; Geijtenbeek, T.B.H. Hepatitis C Virus Targets DC-SIGN and L-SIGN To Escape Lysosomal Degradation. J. Virol. 2004, 78, 8322–8332. [Google Scholar] [CrossRef]
- Ablasser, A.; Chen, Z.J. CGAS in Action: Expanding Roles in Immunity and Inflammation. Science 2019, 363, eaat8657. [Google Scholar] [CrossRef] [PubMed]
- Ablasser, A.; Hur, S. Regulation of CGAS- and RLR-Mediated Immunity to Nucleic Acids. Nat. Immunol. 2020, 21, 17–29. [Google Scholar] [CrossRef]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING Regulates Intracellular DNA-Mediated, Type i Interferon-Dependent Innate Immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef]
- Burdette, D.L.; Monroe, K.M.; Sotelo-Troha, K.; Iwig, J.S.; Eckert, B.; Hyodo, M.; Hayakawa, Y.; Vance, R.E. STING Is a Direct Innate Immune Sensor of Cyclic Di-GMP. Nature 2011, 478, 515–518. [Google Scholar] [CrossRef]
- Ni, G.; Ma, Z.; Damania, B. CGAS and STING: At the Intersection of DNA and RNA Virus-Sensing Networks. PLoS Pathog. 2018, 14, e1007148. [Google Scholar] [CrossRef]
- Civril, F.; Deimling, T.; De Oliveira Mann, C.C.; Ablasser, A.; Moldt, M.; Witte, G.; Hornung, V.; Hopfner, K.P. Structural Mechanism of Cytosolic DNA Sensing by CGAS. Nature 2013, 498, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING Is an Endoplasmic Reticulum Adaptor that Facilitates Innate Immune Signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, B.; Yang, Y.; Li, S.; Wang, Y.Y.; Li, Y.; Diao, F.; Lei, C.; He, X.; Zhang, L.; Tien, P.; et al. The Adaptor Protein MITA Links Virus-Sensing Receptors to IRF3 Transcription Factor Activation. Immunity 2008, 29, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Goulet, M.-L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [PubMed]
- Franz, K.M.; Neidermyer, W.J.; Tan, Y.J.; Whelan, S.P.J.; Kagan, J.C. STING-Dependent Translation Inhibition Restricts RNA Virus Replication. Proc. Natl. Acad. Sci. USA 2018, 115, E2058–E2067. [Google Scholar] [CrossRef]
- Ding, Q.; Cao, X.; Lu, J.; Huang, B.; Liu, Y.J.; Kato, N.; Shu, H.B.; Zhong, J. Hepatitis C Virus NS4B Blocks the Interaction of STING and TBK1 to Evade Host Innate Immunity. J. Hepatol. 2013, 59, 52–58. [Google Scholar] [CrossRef]
- Ma, Z.; Damania, B. The CGAS-STING Defense Pathway and Its Counteraction by Viruses. Cell Host Microbe 2016, 19, 150–158. [Google Scholar] [CrossRef]
- Vincent, J.; Adura, C.; Gao, P.; Luz, A.; Lama, L.; Asano, Y.; Okamoto, R.; Imaeda, T.; Aida, J.; Rothamel, K.; et al. Small Molecule Inhibition of CGAS Reduces Interferon Expression in Primary Macrophages from Autoimmune Mice. Nat. Commun. 2017, 8, 750. [Google Scholar] [CrossRef]
- Hall, J.; Brault, A.; Vincent, F.; Weng, S.; Wang, H.; Dumlao, D.; Aulabaugh, A.; Aivazian, D.; Castro, D.; Chen, M.; et al. Discovery of PF-06928215 as a High Affinity Inhibitor of CGAS Enabled by a Novel Fluorescence Polarization Assay. PLoS ONE 2017, 12, e0184843. [Google Scholar] [CrossRef]
- Lama, L.; Adura, C.; Xie, W.; Tomita, D.; Kamei, T.; Kuryavyi, V.; Gogakos, T.; Steinberg, J.I.; Miller, M.; Ramos-Espiritu, L.; et al. Development of Human CGAS-Specific Small-Molecule Inhibitors for Repression of DsDNA-Triggered Interferon Expression. Nat. Commun. 2019, 10, 2261. [Google Scholar] [CrossRef]
- Padilla-Salinas, R.; Sun, L.; Anderson, R.; Yang, X.; Zhang, S.; Chen, Z.J.; Yin, H. Discovery of Small-Molecule Cyclic GMP-AMP Synthase Inhibitors. J. Org. Chem. 2020, 85, 1579–1600. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Xiong, M.; Yuan, X.; Li, M.; Sun, H.; Xu, Y. In Silico Screening-Based Discovery of Novel Inhibitors of Human Cyclic GMP-AMP Synthase: A Cross-Validation Study of Molecular Docking and Experimental Testing. J. Chem. Inf. Model. 2020, 60, 3265–3276. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Woodward, J.J.; Sasaki, T.; Minie, M.; Elkon, K.B. Cutting Edge: Antimalarial Drugs Inhibit IFN-β Production through Blockade of Cyclic GMP-AMP Synthase–DNA Interaction. J. Immunol. 2015, 194, 4089–4093. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Minie, M.; Sasaki, T.; Woodward, J.J.; Elkon, K.B. Antimalarial Drugs as Immune Modulators: New Mechanisms for Old Drugs. Annu. Rev. Med. 2017, 68, 317–330. [Google Scholar] [CrossRef] [PubMed]
- An, J.; Woodward, J.J.; Lai, W.; Minie, M.; Sun, X.; Tanaka, L.; Snyder, J.M.; Sasaki, T.; Elkon, K.B. Inhibition of Cyclic GMP-AMP Synthase Using a Novel Antimalarial Drug Derivative in Trex1-Deficient Mice. Arthritis Rheumatol. 2018, 70, 1807–1819. [Google Scholar] [CrossRef]
- Steinhagen, F.; Zillinger, T.; Peukert, K.; Fox, M.; Thudium, M.; Barchet, W.; Putensen, C.; Klinman, D.; Latz, E.; Bode, C. Suppressive Oligodeoxynucleotides Containing TTAGGG Motifs Inhibit CGAS Activation in Human Monocytes. Eur. J. Immunol. 2018, 48, 605–611. [Google Scholar] [CrossRef]
- Wang, M.; Sooreshjani, M.A.; Mikek, C.; Opoku-Temeng, C.; Sintim, H.O. Suramin Potently Inhibits CGAMP Synthase, CGAS, in THP1 Cells to Modulate IFN-β Levels. Future Med. Chem. 2018, 10, 1301–1317. [Google Scholar] [CrossRef]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The CGAS–STING Pathway as a Therapeutic Target in Inflammatory Diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Van Herck, S.; Feng, B.; Tang, L. Delivery of STING Agonists for Adjuvanting Subunit Vaccines. Adv. Drug Deliv. Rev. 2021, 179, 114020. [Google Scholar] [CrossRef]
- Tse, S.W.; McKinney, K.; Walker, W.; Nguyen, M.; Iacovelli, J.; Small, C.; Hopson, K.; Zaks, T.; Huang, E. MRNA-Encoded, Constitutively Active STINGV155M Is a Potent Genetic Adjuvant of Antigen-Specific CD8+ T Cell Response. Mol. Ther. 2021, 29, 2227–2238. [Google Scholar] [CrossRef]
- Won, J.K.; Bakhoum, S.F. The Cytosolic DNA-Sensing CGAS–Sting Pathway in Cancer. Cancer Discov. 2020, 10, 26–39. [Google Scholar]
- Miao, L.; Li, L.; Huang, Y.; Delcassian, D.; Chahal, J.; Han, J.; Shi, Y.; Sadtler, K.; Gao, W.; Lin, J.; et al. Delivery of MRNA Vaccines with Heterocyclic Lipids Increases anti-Tumor Efficacy by STING-Mediated Immune Cell Activation. Nat. Biotechnol. 2019, 37, 1174–1185. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Song, H. The Enzymes and Control of Eukaryotic MRNA Turnover. Nat. Struct. Mol. Biol. 2004, 11, 121–127. [Google Scholar] [CrossRef]
- Schoenberg, D.R. Mechanisms of Endonuclease-Mediated MRNA Decay. Wiley Interdiscip. Rev. RNA 2011, 2, 582–600. [Google Scholar] [CrossRef] [PubMed]
- Houseley, J.; LaCava, J.; Tollervey, D. RNA-Quality Control by the Exosome. Nat. Rev. Mol. Cell Biol. 2006, 7, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Guerra, G.M.; May, D.; Kroll, T.; Koch, P.; Groth, M.; Wang, Z.Q.; Li, T.L.; Grigaravičius, P. Cell Type-Specific Role of RNA Nuclease SMG6 in Neurogenesis. Cells 2021, 10, 3365. [Google Scholar] [CrossRef]
- Li, W.M.; Barnes, T.; Lee, C.H. Endoribonucleases-Enzymes Gaining Spotlight in MRNA Metabolism. FEBS J. 2010, 277, 627–641. [Google Scholar] [CrossRef]
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Solinís, M.Á.; Del Pozo-Rodríguez, A. Nanomedicines to Deliver MRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364. [Google Scholar] [CrossRef]
- Noubissi, F.K.; Elcheva, I.; Bhatia, N.; Shakoori, A.; Ougolkov, A.; Liu, J.; Minamoto, T.; Ross, J.; Fuchs, S.Y.; Spiegelman, V.S. CRD-BP Mediates Stabilization of ΒTrCP1 and c-Myc MRNA in Response to β-Catenin Signalling. Nature 2006, 441, 898–901. [Google Scholar] [CrossRef]
- Sparanese, D.; Lee, C.H. CRD-BP Shields c-Myc and MDR-1 RNA from Endonucleolytic Attack by a Mammalian Endoribonuclease. Nucleic Acids Res. 2007, 35, 1209–1221. [Google Scholar] [CrossRef]
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.Y.; Verrier, B. Tailoring MRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Tahtinen, S.; Tong, A.J.; Himmels, P.; Oh, J.; Paler-Martinez, A.; Kim, L.; Wichner, S.; Oei, Y.; McCarron, M.J.; Freund, E.C.; et al. IL-1 and IL-1ra Are Key Regulators of the Inflammatory Response to RNA Vaccines. Nat. Immunol. 2022, 23, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Karpov, T.; Postovalova, A.; Akhmetova, D.; Muslimov, A.R.; Eletskaya, E.; Zyuzin, M.V.; Timin, A.S. Universal Chelator-Free Radiolabeling of Organic and Inorganic-Based Nanocarriers with Diagnostic and Therapeutic Isotopes for Internal Radiotherapy. Chem. Mater. 2022, 34, 6593–6605. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis, E.; Sousa, C. Innate Antiviral Responses by Means of TLR7-Mediated Recognition of Single-Stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-Specific Recognition of Single-Stranded RNA via Till-like Receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzózka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA Is the Ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B.R.; Muramatsu, H.; Nallagatla, S.R.; Bevilacqua, P.C.; Sansing, L.H.; Weissman, D.; Karikó, K. Incorporation of Pseudouridine into MRNA Enhances Translation by Diminishing PKR Activation. Nucleic Acids Res. 2010, 38, 5884–5892. [Google Scholar] [CrossRef]
- Devoldere, J.; Dewitte, H.; De Smedt, S.C.; Remaut, K. Evading Innate Immunity in Nonviral MRNA Delivery: Don’t Shoot the Messenger. Drug Discov. Today 2016, 21, 11–25. [Google Scholar] [CrossRef]
- Baiersdörfer, M.; Boros, G.; Muramatsu, H.; Mahiny, A.; Vlatkovic, I.; Sahin, U.; Karikó, K. A Facile Method for the Removal of DsRNA Contaminant from In Vitro-Transcribed MRNA. Mol. Ther. Nucleic Acids 2019, 15, 26–35. [Google Scholar] [CrossRef]
- Piao, X.; Yadav, V.; Wang, E.; Chang, W.; Tau, L.; Lindenmuth, B.E.; Wang, S.X. Double-Stranded RNA Reduction by Chaotropic Agents during in vitro Transcription of Messenger RNA. Mol. Ther. Nucleic Acids 2022, 29, 618–624. [Google Scholar] [CrossRef]
- Podda, A.; Del Giudice, G. MF59-Adjuvanted Vaccines: Increased Immunogenicity with an Optimal Safety Profile. Expert Rev. Vaccines 2003, 2, 197–204. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. MRNA Vaccines—A New Era in Vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Pardi, N.; Parks, R.; Santra, S.; Mu, Z.; Sutherland, L.; Scearce, R.; Barr, M.; Eaton, A.; Hernandez, G.; et al. Lipid Nanoparticle Encapsulated Nucleoside-Modified MRNA Vaccines Elicit Polyfunctional HIV-1 Antibodies Comparable to Proteins in Nonhuman Primates. Npj Vaccines 2021, 6, 50. [Google Scholar] [CrossRef] [PubMed]
- Ndeupen, S.; Qin, Z.; Jacobsen, S.; Bouteau, A.; Estanbouli, H.; Igyártó, B.Z. The MRNA-LNP Platform’s Lipid Nanoparticle Component Used in Preclinical Vaccine Studies Is Highly Inflammatory. iScience 2021, 24, 103479. [Google Scholar] [CrossRef] [PubMed]
- Parhiz, H.; Brenner, J.S.; Patel, P.N.; Papp, T.E.; Shahnawaz, H.; Li, Q.; Shi, R.; Zamora, M.E.; Yadegari, A.; Marcos-Contreras, O.A.; et al. Added to Pre-Existing Inflammation, MRNA-Lipid Nanoparticles Induce Inflammation Exacerbation (IE). J. Control. Release 2022, 344, 50–61. [Google Scholar] [CrossRef]
- Abrams, M.T.; Koser, M.L.; Seitzer, J.; Williams, S.C.; Dipietro, M.A.; Wang, W.; Shaw, A.W.; Mao, X.; Jadhav, V.; Davide, J.P.; et al. Evaluation of Efficacy, Biodistribution, and Inflammation for a Potent SiRNA Nanoparticle: Effect of Dexamethasone Co-Treatment. Mol. Ther. 2010, 18, 171–180. [Google Scholar] [CrossRef]
- Bevers, S.; Kooijmans, S.A.A.; Van de Velde, E.; Evers, M.J.W.; Seghers, S.; Gitz-Francois, J.J.J.M.; van Kronenburg, N.C.H.; Fens, M.H.A.M.; Mastrobattista, E.; Hassler, L.; et al. MRNA-LNP Vaccines Tuned for Systemic Immunization Induce Strong Antitumor Immunity by Engaging Splenic Immune Cells. Mol. Ther. 2022, 30, 3078–3094. [Google Scholar] [CrossRef]
- Kon, E.; Elia, U.; Peer, D. Principles for Designing an Optimal MRNA Lipid Nanoparticle Vaccine. Curr. Opin. Biotechnol. 2022, 73, 329–336. [Google Scholar] [CrossRef]
- Chatzikleanthous, D.; O’Hagan, D.T.; Adamo, R. Lipid-Based Nanoparticles for Delivery of Vaccine Adjuvants and Antigens: Toward Multicomponent Vaccines. Mol. Pharm. 2021, 18, 2867–2888. [Google Scholar] [CrossRef]
- Fan, J.; Jin, S.; Gilmartin, L.; Toth, I.; Hussein, W.M.; Stephenson, R.J. Advances in Infectious Disease Vaccine Adjuvants. Vaccines 2022, 10, 1120. [Google Scholar] [CrossRef]
- Eusébio, D.; Neves, A.R.; Costa, D.; Biswas, S.; Alves, G.; Cui, Z.; Sousa, Â. Methods to Improve the Immunogenicity of Plasmid DNA Vaccines. Drug Discov. Today 2021, 26, 2575–2592. [Google Scholar] [CrossRef]
- Wu, Z.; Liu, K. Overview of Vaccine Adjuvants. Med. Drug Discov. 2021, 11, 100103. [Google Scholar] [CrossRef]
- Garçon, N.; Leroux-Roels, G.; Cheng, W.-F. Vaccine Adjuvants. Perspect. Vaccinol. 2011, 1, 89–113. [Google Scholar] [CrossRef]
- Miller, S.M.; Cybulski, V.; Whitacre, M.; Bess, L.S.; Livesay, M.T.; Walsh, L.; Burkhart, D.; Bazin, H.G.; Evans, J.T. Novel Lipidated Imidazoquinoline TLR7/8 Adjuvants Elicit Influenza-Specific Th1 Immune Responses and Protect Against Heterologous H3N2 Influenza Challenge in Mice. Front. Immunol. 2020, 11, 406. [Google Scholar] [CrossRef] [PubMed]
- Pifferi, C.; Fuentes, R.; Fernández-Tejada, A. Natural and Synthetic Carbohydrate-Based Vaccine Adjuvants and Their Mechanisms of Action. Nat. Rev. Chem. 2021, 5, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Aucouturier, J.; Dupuis, L.; Ganne, V. Adjuvants Designed for Veterinary and Human Vaccines. Vaccine 2001, 19, 2666–2672. [Google Scholar] [CrossRef]
- Díaz-Dinamarca, D.A.; Salazar, M.L.; Castillo, B.N.; Manubens, A.; Vasquez, A.E.; Salazar, F.; Becker, M.I. Protein-Based Adjuvants for Vaccines as Immunomodulators of the Innate and Adaptive Immune Response: Current Knowledge, Challenges, and Future Opportunities. Pharmaceutics 2022, 14, 1671. [Google Scholar] [CrossRef]
- Draper, S.J.; Angov, E.; Horii, T.; Miller, L.H.; Srinivasan, P.; Theisen, M.; Biswas, S. Recent Advances in Recombinant Protein-Based Malaria Vaccines. Vaccine 2015, 33, 7433–7443. [Google Scholar] [CrossRef] [PubMed]
- Schijns, V.E.; Lavelle, E.C. Trends in Vaccine Adjuvants. Expert Rev. Vaccines 2011, 10, 539–550. [Google Scholar] [CrossRef]
- Nguyen-Contant, P.; Sangster, M.Y.; Topham, D.J. Squalene-Based Influenza Vaccine Adjuvants and Their Impact on the Hemagglutinin-Specific B Cell Response. Pathogens 2021, 10, 355. [Google Scholar] [CrossRef]
- Brito, L.A.; Chan, M.; Shaw, C.A.; Hekele, A.; Carsillo, T.; Schaefer, M.; Archer, J.; Seubert, A.; Otten, G.R.; Beard, C.W.; et al. A Cationic Nanoemulsion for the Delivery of Next-Generation RNA Vaccines. Mol. Ther. 2014, 22, 2118–2129. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Petrovsky, N. Molecular Adjuvants for DNA Vaccines. Curr. Issues Mol. Biol. 2017, 22, 17–40. [Google Scholar] [CrossRef] [PubMed]
- Kobiyama, K.; Jounai, N.; Aoshi, T.; Tozuka, M.; Takeshita, F.; Coban, C.; Ishii, K. Innate Immune Signaling by, and Genetic Adjuvants for DNA Vaccination. Vaccines 2013, 1, 278–292. [Google Scholar] [CrossRef]
- Floros, T.; Tarhini, A.A. Anticancer Cytokines: Biology and Clinical Effects of Interferon-A2, Interleukin (IL)-2, IL-15, IL-21, and IL-12. In Seminars in Oncology; WB Saunders: Philadelphia, PA, USA, 2015; Volume 42. [Google Scholar]
- Konjević, G.M.; Vuletić, A.M.; Mirjačić Martinović, K.M.; Larsen, A.K.; Jurišić, V.B. The Role of Cytokines in the Regulation of NK Cells in the Tumor Environment. Cytokine 2019, 117, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Liu, C.H.; Roberts, A.I.; Das, J.; Xu, G.; Ren, G.; Zhang, Y.; Zhang, L.; Zeng, R.Y.; Tan, H.S.W.; et al. Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and T-Cell Responses: What We Do and Don’t Know. Cell Res. 2006, 16, 126–133. [Google Scholar] [CrossRef]
- Luster, A.D. The Role of Chemokines in Linking Innate and Adaptive Immunity. Curr. Opin. Immunol. 2002, 14, 129–135. [Google Scholar] [CrossRef]
- Van Kooten, C.; Banchereau, J. Functions of CD40 on B Cells, Dendritic Cells and Other Cells. Curr. Opin. Immunol. 1997, 9, 330–337. [Google Scholar] [CrossRef]
- Gool, S.W.; Vandenberghe, P.; de Boer, M.; Ceuppens, J.L. CD80, CD86 and CD40 Provide Accessory Signals in a Multiple-Step T-Cell Activation Model. Immunol. Rev. 1996, 153, 47–83. [Google Scholar] [CrossRef]
- Applequist, S.E.; Rollman, E.; Wareing, M.D.; Lidén, M.; Rozell, B.; Hinkula, J.; Ljunggren, H.-G. Activation of Innate Immunity, Inflammation, and Potentiation of DNA Vaccination through Mammalian Expression of the TLR5 Agonist Flagellin. J. Immunol. 2005, 175, 3882–3891. [Google Scholar] [CrossRef]
- Leal, L.; Guardo, A.C.; Moron-Lopez, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; Van Den Ham, H.J.; Gruters, R.; et al. Phase i Clinical Trial of an Intranodally Administered MRNA-Based Therapeutic Vaccine against HIV-1 Infection. AIDS 2018, 32, 2533–2545. [Google Scholar] [CrossRef]
- Bonehill, A.; Tuyaerts, S.; Van Nuffel, A.M.T.; Heirman, C.; Bos, T.J.; Fostier, K.; Neyns, B.; Thielemans, K. Enhancing the T-Cell Stimulatory Capacity of Human Dendritic Cells by Co-Electroporation with CD40L, CD70 and Constitutively Active TLR4 Encoding MRNA. Mol. Ther. 2008, 16, 1170–1180. [Google Scholar] [CrossRef]
- Van Lint, S.; Goyvaerts, C.; Maenhout, S.; Goethals, L.; Disy, A.; Benteyn, D.; Pen, J.; Bonehill, A.; Heirman, C.; Breckpot, K.; et al. Preclinical Evaluation of TriMix and Antigen MRNA-Based Antitumor Therapy. Cancer Res. 2012, 72, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, F.; Tanaka, T.; Matsuda, T.; Tozuka, M.; Kobiyama, K.; Saha, S.; Matsui, K.; Ishii, K.J.; Coban, C.; Akira, S.; et al. Toll-Like Receptor Adaptor Molecules Enhance DNA-Raised Adaptive Immune Responses against Influenza and Tumors through Activation of Innate Immunity. J. Virol. 2006, 80, 6218–6224. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Cai, S.; Pang, H.; Jian, J.; Wu, Z. Immunogenicity and Efficacy of DNA Vaccine Encoding Antigenic AcfA via Addition of the Molecular Adjuvant Myd88 against Vibrio Alginolyticus in Epinephelus Coioides. Fish Shellfish. Immunol. 2017, 66, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.; Yi, L.; Yang, Z.; Yang, J.; Shao, H.; Zhang, C.; Pan, Z. The Toll-like Receptor Adaptor Molecule TRIF Enhances DNA Vaccination against Classical Swine Fever. Vet. Immunol. Immunopathol. 2010, 137, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, S.; Amara, R.R.; Yeow, W.-S.; Pitha, P.M.; Robinson, H.L. Regulation of DNA-Raised Immune Responses by Cotransfected Interferon Regulatory Factors. J. Virol. 2002, 76, 6652–6659. [Google Scholar] [CrossRef]
- Castaldello, A.; Sgarbanti, M.; Marsili, G.; Brocca-Cofano, E.; Remoli, A.L.; Caputo, A.; Battistini, A. Interferon Regulatory Factor-1 Acts as a Powerful Adjuvant in Tat DNA Based Vaccination. J. Cell. Physiol. 2010, 224, 702–709. [Google Scholar] [CrossRef]
- Bramson, J.L.; Dayball, K.; Hall, J.R.; Millar, J.B.; Miller, M.; Wan, Y.H.; Lin, R.; Hiscott, J. Super-Activated Interferon-Regulatory Factors Can Enhance Plasmid Immunization. Vaccine 2003, 21, 1363–1370. [Google Scholar] [CrossRef]
- Chu, D.; Moroda, M.; Piao, L.X.; Aosai, F. CTL Induction by DNA Vaccine with Toxoplasma Gondii-HSP70 Gene. Parasitol. Int. 2014, 63, 408–416. [Google Scholar] [CrossRef]
- Zhou, J.; Cheung, A.K.L.; Tan, Z.; Wang, H.; Yu, W.; Du, Y.; Kang, Y.; Lu, X.; Liu, L.; Yuen, K.Y.; et al. PD1-Based DNA Vaccine Amplifies HIV-1 GAG-Specific CD8+ T Cells in Mice. J. Clin. Investig. 2013, 123, 2629–2642. [Google Scholar] [CrossRef]
- Liniger, M.; Summerfield, A.; Ruggli, N. MDA5 Can Be Exploited as Efficacious Genetic Adjuvant for DNA Vaccination against Lethal H5N1 Influenza Virus Infection in Chickens. PLoS ONE 2012, 7, e49952. [Google Scholar] [CrossRef]
- Shedlock, D.J.; Tingey, C.; Mahadevan, L.; Hutnick, N.; Reuschel, E.L.; Kudchodkar, S.; Flingai, S.; Yan, J.; Kim, J.J.; Ugen, K.E.; et al. Co-Administration of Molecular Adjuvants Expressing NF-Kappa B Subunit P65/RelA or Type-1 Transactivator T-Bet Enhance Antigen Specific DNA Vaccine-Induced Immunity. Vaccines 2014, 2, 196–215. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Wu, J.; Zhang, R.; Chen, L. T-Bet Acts as a Powerful Adjuvant in Ag85B DNA-Based Vaccination against Tuberculosis. Mol. Med. Rep. 2012, 6, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Coban, C.; Kobiyama, K.; Aoshi, T.; Takeshita, F.; Horii, T.; Akira, S.; Ishii, K.J. Novel Strategies to Improve DNA Vaccine Immunogenicity. Curr. Gene Ther. 2011, 11, 479–484. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, G.; Laddy, D.J.; Sundaram, S.G.; Fagone, P.; Shedlock, D.J.; Kannan, S.; Wu, L.; Chung, C.W.; Lankaraman, K.M.; Burns, J.; et al. Co-Immunization with an Optimized Plasmid-Encoded Immune Stimulatory Interleukin, High-Mobility Group Box 1 Protein, Results in Enhanced Interferon-β Secretion by Antigen-Specific CD8 T Cells. Immunology 2009, 128, e612–e620. [Google Scholar] [CrossRef] [PubMed]
- Fagone, P.; Shedlock, D.J.; Bao, H.; Kawalekar, O.U.; Yan, J.; Gupta, D.; Morrow, M.P.; Patel, A.; Kobinger, G.P.; Muthumani, K.; et al. Molecular Adjuvant HMGB1 Enhances Anti-Influenza Immunity during DNA Vaccination. Gene Ther. 2011, 18, 1070–1077. [Google Scholar] [CrossRef] [PubMed]
- Lladser, A.; Mougiakakos, D.; Tufvesson, H.; Ligtenberg, M.A.; Quest, A.F.G.; Kiessling, R.; Ljungberg, K. DAI (DLM-1/ZBP1) as a Genetic Adjuvant for DNA Vaccines That Promotes Effective Antitumor CTL Immunity. Mol. Ther. 2011, 19, 594–601. [Google Scholar] [CrossRef]
- Siddiqui, A.A.; Phillips, T.; Charest, H.; Podesta, R.B.; Quinlin, M.L.; Pinkston, J.R.; Lloyd, J.D.; Paz, M.; Villalovos, R.M.; Pompa, J. Induction of Protective Immunity against Schistosoma Mansoni via DNA Priming and Boosting with the Large Subunit of Calpain (Sm-P80): Adjuvant Effects of Granulocyte-Macrophage Colony-Stimulating Factor and Interleukin-4. Infect. Immun. 2003, 71, 3844–3851. [Google Scholar] [CrossRef]
- Sedegah, M.; Weiss, W.; Sacci, J.B.; Charoenvit, Y.; Hedstrom, R.; Gowda, K.; Majam, V.F.; Tine, J.; Kumar, S.; Hobart, P.; et al. Improving Protective Immunity Induced by DNA-Based Immunization: Priming with Antigen and GM-CSF-Encoding Plasmid DNA and Boosting with Antigen-Expressing Recombinant Poxvirus. J. Immunol. 2000, 164, 5905–5912. [Google Scholar] [CrossRef]
- Norell, H.; Poschke, I.; Charo, J.; Wei, W.Z.; Erskine, C.; Piechocki, M.P.; Knutson, K.L.; Bergh, J.; Lidbrink, E.; Kiessling, R. Vaccination with a Plasmid DNA Encoding HER-2/Neu Together with Low Doses of GM-CSF and IL-2 in Patients with Metastatic Breast Carcinoma: A Pilot Clinical Trial. J. Transl. Med. 2010, 8, 53. [Google Scholar] [CrossRef]
- Perales, M.A.; Yuan, J.; Powel, S.; Gallardo, H.F.; Rasalan, T.S.; Gonzalez, C.; Manukian, G.; Wang, J.; Zhang, Y.; Chapman, P.B.; et al. Phase I/II Study of GM-CSF DNA as an Adjuvant for a Multipeptide Cancer Vaccine in Patients with Advanced Melanoma. Mol. Ther. 2008, 16, 2022–2029. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.A.; Phillips, T.; Charest, H.; Podesta, R.B.; Quinlin, M.L.; Pinkston, J.R.; Lloyd, J.D.; Pompa, J.; Villalovos, R.M.; Paz, M. Enhancement of Sm-P80 (Large Subunit of Calpain) Induced Protective Immunity against Schistosoma Mansoni through Co-Delivery of Interleukin-2 and Interleukin-12 in a DNA Vaccine Formulation. Vaccine 2003, 21, 2882–2889. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Taylor, M.G.; Johansen, M.V.; Bickle, Q.D. Vaccination of Mice with a Cocktail DNA Vaccine Induces a Th1-Type Immune Response and Partial Protection against Schistosoma Japonicum Infection. Vaccine 2001, 20, 724–730. [Google Scholar] [CrossRef] [PubMed]
- Khosroshahi, K.H.; Ghaffarifar, F.; Sharifi, Z.; D’Souza, S.; Dalimi, A.; Hassan, Z.M.; Khoshzaban, F. Comparing the Effect of IL-12 Genetic Adjuvant and Alum Non-Genetic Adjuvant on the Efficiency of the Cocktail DNA Vaccine Containing Plasmids Encoding SAG-1 and ROP-2 of Toxoplasma Gondii. Parasitol. Res. 2012, 111, 403–411. [Google Scholar] [CrossRef]
- Maspi, N.; Ghaffarifar, F.; Sharifi, Z.; Dalimi, A. Codelivery of DNA Vaccination Encoding LeIF Gene and IL-12 Increases Protection against Leishmania Major Infection in BALB/c Mice. Parasite Immunol. 2016, 38, 228–235. [Google Scholar] [CrossRef]
- Naderi, M.; Saeedi, A.; Moradi, A.; Kleshadi, M.; Zolfaghari, M.R.; Gorji, A.; Ghaemi, A. Interleukin-12 as a Genetic Adjuvant Enhances Hepatitis C Virus NS3 DNA Vaccine Immunogenicity. Virol. Sin. 2013, 28, 167–173. [Google Scholar] [CrossRef]
- Yang, S.H.; Lee, C.G.; Park, S.H.; Im, S.J.; Kim, Y.M.; Son, J.M.; Wang, J.S.; Yoon, S.K.; Song, M.K.; Ambrozaitis, A.; et al. Correlation of Antiviral T-Cell Responses with Suppression of Viral Rebound in Chronic Hepatitis B Carriers: A Proof-of-Concept Study. Gene Ther. 2006, 13, 1110–1117. [Google Scholar] [CrossRef]
- Yang, Y.; Shao, Z.; Gao, J. Antitumor Effect of a DNA Vaccine Harboring Prostate Cancer-Specific Antigen with IL-12 as an Intramolecular Adjuvant. J. Mol. Microbiol. Biotechnol. 2017, 27, 168–174. [Google Scholar] [CrossRef]
- Tian, D.Y.; Sun, Y.; Wai, S.F.; Lee, F.K.; Meng, Q.L.; Suen, K.M.; Wang, N.; Han, W.; Li, S.; Li, Y.F.; et al. Enhancement of the Immunogenicity of an Alphavirus Replicon-Based DNA Vaccine against Classical Swine Fever by Electroporation and Coinjection with a Plasmid Expressing Porcine Interleukin 2. Vaccine 2012, 30, 3587–3594. [Google Scholar] [CrossRef]
- Zhao, G.W.; Yan, R.F.; Muleke, C.I.; Sun, Y.M.; Xu, L.X.; Li, X.R. Vaccination of Goats with DNA Vaccines Encoding H11 and IL-2 Induces Partial Protection against Haemonchus Contortus Infection. Vet. J. 2012, 191, 94–100. [Google Scholar] [CrossRef]
- Passardi, A.; Cecconetto, L.; Dall’Agata, M.; Dazzi, C.; Pasquini, E.; Oliverio, G.; Zumaglini, F.; Zoli, W.; Nanni, O.; Milandri, C.; et al. Randomized Phase II Study with Two Gemcitabine- and Docetaxel-Based Combinations as First-Line Chemotherapy for Metastatic Non-Small Cell Lung Cancer. J. Transl. Med. 2008, 6, 65. [Google Scholar] [CrossRef] [PubMed]
- Hezarjaribi, H.Z.; Ghaffarifar, F.; Dalimi, A.; Sharifi, Z.; Jorjani, O. Effect of IL-22 on DNA Vaccine Encoding LACK Gene of Leishmania Major in BALB/c Mice. Exp. Parasitol. 2013, 134, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Chen, J.; Petersen, E.; Zhou, D.H.; Huang, S.Y.; Song, H.Q.; Zhu, X.Q. Synergy of MIL-21 and MIL-15 in Enhancing DNA Vaccine Efficacy against Acute and Chronic Toxoplasma Gondii Infection in Mice. Vaccine 2014, 32, 3058–3065. [Google Scholar] [CrossRef]
- Chen, J.; Li, Z.Y.; Huang, S.Y.; Petersen, E.; Song, H.Q.; Zhou, D.H.; Zhu, X.Q. Protective Efficacy of Toxoplasma Gondii Calcium-Dependent Protein Kinase 1 (TgCDPK1) Adjuvated with Recombinant IL-15 and IL-21 against Experimental Toxoplasmosis in Mice. BMC Infect. Dis. 2014, 14, 487. [Google Scholar] [CrossRef] [PubMed]
- Eickhoff, C.S.; Vasconcelos, J.R.; Sullivan, N.L.; Blazevic, A.; Bruna-Romero, O.; Rodrigues, M.M.; Hoft, D.F. Co-Administration of a Plasmid DNA Encoding IL-15 Improves Long-Term Protection of a Genetic Vaccine against Trypanosoma Cruzi. PLoS Negl. Trop. Dis. 2011, 5, e983. [Google Scholar] [CrossRef]
- Kutzler, M.A.; Robinson, T.M.; Chattergoon, M.A.; Choo, D.K.; Choo, A.Y.; Choe, P.Y.; Ramanathan, M.P.; Parkinson, R.; Kudchodkar, S.; Tamura, Y.; et al. Coimmunization with an Optimized IL-15 Plasmid Results in Enhanced Function and Longevity of CD8 T Cells That Are Partially Independent of CD4 T Cell Help. J. Immunol. 2005, 175, 112–123. [Google Scholar] [CrossRef]
- Su, B.; Wang, J.; Zhao, G.; Wang, X.; Li, J.; Wang, B. Sequential Administration of Cytokine Genes to Enhance Cellular Immune Responses and CD4+ T Memory Cells during DNA Vaccination. Hum. Vaccines Immunother. 2012, 8, 1659–1667. [Google Scholar] [CrossRef]
- Williman, J.; Young, S.; Buchan, G.; Slobbe, L.; Wilson, M.; Pang, P.; Austyn, J.; Preston, S.; Baird, M. DNA Fusion Vaccines Incorporating IL-23 or RANTES for Use in Immunization against Influenza. Vaccine 2008, 26, 5153–5158. [Google Scholar] [CrossRef]
- Song, R.; Liu, S.; Leong, K.W. Effects of MIP-1α, MIP-3α, and MIP-3β on the Induction of HIV Gag-Specific Immune Response with DNA Vaccines. Mol. Ther. 2007, 15, 1007–1015. [Google Scholar] [CrossRef]
- Westermann, J.; Nguyen-Hoai, T.; Baldenhofer, G.; Höpken, U.E.; Lipp, M.; Dörken, B.; Pezzutto, A. CCL19 (ELC) as an Adjuvant for DNA Vaccination: Induction of a TH1-Type T-Cell Response and Enhancement of Antitumor Immunity. Cancer Gene Ther. 2007, 14, 523–532. [Google Scholar] [CrossRef]
- Kang, T.H.; Kim, K.W.; Bae, H.C.; Seong, S.Y.; Kim, T.W. Enhancement of DNA Vaccine Potency by Antigen Linkage to IFN-Iγ-Inducible Protein-10. Int. J. Cancer 2011, 128, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.W.; Aleyas, A.G.; George, J.A.; Kim, S.J.; Kim, H.K.; Yoo, D.J.; Kang, S.H.; Eo, S.K. Genetic Co-Transfer of CCR7 Ligands Enhances Immunity and Prolongs Survival against Virulent Challenge of Pseudorabies Virus. Immunol. Cell Biol. 2009, 87, 91–99. [Google Scholar] [CrossRef]
- Toka, F.N.; Gierynska, M.; Rouse, B.T. Codelivery of CCR7 Ligands as Molecular Adjuvants Enhances the Protective Immune Response against Herpes Simplex Virus Type 1. J. Virol. 2003, 77, 12742–12752. [Google Scholar] [CrossRef] [PubMed]
- Yamano, T.; Kaneda, Y.; Huang, S.; Hiramatsu, S.H.; Hoon, D.S.B. Enhancement of Immunity by a DNA Melanoma Vaccine against TRP2 with CCL21 as an Adjuvant. Mol. Ther. 2006, 13, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Fló, J.; Tisminetzky, S.; Baralle, F. Modulation of the Immune Response to DNA Vaccine by Co-Delivery of Costimulatory Molecules. Immunology 2000, 100, 259–267. [Google Scholar] [CrossRef] [PubMed]
- de Andrés, X.; Reina, R.; Ciriza, J.; Crespo, H.; Glaria, I.; Ramírez, H.; Grilló, M.J.; Pérez, M.M.; Andrésdóttir, V.; Rosati, S.; et al. Use of B7 Costimulatory Molecules as Adjuvants in a Prime-Boost Vaccination against Visna/Maedi Ovine Lentivirus. Vaccine 2009, 27, 4591–4600. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.; Liang, F.; Liu, N.; Song, X.; Yuan, F.; Luo, Y.; Zhao, X.; Long, J.; Sun, Y.; Xi, Y. Potent Antirheumatic Activity of a New DNA Vaccine Targeted to B7-2/CD28 Costimulatory Signaling Pathway in Autoimmune Arthritis. Hum. Gene Ther. 2011, 22, 65–76. [Google Scholar] [CrossRef]
- Stone, G.W.; Barzee, S.; Snarsky, V.; Kee, K.; Spina, C.A.; Yu, X.-F.; Kornbluth, R.S. Multimeric Soluble CD40 Ligand and GITR Ligand as Adjuvants for Human Immunodeficiency Virus DNA Vaccines. J. Virol. 2006, 80, 1762–1772. [Google Scholar] [CrossRef]
- Dybul, M.; Mercier, G.; Belson, M.; Hallahan, C.W.; Liu, S.; Perry, C.; Herpin, B.; Ehler, L.; Davey, R.T.; Metcalf, J.A.; et al. CD40 Ligand Trimer and IL-12 Enhance Peripheral Blood Mononuclear Cells and CD4 + T Cell Proliferation and Production of IFN-γ in Response to P24 Antigen in HIV-Infected Individuals: Potential Contribution of Anergy to HIV-Specific Unresponsiveness. J. Immunol. 2000, 165, 1685–1691. [Google Scholar] [CrossRef]
- Elia, U.; Ramishetti, S.; Rosenfeld, R.; Dammes, N.; Bar-Haim, E.; Naidu, G.S.; Makdasi, E.; Yahalom-Ronen, Y.; Tamir, H.; Paran, N.; et al. Design of SARS-CoV-2 HFc-Conjugated Receptor-Binding Domain MRNA Vaccine Delivered via Lipid Nanoparticles. ACS Nano 2021, 15, 9627–9637. [Google Scholar] [CrossRef]
- Elia, U.; Rotem, S.; Bar-Haim, E.; Ramishetti, S.; Naidu, G.S.; Gur, D.; Aftalion, M.; Israeli, M.; Bercovich-Kinori, A.; Alcalay, R.; et al. Lipid Nanoparticle RBD-HFc MRNA Vaccine Protects HACE2 Transgenic Mice against a Lethal SARS-CoV-2 Infection. Nano Lett. 2021, 21, 4774–4779. [Google Scholar] [CrossRef] [PubMed]
- Duivelshof, B.L.; Murisier, A.; Camperi, J.; Fekete, S.; Beck, A.; Guillarme, D.; D’Atri, V. Therapeutic Fc-Fusion Proteins: Current Analytical Strategies. J. Sep. Sci. 2021, 44, 35–62. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.; Chatzikleanthous, D.; Lou, G.; Giusti, F.; Bonci, A.; Taccone, M.; Brazzoli, M.; Gallorini, S.; Ferlenghi, I.; Berti, F.; et al. Mannosylation of LNP Results in Improved Potency for Self-Amplifying RNA (SAM) Vaccines. ACS Infect. Dis. 2019, 5, 1546–1558. [Google Scholar] [CrossRef] [PubMed]
- Satija, R.; Shalek, A.K. Heterogeneity in Immune Responses: From Populations to Single Cells. Trends Immunol. 2014, 35, 219–229. [Google Scholar] [CrossRef]
- Hervé, C.; Laupèze, B.; Del Giudice, G.; Didierlaurent, A.M.; Da Silva, F.T. The How’s and What’s of Vaccine Reactogenicity. Npj Vaccines 2019, 4, 39. [Google Scholar] [CrossRef]
- Dabo, S.; Meurs, E.F. DsRNA-Dependent Protein Kinase PKR and Its Role in Stress, Signaling and HCV Infection. Viruses 2012, 4, 2598–2635. [Google Scholar] [CrossRef]
- Kim, Y.; Park, J.; Kim, S.; Kim, M.A.; Kang, M.G.; Kwak, C.; Kang, M.; Kim, B.; Rhee, H.W.; Kim, V.N. PKR Senses Nuclear and Mitochondrial Signals by Interacting with Endogenous Double-Stranded RNAs. Mol. Cell 2018, 71, 1051–1063.e6. [Google Scholar] [CrossRef]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L Pathway by Viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef]
- Shevyrev, D.; Blinova, E.A.; Kozlov, V.A. The influence of humoral factors of homeostatistic proliferation on t-regulatory cells in vitro. Bull. Sib. Med. 2019, 18, 286–293. [Google Scholar] [CrossRef]
- Bolze, A.; Neveux, I.; Schiabor Barrett, K.M.; White, S.; Isaksson, M.; Dabe, S.; Lee, W.; Grzymski, J.J.; Washington, N.L.; Cirulli, E.T. HLA-A*03:01 Is Associated with Increased Risk of Fever, Chills, and Stronger Side Effects from Pfizer-BioNTech COVID-19 Vaccination. Hum. Genet. Genom. Adv. 2022, 3, 100084. [Google Scholar] [CrossRef]
- Mouliou, D.S.; Dardiotis, E. Current Evidence in SARS-CoV-2 MRNA Vaccines and Post-Vaccination Adverse Reports: Knowns and Unknowns. Diagnostics 2022, 12, 1555. [Google Scholar] [CrossRef] [PubMed]
- Seneff, S.; Nigh, G.; Kyriakopoulos, A.M.; McCullough, P.A. Innate Immune Suppression by SARS-CoV-2 MRNA Vaccinations: The Role of G-Quadruplexes, Exosomes, and MicroRNAs. Food Chem. Toxicol. 2022, 164, 113008. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhao, M.; Zhou, W.; Chen, L.; Li, M.; Jiang, B.; Zhao, X.; Wu, H.; Shen, L.; Zhang, N.; et al. The Link between Genetic Variation and Variability in Vaccine Responses: A Narrative Review. J. Bio-X Res. 2022, 5, 49–54. [Google Scholar] [CrossRef]
- Newport, M.J. The Genetic Regulation of Infant Immune Responses to Vaccination. Front. Immunol. 2015, 6, 18. [Google Scholar] [CrossRef]
- Nishibayashi, H.; Kishimoto, S.; Sekiguchi, K.; Okada, S.; Ihara, K. Myocarditis in 13-Year-Old Monochorionic Diamniotic Twins After COVID-19 Vaccination. J. Clin. Immunol. 2022, 42, 1351–1353. [Google Scholar] [CrossRef]
- Heymans, S.; Cooper, L.T. Myocarditis after COVID-19 MRNA Vaccination: Clinical Observations and Potential Mechanisms. Nat. Rev. Cardiol. 2022, 19, 75–77. [Google Scholar] [CrossRef]
- Monge, S.; Pastor-Barriuso, R.; Hernán, M.A. The imprinting effect of COVID-19 vaccines: An expected selection bias in observational studies. BMJ 2023, 381, e074404. [Google Scholar] [CrossRef]
- Jankowsky, E.; Fairman, M.E. RNA helicases—One fold for many functions. Curr. Opin. Struct. Biol. 2007, 17, 316–324. [Google Scholar] [CrossRef]
- Chavda, V.P.; Hossain, M.K.; Beladiya, J.; Apostolopoulos, V. Nucleic Acid Vaccines for COVID-19: A Paradigm Shift in the Vaccine Development Arena. Biologics 2021, 1, 337–356. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).