
Inflammatory Blood Biomarkers Are Associated with Long-Term Clinical Disease Severity in Parkinson’s Disease


Abstract
:1. Introduction
2. Results
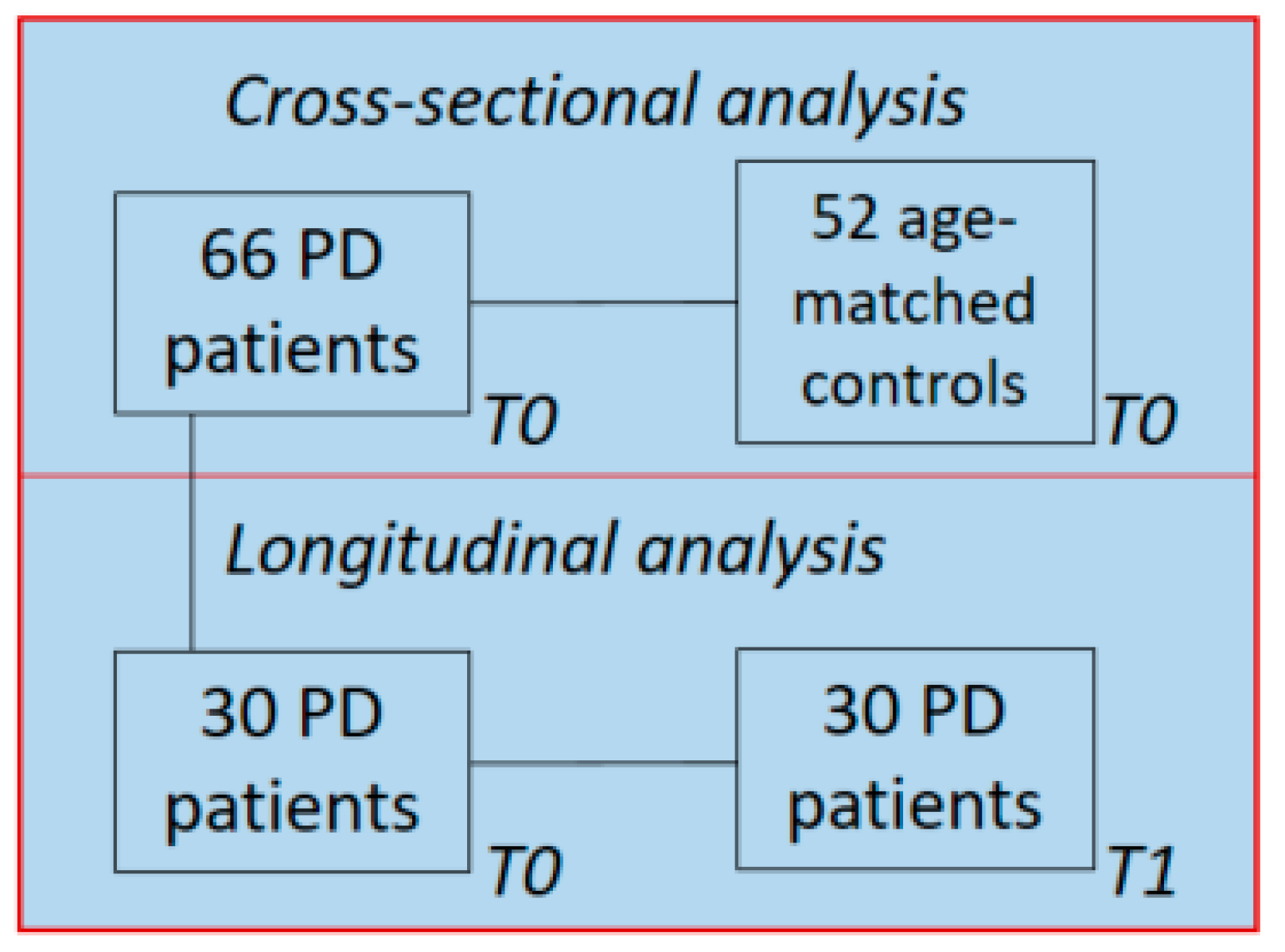
2.1. Demographics and Clinical Data
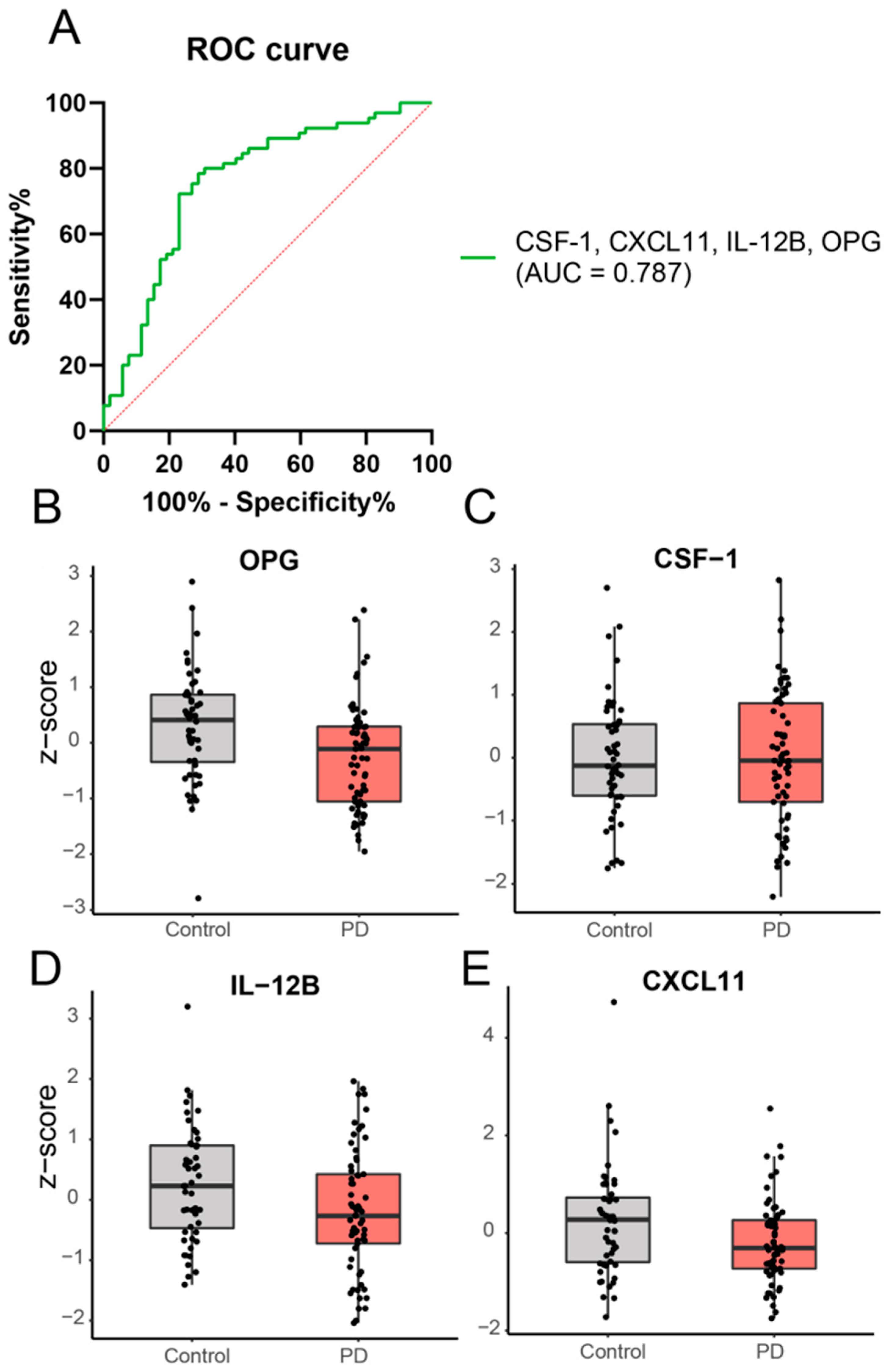
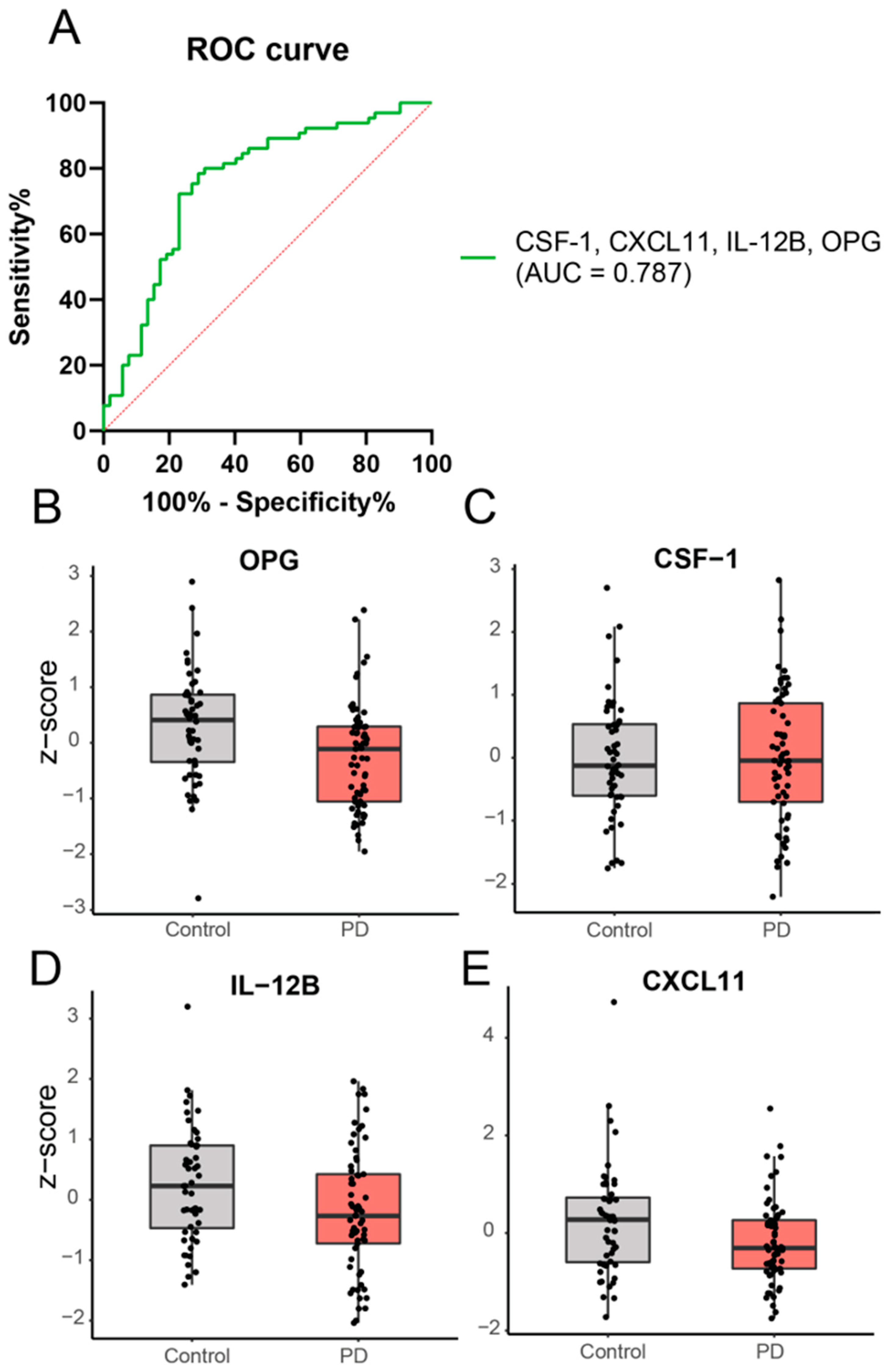
2.2. Cross-Sectional Analysis: A Panel of Four Inflammatory Proteins Can Discriminate PD from Controls
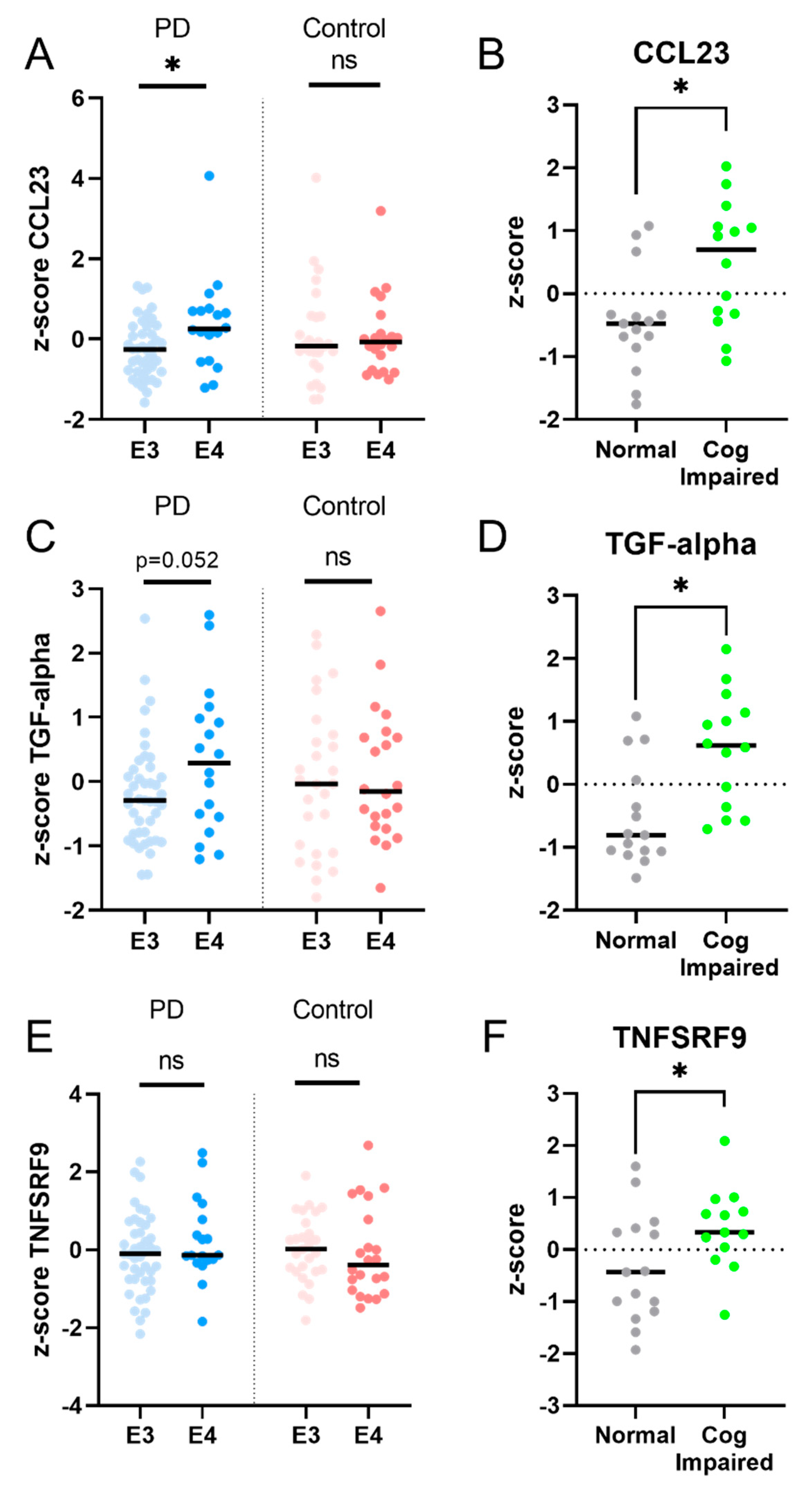
2.3. Longitudinal Analysis: Five Inflammatory Proteins Increase over Time and Clinical Correlations
2.4. Independent Validation in AMP-PD Program
3. Discussion
4. Conclusions and Future Directions
5. Materials and Methods
5.1. Study Population and Clinical Assessment
5.2. Validation in PDBP and PPMI Cohorts
Plasma samples and Proximity Extension Technology (PEA) by OLINK
5.3. APOE Genotyping
5.4. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef] [PubMed]
- Bartl, M.; Xylaki, M.; Bähr, M.; Weber, S.; Trenkwalder, C.; Mollenhauer, B. Evidence for immune system alterations in peripheral biological fluids in Parkinson’s disease. Neurobiol. Dis. 2022, 170, 105744. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef] [PubMed]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bove, J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 2020, 143, 3717–3733. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Alcalay, R.N.; Garretti, F.; Cote, L.; Kanter, E.; Agin-Liebes, J.; Liong, C.; McMurtrey, C.; Hildebrand, W.H.; Mao, X.; et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature 2017, 546, 656–661. [Google Scholar] [CrossRef]
- Reale, M.; Iarlori, C.; Thomas, A.; Gambi, D.; Perfetti, B.; Di Nicola, M.; Onofrj, M. Peripheral cytokines profile in Parkinson’s disease. Brain Behav. Immun. 2009, 23, 55–63. [Google Scholar] [CrossRef]
- Brodacki, B.; Staszewski, J.; Toczylowska, B.; Kozlowska, E.; Drela, N.; Chalimoniuk, M.; Stepien, A. Serum interleukin (IL-2, IL-10, IL-6, IL-4), TNFalpha, and INFgamma concentrations are elevated in patients with atypical and idiopathic parkinsonism. Neurosci. Lett. 2008, 441, 158–162. [Google Scholar] [CrossRef]
- Rathnayake, D.; Chang, T.; Udagama, P. Selected serum cytokines and nitric oxide as potential multi-marker biosignature panels for Parkinson disease of varying durations: A case-control study. BMC Neurol. 2019, 19, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.W.; Chen, C.M.; Chang, K.H. Biomarker of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4148. [Google Scholar] [CrossRef]
- Usenko, T.S.; Nikolaev, M.A.; Miliukhina, I.V.; Bezrukova, A.I.; Senkevich, K.A.; Gomzyakova, N.A.; Beltceva, Y.A.; Zalutskaya, N.M.; Gracheva, E.V.; Timofeeva, A.A.; et al. Plasma cytokine profile in synucleinophaties with dementia. J. Clin. Neurosci. 2020, 78, 323–326. [Google Scholar] [CrossRef] [PubMed]
- Brockmann, K.; Schulte, C.; Schneiderhan-Marra, N.; Apel, A.; Pont-Sunyer, C.; Vilas, D.; Ruiz-Martinez, J.; Langkamp, M.; Corvol, J.C.; Cormier, F.; et al. Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur. J. Neurol. 2017, 24, 427-e6. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.; Yarnall, A.J.; Lawson, R.A.; Breen, D.P.; Evans, J.R.; Cummins, G.A.; Duncan, G.W.; Khoo, T.K.; Burn, D.J.; et al. Serum immune markers and disease progression in an incident Parkinson’s disease cohort (ICICLE-PD). Mov. Disord. 2016, 31, 995–1003. [Google Scholar] [CrossRef] [PubMed]
- Bartl, M.; Dakna, M.; Schade, S.; Otte, B.; Wicke, T.; Lang, E.; Starke, M.; Ebentheuer, J.; Weber, S.; Toischer, K.; et al. Blood Markers of Inflammation, Neurodegeneration, and Cardiovascular Risk in Early Parkinson’s Disease. Mov. Disord. 2023, 38, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Abdi, I.Y.; Bartl, M.; Dakna, M.; Abdesselem, H.; Majbour, N.; Trenkwalder, C.; El-Agnaf, O.; Mollenhauer, B. Cross-sectional proteomic expression in Parkinson’s disease-related proteins in drug-naive patients vs healthy controls with longitudinal clinical follow-up. Neurobiol. Dis. 2023, 177, 105997. [Google Scholar] [CrossRef]
- Winchester, L.; Barber, I.; Lawton, M.; Ash, J.; Liu, B.; Evetts, S.; Hopkins-Jones, L.; Lewis, S.; Bresner, C.; Malpartida, A.B.; et al. Identification of a possible proteomic biomarker in Parkinson’s disease: Discovery and replication in blood, brain and cerebrospinal fluid. Brain Commun. 2023, 5, fcac343. [Google Scholar] [CrossRef]
- Parkinson Progression Marker, I. The Parkinson Progression Marker Initiative (PPMI). Prog. Neurobiol. 2011, 95, 629–635. [Google Scholar] [CrossRef]
- Neal, M.L.; Fleming, S.M.; Budge, K.M.; Boyle, A.M.; Kim, C.; Alam, G.; Beier, E.E.; Wu, L.J.; Richardson, J.R. Pharmacological inhibition of CSF1R by GW2580 reduces microglial proliferation and is protective against neuroinflammation and dopaminergic neurodegeneration. FASEB J. 2020, 34, 1679–1694. [Google Scholar] [CrossRef]
- Chandrasekaran, S.; Bonchev, D. A network view on Parkinson’s disease. Comput. Struct. Biotechnol. J. 2013, 7, e201304004. [Google Scholar] [CrossRef]
- Yun, H.M.; Kim, J.A.; Hwang, C.J.; Jin, P.; Baek, M.K.; Lee, J.M.; Hong, J.E.; Lee, S.M.; Han, S.B.; Oh, K.W.; et al. Neuroinflammatory and Amyloidogenic Activities of IL-32beta in Alzheimer’s Disease. Mol. Neurobiol. 2015, 52, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Alrafiah, A.; Al-Ofi, E.; Obaid, M.T.; Alsomali, N. Assessment of the Levels of Level of Biomarkers of Bone Matrix Glycoproteins and Inflammatory Cytokines from Saudi Parkinson Patients. Biomed. Res. Int. 2019, 2019, 2690205. [Google Scholar] [CrossRef] [PubMed]
- Koper, O.M.; Kaminska, J.; Sawicki, K.; Kemona, H. CXCL9, CXCL10, CXCL11, and their receptor (CXCR3) in neuroinflammation and neurodegeneration. Adv. Clin. Exp. Med. 2018, 27, 849–856. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, M.; Dai, W.; Guo, W.; Qiu, J.; Zhang, Z.; Mo, M.; Ding, L.; Ye, P.; Wu, Y.; et al. Bone-Derived Factors as Potential Biomarkers for Parkinson’s Disease. Front. Aging Neurosci. 2021, 13, 634213. [Google Scholar] [CrossRef] [PubMed]
- Lindestam Arlehamn, C.S.; Dhanwani, R.; Pham, J.; Kuan, R.; Frazier, A.; Rezende Dutra, J.; Phillips, E.; Mallal, S.; Roederer, M.; Marder, K.S.; et al. alpha-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 2020, 11, 1875. [Google Scholar] [CrossRef]
- Stevens, C.H.; Rowe, D.; Morel-Kopp, M.C.; Orr, C.; Russell, T.; Ranola, M.; Ward, C.; Halliday, G.M. Reduced T helper and B lymphocytes in Parkinson’s disease. J. Neuroimmunol. 2012, 252, 95–99. [Google Scholar] [CrossRef]
- Cleary, A.M.; Tu, W.; Enright, A.; Giffon, T.; Dewaal-Malefyt, R.; Gutierrez, K.; Lewis, D.B. Impaired accumulation and function of memory CD4 T cells in human IL-12 receptor beta 1 deficiency. J. Immunol. 2003, 170, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.C.; Choi, Y. Regulation of T cell-associated tissues and T cell activation by RANKL-RANK-OPG. J. Bone Miner. Metab. 2021, 39, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Carter, S.; Hofer, M.J.; Campbell, I.L. Review: The chemokine receptor CXCR3 and its ligands CXCL9, CXCL10 and CXCL11 in neuroimmunity—A tale of conflict and conundrum. Neuropathol. Appl. Neurobiol. 2010, 36, 368–387. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.D.; Banerjee, R.; Liu, J.; Gendelman, H.E.; Mosley, R.L. Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson’s disease. J. Leukoc. Biol. 2007, 82, 1083–1094. [Google Scholar] [CrossRef]
- Simats, A.; Garcia-Berrocoso, T.; Penalba, A.; Giralt, D.; Llovera, G.; Jiang, Y.; Ramiro, L.; Bustamante, A.; Martinez-Saez, E.; Canals, F.; et al. CCL23: A new CC chemokine involved in human brain damage. J. Intern. Med. 2018, 283, 461–475. [Google Scholar] [CrossRef] [PubMed]
- Faura, J.; Bustamante, A.; Penalba, A.; Giralt, D.; Simats, A.; Martinez-Saez, E.; Alcolea, D.; Fortea, J.; Lleo, A.; Teunissen, C.E.; et al. CCL23: A Chemokine Associated with Progression from Mild Cognitive Impairment to Alzheimer’s Disease. J. Alzheimer’s Dis. 2020, 73, 1585–1595. [Google Scholar] [CrossRef]
- Umeh, C.C.; Mahajan, A.; Mihailovic, A.; Pontone, G.M. APOE4 Allele, Sex, and Dementia Risk in Parkinson’s Disease: Lessons From a Longitudinal Cohort. J. Geriatr. Psychiatry Neurol. 2022, 35, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.T.; Tan, L.; Hardy, J. Apolipoprotein E in Alzheimer’s disease: An update. Annu. Rev. Neurosci. 2014, 37, 79–100. [Google Scholar] [CrossRef]
- Yeo, Y.A.; Martinez Gomez, J.M.; Croxford, J.L.; Gasser, S.; Ling, E.A.; Schwarz, H. CD137 ligand activated microglia induces oligodendrocyte apoptosis via reactive oxygen species. J. Neuroinflamm. 2012, 9, 173. [Google Scholar] [CrossRef] [PubMed]
- Wong, H.Y.; Prasad, A.; Gan, S.U.; Chua, J.J.E.; Schwarz, H. Identification of CD137-Expressing B Cells in Multiple Sclerosis Which Secrete IL-6 Upon Engagement by CD137 Ligand. Front. Immunol. 2020, 11, 571964. [Google Scholar] [CrossRef] [PubMed]
- Guler, S.; Gul, T.; Guler, S.; Haerle, M.C.; Basak, A.N. Early-Onset Parkinson’s Disease: A Novel Deletion Comprising the DJ-1 and TNFRSF9 Genes. Mov. Disord. 2021, 36, 2973–2976. [Google Scholar] [CrossRef]
- Ma, Y.J.; Berg-von der Emde, K.; Moholt-Siebert, M.; Hill, D.F.; Ojeda, S.R. Region-specific regulation of transforming growth factor alpha (TGF alpha) gene expression in astrocytes of the neuroendocrine brain. J. Neurosci. 1994, 14, 5644–5651. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Gigase, F.A.J.; Smith, E.; Collins, B.; Moore, K.; Snijders, G.; Katz, D.; Bergink, V.; Perez-Rodriquez, M.M.; De Witte, L.D. The association between inflammatory markers in blood and cerebrospinal fluid: A systematic review and meta-analysis. Mol. Psychiatry 2023, 28, 1502–1515. [Google Scholar] [CrossRef]
- Lan, G.; Wang, P.; Chan, R.B.; Liu, Z.; Yu, Z.; Liu, X.; Yang, Y.; Zhang, J. Astrocytic VEGFA: An essential mediator in blood-brain-barrier disruption in Parkinson’s disease. Glia 2022, 70, 337–353. [Google Scholar] [CrossRef]
- Wada, K.; Arai, H.; Takanashi, M.; Fukae, J.; Oizumi, H.; Yasuda, T.; Mizuno, Y.; Mochizuki, H. Expression levels of vascular endothelial growth factor and its receptors in Parkinson’s disease. Neuroreport 2006, 17, 705–709. [Google Scholar] [CrossRef] [PubMed]
- Trares, K.; Bhardwaj, M.; Perna, L.; Stocker, H.; Petrera, A.; Hauck, S.M.; Beyreuther, K.; Brenner, H.; Schottker, B. Association of the inflammation-related proteome with dementia development at older age: Results from a large, prospective, population-based cohort study. Alzheimer’s Res. Ther. 2022, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A.; Carlsson, L.; Gordh, T.; Lind, A.L.; Thulin, M.; Kamali-Moghaddam, M. The effects of age and gender on plasma levels of 63 cytokines. J. Immunol. Methods 2015, 425, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Cerri, S.; Mus, L.; Blandini, F. Parkinson’s Disease in Women and Men: What’s the Difference? J. Park. Dis. 2019, 9, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Espay, A.J.; Schwarzschild, M.A.; Tanner, C.M.; Fernandez, H.H.; Simon, D.K.; Leverenz, J.B.; Merola, A.; Chen-Plotkin, A.; Brundin, P.; Kauffman, M.A.; et al. Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Mov. Disord. 2017, 32, 319–324. [Google Scholar] [CrossRef]
- van Dijk, K.D.; Jongbloed, W.; Heijst, J.A.; Teunissen, C.E.; Groenewegen, H.J.; Berendse, H.W.; van de Berg, W.D.; Veerhuis, R. Cerebrospinal fluid and plasma clusterin levels in Parkinson’s disease. Park. Relat. Disord. 2013, 19, 1079–1083. [Google Scholar] [CrossRef]
- Hughes, A.J.; Daniel, S.E.; Kilford, L.; Lees, A.J. Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J. Neurol. Neurosurg. Psychiatry 1992, 55, 181–184. [Google Scholar] [CrossRef]
- Dalrymple-Alford, J.C.; Livingston, L.; MacAskill, M.R.; Graham, C.; Melzer, T.R.; Porter, R.J.; Watts, R.; Anderson, T.J. Characterizing mild cognitive impairment in Parkinson’s disease. Mov. Disord. 2011, 26, 629–636. [Google Scholar] [CrossRef]
- Litvan, I.; Goldman, J.G.; Troster, A.I.; Schmand, B.A.; Weintraub, D.; Petersen, R.C.; Mollenhauer, B.; Adler, C.H.; Marder, K.; Williams-Gray, C.H.; et al. Diagnostic criteria for mild cognitive impairment in Parkinson’s disease: Movement Disorder Society Task Force guidelines. Mov. Disord. 2012, 27, 349–356. [Google Scholar] [CrossRef]
- Olde Dubbelink, K.T.; Stoffers, D.; Deijen, J.B.; Twisk, J.W.; Stam, C.J.; Berendse, H.W. Cognitive decline in Parkinson’s disease is associated with slowing of resting-state brain activity: A longitudinal study. Neurobiol. Aging 2013, 34, 408–418. [Google Scholar] [CrossRef]
- Wik, L.; Nordberg, N.; Broberg, J.; Bjorkesten, J.; Assarsson, E.; Henriksson, S.; Grundberg, I.; Pettersson, E.; Westerberg, C.; Liljeroth, E.; et al. Proximity Extension Assay in Combination with Next-Generation Sequencing for High-throughput Proteome-wide Analysis. Mol. Cell Proteom. 2021, 20, 100168. [Google Scholar] [CrossRef]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Faghri, F.; Pihlstrom, L.; Geiger, J.T.; Elbaz, A.; Lesage, S.; Corvol, J.C.; May, P.; Nicolas, A.; Abramzon, Y.; et al. NeuroChip, an updated version of the NeuroX genotyping platform to rapidly screen for variants associated with neurological diseases. Neurobiol. Aging 2017, 57, 247.e9–247.e13. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.; Jones, K.; Manley, D. Confounding and collinearity in regression analysis: A cautionary tale and an alternative procedure, illustrated by studies of British voting behaviour. Qual. Quant. 2018, 52, 1957–1976. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PD Patients | Healthy Controls | |
|---|---|---|
| N | 66 | 52 |
| Male nr. (%) | 44 (67%) * | 18 (35%) |
| Age (years) | 62 ± 10 | 63 ± 7 |
| Verhage education score (1/2/3/4/5/6/7) | 0/1/1/7/14/7/10 | n/a |
| Disease duration (years) | 6 ± 5 | n/a |
| H and Y stage (1/1.5/2/2.5/3/4/5) | 5/5/32/17/7/0/0 | n/a |
| UPDRS-III score | 22 ± 8 | n/a |
| MMSE score (range) | 28 ± 2 (23–30) * | 29 ± 1 (25–30) |
| LEDD | 417 ± 453 | n/a |
| Baseline | Follow-up | |
|---|---|---|
| Male nr. (%) | 23 (77%) | n/a |
| Age (years) | 57 ± 9 | 65 ± 9 |
| Verhage education score (1/2/3/4/5/6/7) | 0/1/0/4/11/3/11 | n/a |
| Disease duration (years) | 5 ± 5 | 14 ± 5 |
| H and Y stage (1/1.5/2/2.5/3/4/5) | 4/5/16/4/1/0/0 | 0/0/13/7/4/6/0 ** |
| UPDRS-III score | 20 ± 10 | 26 ± 14 * |
| MMSE score | 29 ± 2 | 29 ± 2 |
| LEDD | 548 ± 481 | 1075 ± 455 |
| Cognitive status (normal/MCI/PDD) | 29/0/1 | 16/5/9 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hepp, D.H.; van Wageningen, T.A.; Kuiper, K.L.; van Dijk, K.D.; Oosterveld, L.P.; Berendse, H.W.; van de Berg, W.D.J. Inflammatory Blood Biomarkers Are Associated with Long-Term Clinical Disease Severity in Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 14915. https://doi.org/10.3390/ijms241914915
Hepp DH, van Wageningen TA, Kuiper KL, van Dijk KD, Oosterveld LP, Berendse HW, van de Berg WDJ. Inflammatory Blood Biomarkers Are Associated with Long-Term Clinical Disease Severity in Parkinson’s Disease. International Journal of Molecular Sciences. 2023; 24(19):14915. https://doi.org/10.3390/ijms241914915
Chicago/Turabian StyleHepp, Dagmar H., Thecla A. van Wageningen, Kirsten L. Kuiper, Karin D. van Dijk, Linda P. Oosterveld, Henk W. Berendse, and Wilma D. J. van de Berg. 2023. "Inflammatory Blood Biomarkers Are Associated with Long-Term Clinical Disease Severity in Parkinson’s Disease" International Journal of Molecular Sciences 24, no. 19: 14915. https://doi.org/10.3390/ijms241914915
APA StyleHepp, D. H., van Wageningen, T. A., Kuiper, K. L., van Dijk, K. D., Oosterveld, L. P., Berendse, H. W., & van de Berg, W. D. J. (2023). Inflammatory Blood Biomarkers Are Associated with Long-Term Clinical Disease Severity in Parkinson’s Disease. International Journal of Molecular Sciences, 24(19), 14915. https://doi.org/10.3390/ijms241914915