
Frontotemporal Dementia P301L Mutation Potentiates but Is Not Sufficient to Cause the Formation of Cytotoxic Fibrils of Tau
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
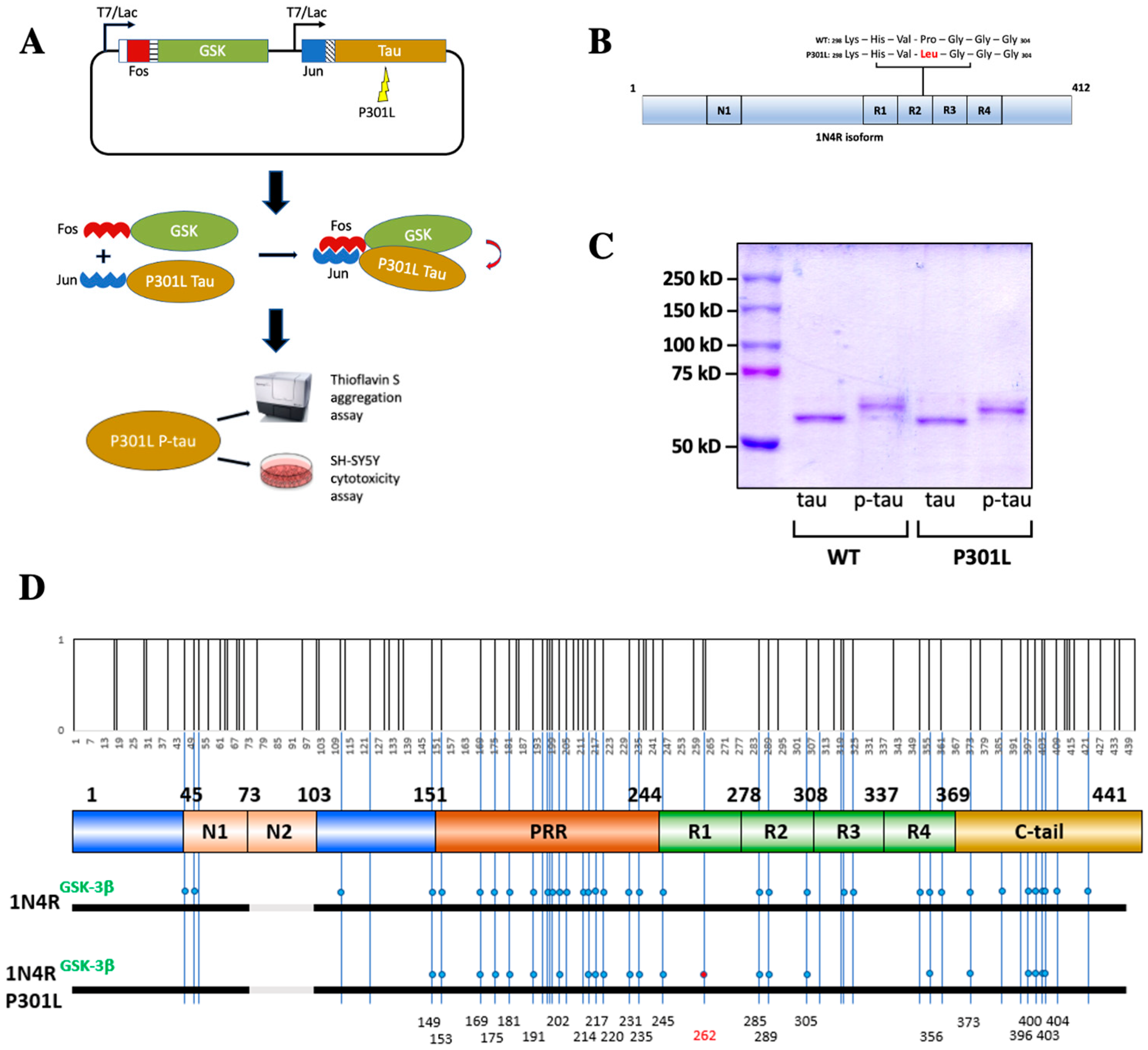
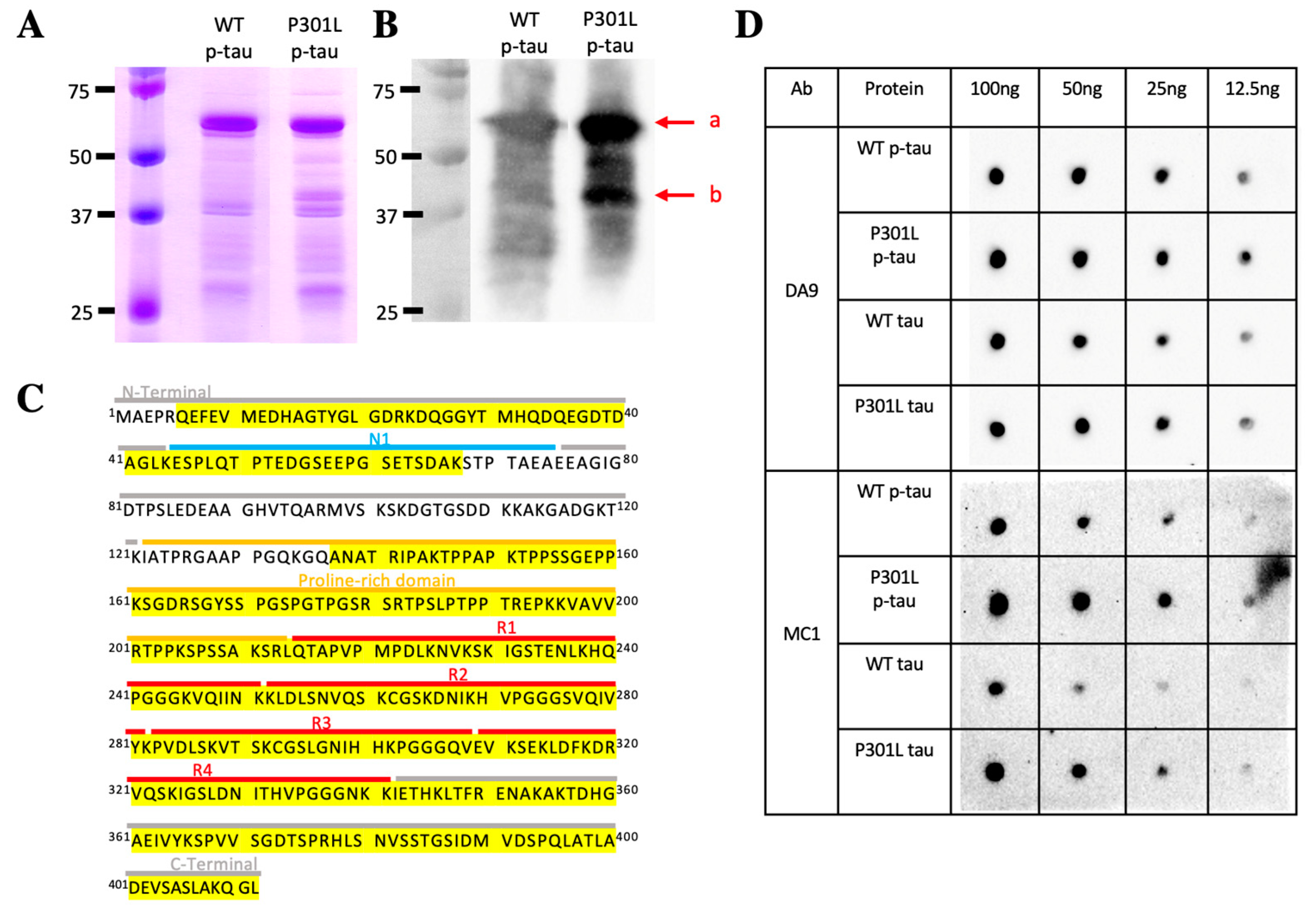
2.1. Production of Hyperphosphorylated Tau Using the PIMAX Approach
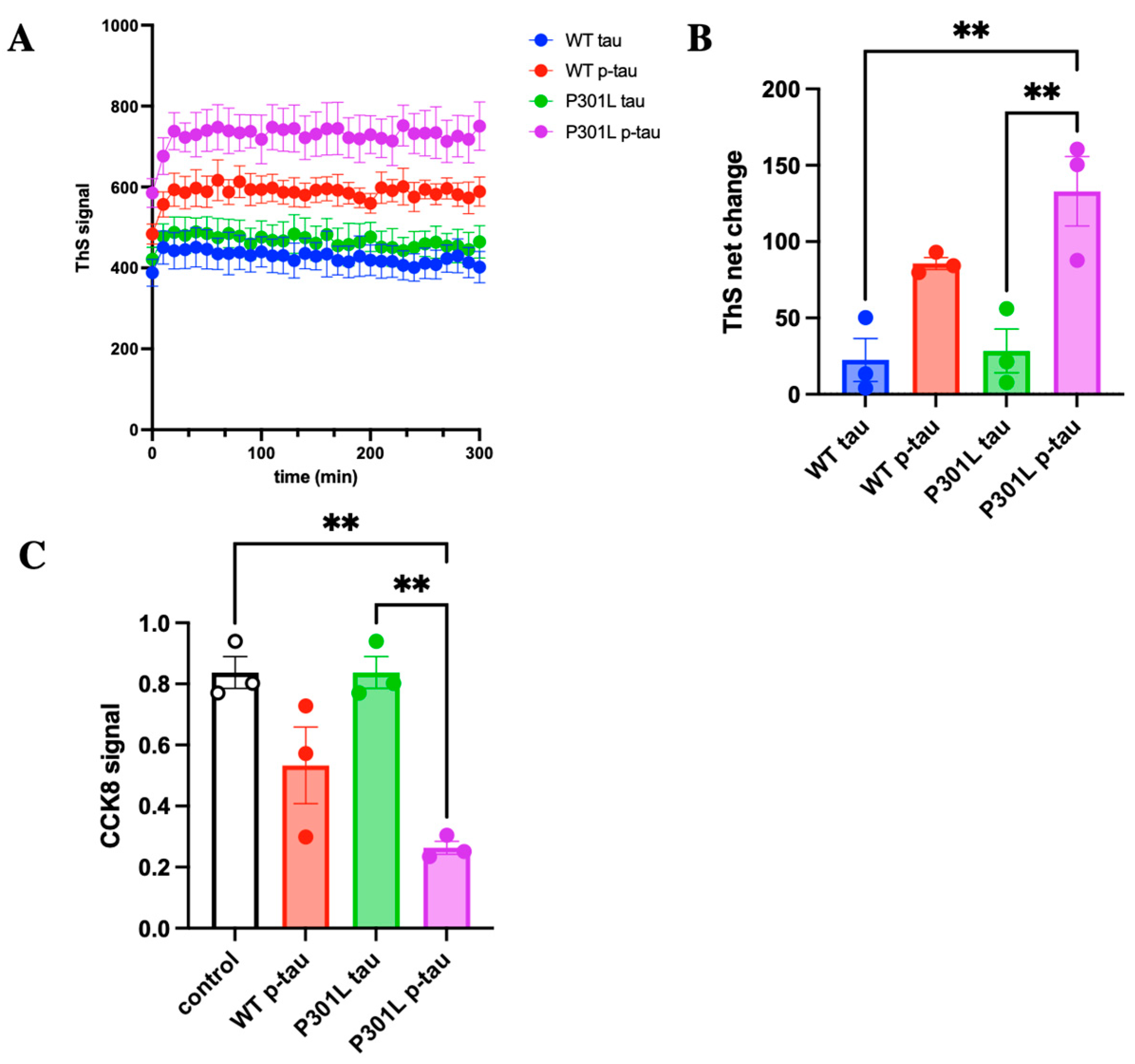
2.2. P301L Mutation Enhances the Propensity to Aggregate and to Damage Cells by Hyperphosphorylated Tau
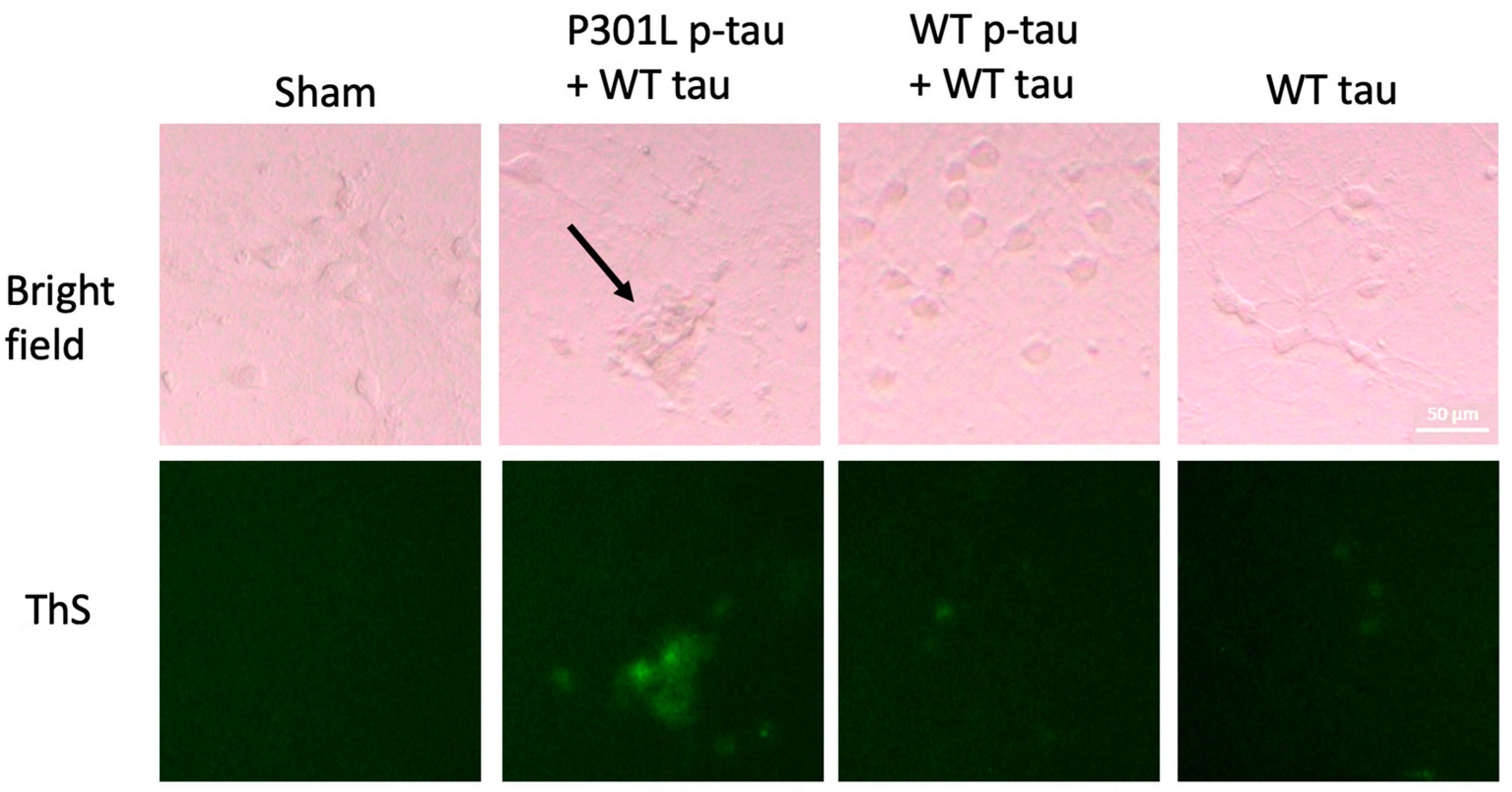
2.3. P301L p-Tau Has Enhanced Seeding Activity That Induces Tau Aggregation in Primary Neurons
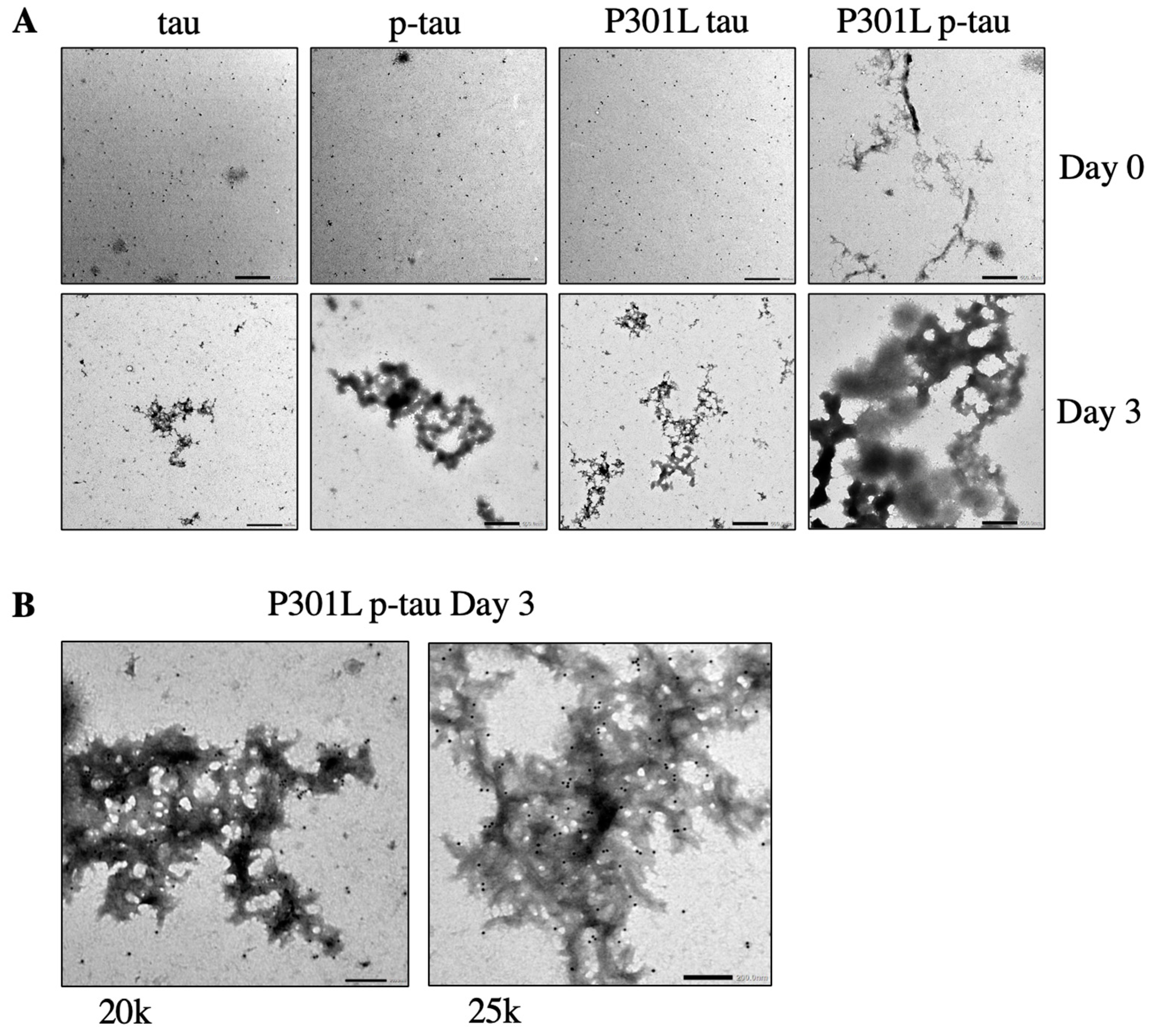
2.4. P301L p-Tau Forms Aggregate Structures More Rapidly Than WT p-Tau and P301L Tau
2.5. P301L p-Tau Adopts a Specific Conformation Sensitive to Bacterial Endopeptidases
2.6. P301L p-Tau Exhibits the Strongest Induction of ER Stress-Associated Pro-Apoptosis
2.7. Apomorphine Antagonizes Both the Aggregation and Cytotoxicity of P301L p-Tau
2.8. P301L p-Tau, but Not P301L Tau, Can Disrupt the Cellular Cytoskeleton
3. Discussion
4. Materials and Methods
4.1. Mass Spectrometry
4.2. Coomassie Blue Staining
4.3. Aggregation Assay
4.4. Cytotoxicity Assay
4.5. Primary Neurons Isolation
4.6. Thioflavin S Staining
4.7. Transmission Electron Microscopy
4.8. Western Blotting
4.9. Dot-Blot Assay
4.10. qPCR
4.11. Cytoskeleton Staining
4.12. Statistical Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ballatore, C.; Lee, V.M.Y.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672. [Google Scholar]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [PubMed]
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189. [Google Scholar] [CrossRef]
- Goedert, M.; Ghetti, B.; Spillantini, M.G. Frontotemporal dementia: Implications for understanding Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006254. [Google Scholar] [CrossRef]
- Kovacs, G.G. Invited review: Neuropathology of tauopathies: Principles and practice. Neuropathol. Appl. Neurobiol. 2015, 41, 3–23. [Google Scholar] [CrossRef]
- Zhang, Y.; Wu, K.-M.; Yang, L.; Dong, Q.; Yu, J.-T. Tauopathies: New perspectives and challenges. Mol. Neurodegener. 2022, 17, 28. [Google Scholar]
- Cacace, R.; Sleegers, K.; Van Broeckhoven, C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016, 12, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Reitz, C.; Pericak-Vance, M.A.; Foroud, T.; Mayeux, R. A global view of the genetic basis of Alzheimer disease. Nat. Rev. Neurol. 2023, 19, 261–277. [Google Scholar]
- Spillantini, M.G.; Goedert, M. Tau pathology and neurodegeneration. Lancet Neurol. 2013, 12, 609–622. [Google Scholar]
- Khatoon, S.; Grundke-Iqbal, I.; Iqbal, K. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett. 1994, 351, 80–84. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nat. Neurosci. 2018, 21, 1341–1349. [Google Scholar] [PubMed]
- Clavaguera, F.; Bolmont, T.; Crowther, R.A.; Abramowski, D.; Frank, S.; Probst, A.; Fraser, G.; Stalder, A.K.; Beibel, M.; Staufenbiel, M.; et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat. Cell Biol. 2009, 11, 909–913. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef]
- Clavaguera, F.; Akatsu, H.; Fraser, G.; Crowther, R.A.; Frank, S.; Hench, J.; Probst, A.; Winkler, D.T.; Reichwald, J.; Staufenbiel, M.; et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc. Natl. Acad. Sci. USA 2013, 110, 9535–9540. [Google Scholar] [CrossRef]
- Iqbal, K.; Liu, F.; Gong, C.-X.; Grundke-Iqbal, I. Tau in Alzheimer disease and related tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Kuchibhotla, K.V.; Wegmann, S.; Kopeikina, K.J.; Hawkes, J.; Rudinskiy, N.; Andermann, M.L.; Spires-Jones, T.L.; Bacskai, B.J.; Hyman, B.T. Neurofibrillary tangle-bearing neurons are functionally integrated in cortical circuits in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Cairns, N.J.; Bigio, E.H.; Mackenzie, I.R.A.; Neumann, M.; Lee, V.M.-Y.; Hatanpaa, K.J.; White, C.L.; Schneider, J.A.; Grinberg, L.T.; Halliday, G.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef]
- Rizzu, P.; Van Swieten, J.C.; Joosse, M.; Hasegawa, M.; Stevens, M.; Tibben, A.; Niermeijer, M.F.; Hillebrand, M.; Ravid, R.; Oostra, B.A.; et al. High prevalence of mutations in the microtubule-associated protein tau in a population study of frontotemporal dementia in the Netherlands. Am. J. Hum. Genet. 1999, 64, 414–421. [Google Scholar] [CrossRef]
- Dayanandan, R.; Van Slegtenhorst, M.; Mack, T.; Ko, L.; Yen, S.-H.; Leroy, K.; Brion, J.-P.; Anderton, B.; Hutton, M.; Lovestone, S. Mutations in tau reduce its microtubule binding properties in intact cells and affect its phosphorylation. FEBS Lett. 1999, 446, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Weissmann, C.; Golovyashkina, N.; Sebö-Lemke, Z.; Drewes, G.; Gerke, V.; Heinisch, J.J.; Brandt, R. The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2-dependent manner. J. Cell Biol. 2011, 192, 647–661. [Google Scholar] [CrossRef]
- Götz, J.; Ittner, L.M. Animal models of Alzheimer’s disease and frontotemporal dementia. Nat. Rev. Neurosci. 2008, 9, 532–544. [Google Scholar] [CrossRef]
- Yu, D.; Feinstein, S.C.; Valentine, M.T. Effects of wild type tau and disease-linked tau mutations on microtubule organization and intracellular trafficking. J. Biomech. 2016, 49, 1280–1285. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Zheng, H. Practical considerations for choosing a mouse model of Alzheimer’s disease. Mol. Neurodegener. 2017, 12, 89. [Google Scholar]
- Ajoolabady, A.; Lindholm, D.; Ren, J.; Pratico, D. ER stress and UPR in Alzheimer’s disease: Mechanisms, pathogenesis, treatments. Cell Death Dis. 2022, 13, 706. [Google Scholar]
- Hetz, C.; Zhang, K.; Kaufman, R.J. Mechanisms, regulation and functions of the unfolded protein response. Nat. Rev. Mol. Cell Biol. 2020, 21, 421–438. [Google Scholar]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [PubMed]
- Zhang, K.; Kaufman, R.J. Identification and characterization of endoplasmic reticulum stress-induced apoptosis in vivo. Methods Enzymol. 2008, 442, 395–419. [Google Scholar] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar]
- Sui, D.; Xu, X.; Ye, X.; Liu, M.; Mianecki, M.; Rattanasinchai, C.; Buehl, C.; Deng, X.; Kuo, M.H. Protein Interaction Module-assisted Function X (PIMAX) Approach to Producing Challenging Proteins Including Hyperphosphorylated Tau and Active CDK5/p25 Kinase Complex. Mol. Cell Proteom. 2015, 14, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sui, D.; Dexheimer, T.; Hovde, S.; Deng, X.; Wang, K.-W.; Lin, H.L.; Chien, H.-T.; Kweon, H.K.; Kuo, N.S.; et al. Hyperphosphorylation Renders Tau Prone to Aggregate and to Cause Cell Death. Mol. Neurobiol. 2020, 57, 4704–4719. [Google Scholar]
- Pooler, A.M.; Polydoro, M.; Wegmann, S.; Nicholls, S.B.; Spires-Jones, T.L.; Hyman, B.T. Propagation of tau pathology in Alzheimer’s disease: Identification of novel therapeutic targets. Alzheimers Res. Ther. 2013, 5, 49. [Google Scholar] [CrossRef]
- Dumanchin, C.; Camuzat, A.; Campion, D.; Verpillat, P.; Hannequin, D.; Dubois, B.; Saugier-Veber, P.; Martin, C.; Penet, C.; Charbonnier, F.; et al. Segregation of a missense mutation in the microtubule-associated protein tau gene with familial frontotemporal dementia and parkinsonism. Hum. Mol. Genet. 1998, 7, 1825–1829. [Google Scholar] [CrossRef] [PubMed]
- Steinhilb, M.L.; Dias-Santagata, D.; Fulga, T.A.; Felch, D.L.; Feany, M.B. Tau Phosphorylation Sites Work in Concert to Promote Neurotoxicity In Vivo. Mol. Biol. Cell 2007, 18, 5060–5068. [Google Scholar] [CrossRef]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar]
- Shafiei, S.S.; Guerrero-Muñoz, M.J.; Castillo-Carranza, D.L. Tau Oligomers: Cytotoxicity, Propagation, and Mitochondrial Damage. Front. Aging Neurosci. 2017, 9, 83. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Prokop, S.; Giasson, B.I. “Don’t Phos Over Tau”: Recent developments in clinical biomarkers and therapies targeting tau phosphorylation in Alzheimer’s disease and other tauopathies. Mol. Neurodegener. 2021, 16, 37. [Google Scholar]
- Sui, D.; Liu, M.; Kuo, M.H. In vitro aggregation assays using hyperphosphorylated tau protein. J. Vis. Exp. 2015, 95, e51537. [Google Scholar]
- Köpke, E.; Tung, Y.; Shaikh, S.; Alonso, A.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J. Biol. Chem. 2011, 286, 15317–15331. [Google Scholar] [PubMed]
- Huseby, C.J.; Kuret, J. Analyzing Tau Aggregation with Electron Microscopy. Methods Mol. Biol. 2016, 1345, 101–112. [Google Scholar]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Commins, C.; Lathuiliere, A.; Beerepoot, P.; Fernandes, A.R.; Kamath, T.V.; De Los Santos, M.B.; Klickstein, N.; Corjuc, D.L.; Corjuc, B.T.; et al. Tau molecular diversity contributes to clinical heterogeneity in Alzheimer’s disease. Nat. Med. 2020, 26, 1256–1263. [Google Scholar] [CrossRef]
- Petry, F.R.; Pelletier, J.; Bretteville, A.; Morin, F.; Calon, F.; Hébert, S.S.; Whittington, R.A.; Planel, E. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: Problems and solutions. PLoS ONE 2014, 9, e94251. [Google Scholar]
- Jicha, G.A.; Bowser, R.; Kazam, I.G.; Davies, P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J. Neurosci. Res. 1997, 48, 128–132. [Google Scholar] [CrossRef]
- Weaver, C.L.; Espinoza, M.; Kress, Y.; Davies, P. Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol. Aging 2000, 21, 719–727. [Google Scholar] [CrossRef]
- Kaufman, R.J. Stress signaling from the lumen of the endoplasmic reticulum: Coordination of gene transcriptional and translational controls. Genes. Dev. 1999, 13, 1211–1233. [Google Scholar] [CrossRef]
- Carbone, F.; Djamshidian, A.; Seppi, K.; Poewe, W. Apomorphine for Parkinson’s Disease: Efficacy and Safety of Current and New Formulations. CNS Drugs 2019, 33, 905–918. [Google Scholar] [PubMed]
- Liu, M.; Dexheimer, T.; Sui, D.; Hovde, S.; Deng, X.; Kwok, R.; Bochar, D.A.; Kuo, M.H. Hyperphosphorylated tau aggregation and cytotoxicity modulators screen identified prescription drugs linked to Alzheimer’s disease and cognitive functions. Sci. Rep. 2020, 10, 16551. [Google Scholar]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: A review of the literature. J. Neuropathol. Exp. Neurol. 2012, 71, 362–381. [Google Scholar]
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082. [Google Scholar]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef]
- Grossman, M.; Seeley, W.W.; Boxer, A.L.; Hillis, A.E.; Knopman, D.S.; Ljubenov, P.A.; Miller, B.; Piguet, O.; Rademakers, R.; Whitwell, J.L.; et al. Frontotemporal lobar degeneration. Nat. Rev. Dis. Primers 2023, 9, 40. [Google Scholar] [PubMed]
- Wenger, K.; Viode, A.; Schlaffner, C.N.; van Zalm, P.; Cheng, L.; Dellovade, T.; Langlois, X.; Bannon, A.; Chang, R.; Connors, T.R.; et al. Common mouse models of tauopathy reflect early but not late human disease. Mol. Neurodegener. 2023, 18, 10. [Google Scholar] [CrossRef]
- Adamec, E.; Murrell, J.R.; Takao, M.; Hobbs, W.; Nixon, R.A.; Ghetti, B.; Vonsattel, J.P. P301L tauopathy: Confocal immunofluorescence study of perinuclear aggregation of the mutated protein. J. Neurol. Sci. 2002, 200, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Rösler, T.W.; Carlsson, T.; de Andrade, A.; Bruch, J.; Höllerhage, M.; Oertel, W.H.; Höglinger, G.U. Memory deficits correlate with tau and spine pathology in P301S MAPT transgenic mice. Neuropathol. Appl. Neurobiol. 2014, 40, 833–843. [Google Scholar] [CrossRef]
- Wen, J.; Hong, L.; Krainer, G.; Yao, Q.-Q.; Knowles, T.P.J.; Wu, S.; Perrett, S. Conformational Expansion of Tau in Condensates Promotes Irreversible Aggregation. J. Am. Chem. Soc. 2021, 143, 13056–13064. [Google Scholar] [CrossRef]
- Lu, P.J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Fiol, C.J.; Mahrenholz, A.M.; Wang, Y.; Roeske, R.W.; Roach, P.J. Formation of protein kinase recognition sites by covalent modification of the substrate. Molecular mechanism for the synergistic action of casein kinase II and glycogen synthase kinase 3. J. Biol. Chem. 1987, 262, 14042–14048. [Google Scholar] [CrossRef] [PubMed]
- Kaidanovich-Beilin, O.; Woodgett, J.R. GSK-3: Functional Insights from Cell Biology and Animal Models. Front. Mol. Neurosci. 2011, 4, 40. [Google Scholar] [CrossRef]
- Song, L.; Oseid, D.E.; Wells, E.A.; Robinson, A.S. The Interplay between GSK3β and Tau Ser262 Phosphorylation during the Progression of Tau Pathology. Int. J. Mol. Sci. 2022, 23, 11610. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, N.; Ohyagi, Y.; Imamura, T.; Yanagihara, Y.T.; Iinuma, K.M.; Soejima, N.; Murai, H.; Yamasaki, R.; Kira, J.-I. Apomorphine Therapy for Neuronal Insulin Resistance in a Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 1151–1161. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, K.-W.; Zhang, G.; Kuo, M.-H. Frontotemporal Dementia P301L Mutation Potentiates but Is Not Sufficient to Cause the Formation of Cytotoxic Fibrils of Tau. Int. J. Mol. Sci. 2023, 24, 14996. https://doi.org/10.3390/ijms241914996
Wang K-W, Zhang G, Kuo M-H. Frontotemporal Dementia P301L Mutation Potentiates but Is Not Sufficient to Cause the Formation of Cytotoxic Fibrils of Tau. International Journal of Molecular Sciences. 2023; 24(19):14996. https://doi.org/10.3390/ijms241914996
Chicago/Turabian StyleWang, Kuang-Wei, Gary Zhang, and Min-Hao Kuo. 2023. "Frontotemporal Dementia P301L Mutation Potentiates but Is Not Sufficient to Cause the Formation of Cytotoxic Fibrils of Tau" International Journal of Molecular Sciences 24, no. 19: 14996. https://doi.org/10.3390/ijms241914996