
Blood-Based Transcriptomic Biomarkers Are Predictive of Neurodegeneration Rather Than Alzheimer’s Disease
 , , , ,
, , , ,  , ,
, ,  , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Characteristics of the Included Datasets
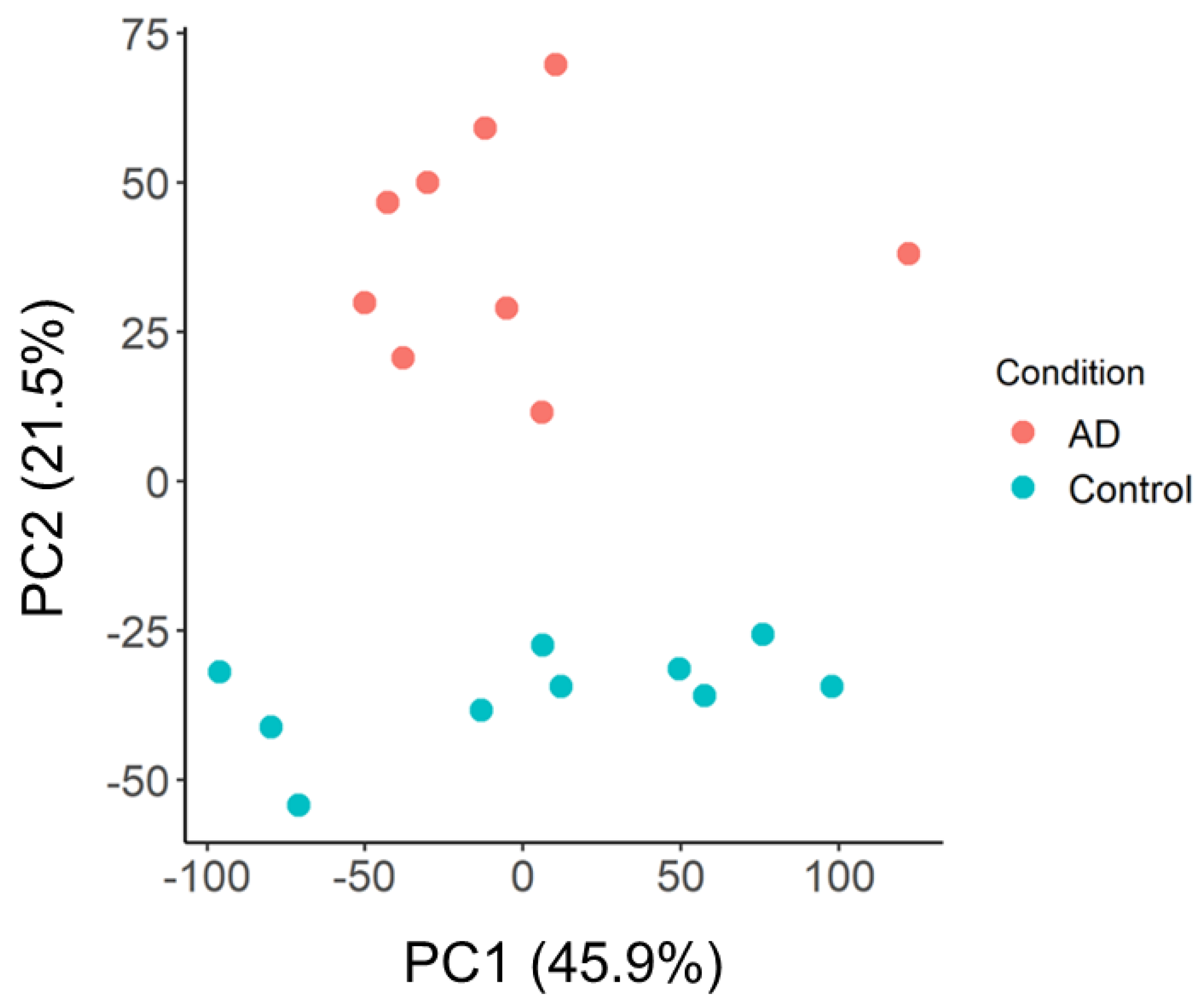
2.2. Principal Component and Functional Network Analyses of GSE97760 Indicate Dysregulated Pathways and Central Gene Nodes in Whole Blood of AD Patients
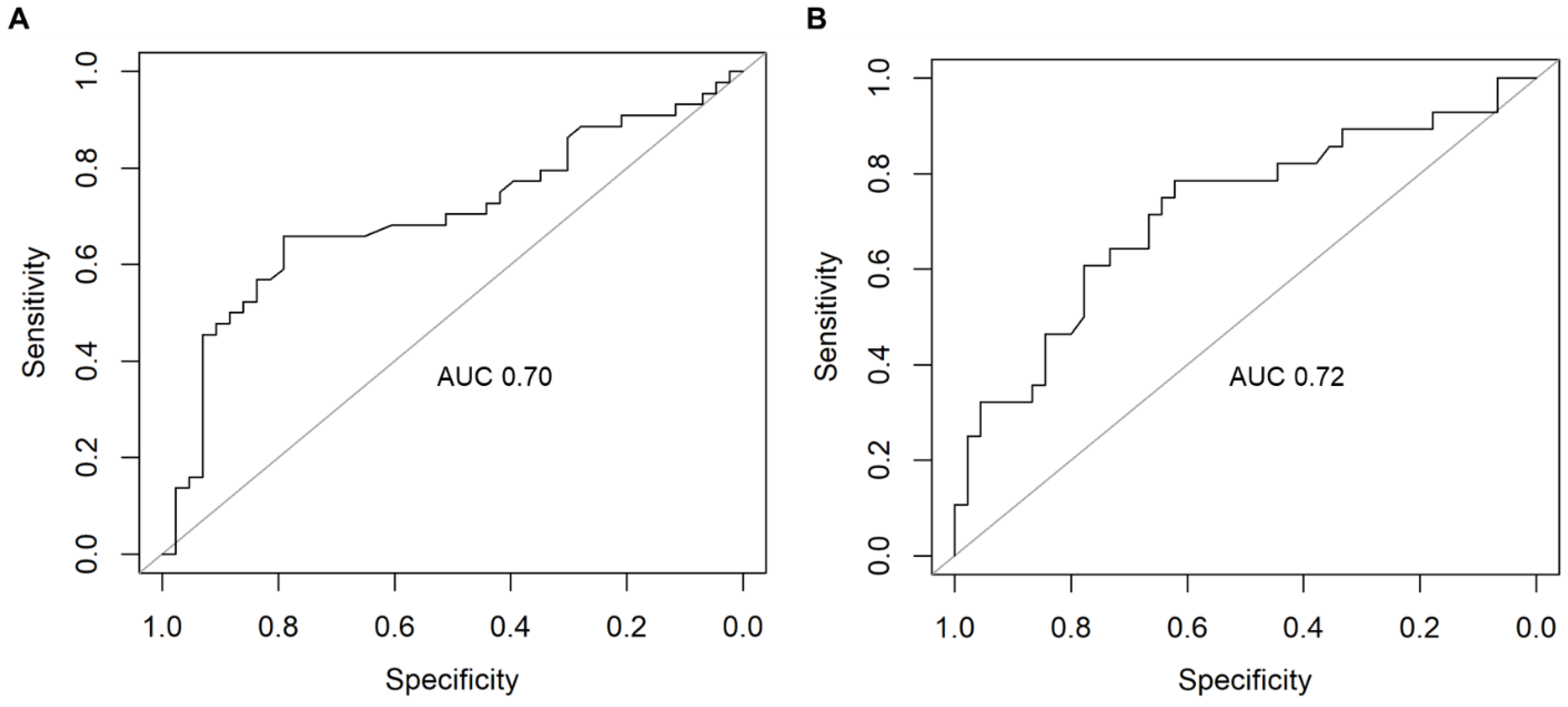
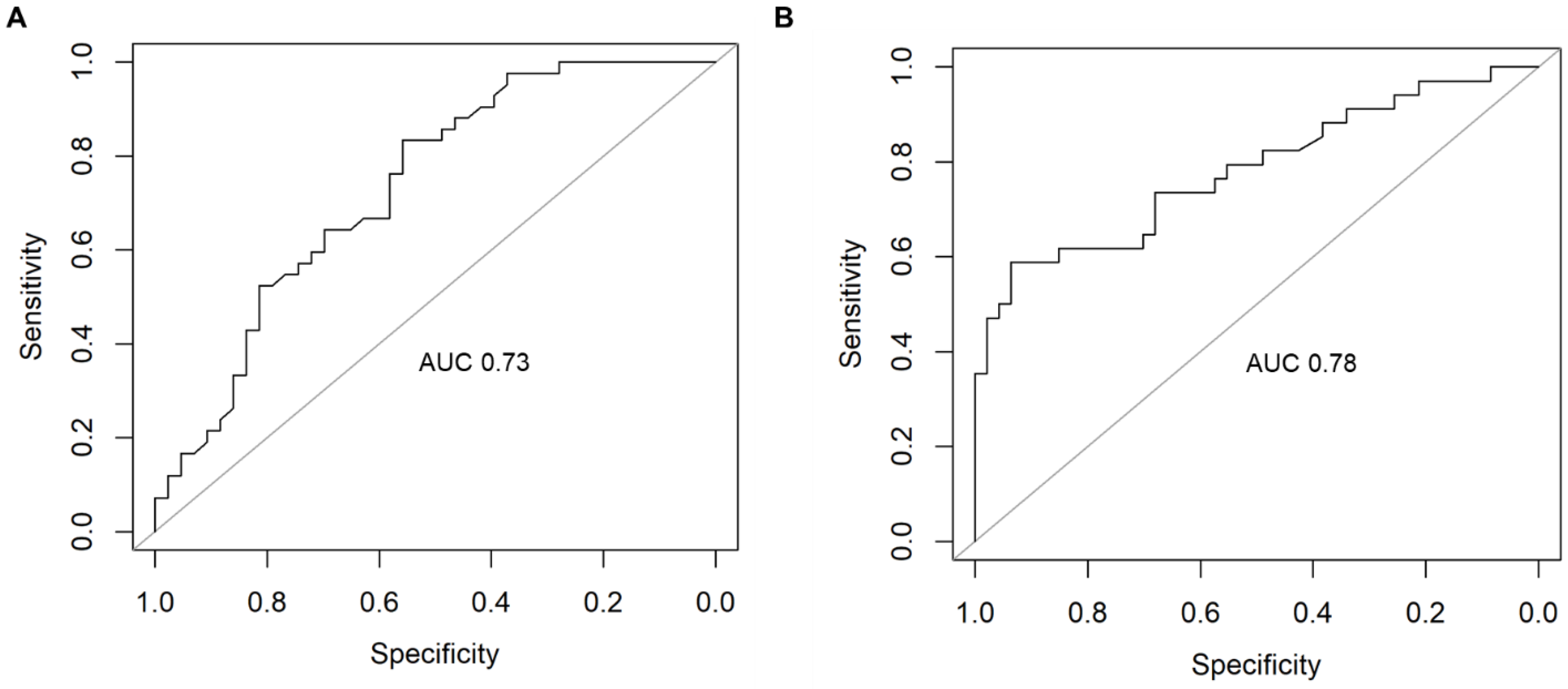
2.3. Supervised Machine Learning Identifies Dysregulated Pathways That Can Predict AD
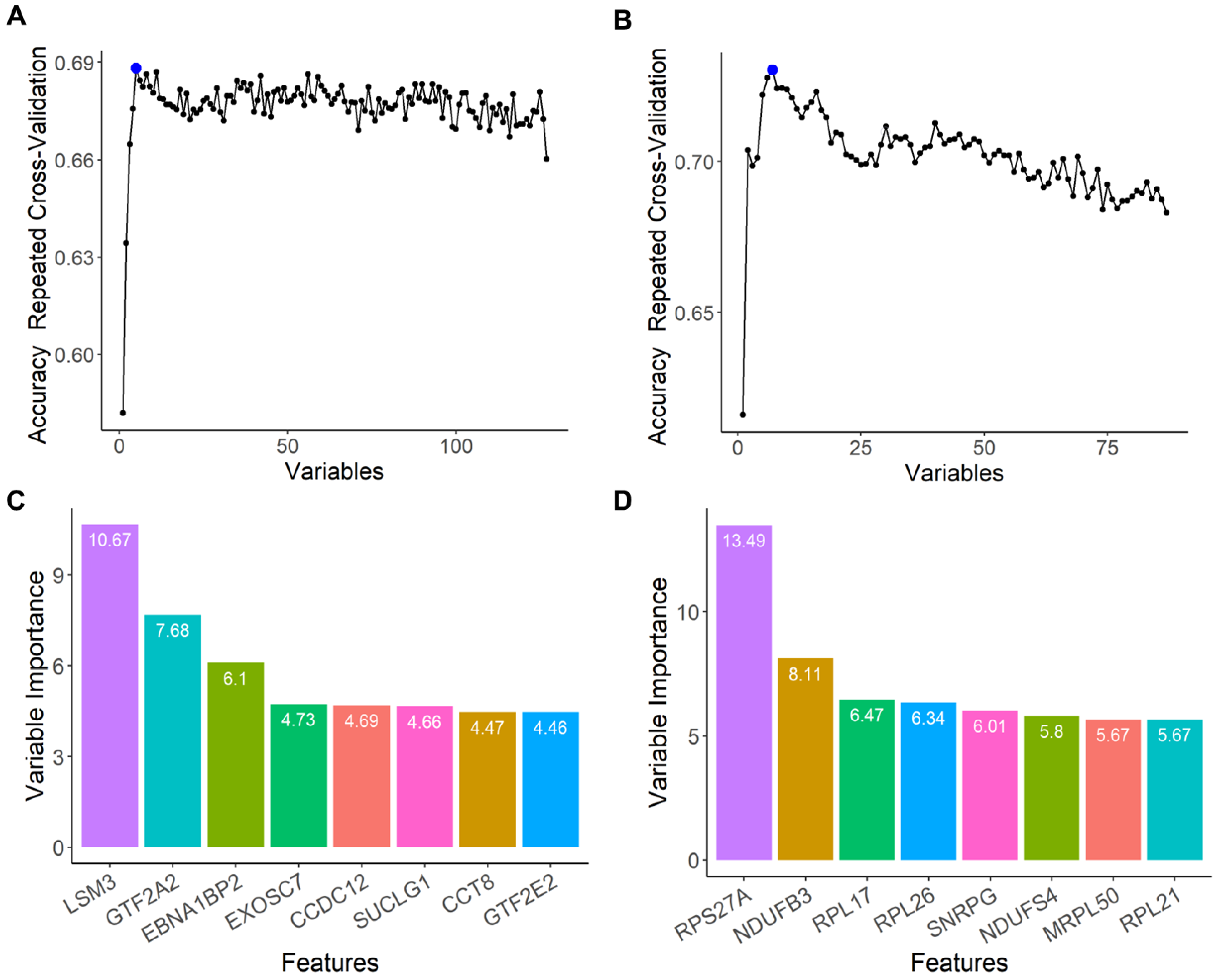
2.4. Feature Selection of the Top-Performing Genes That Contribute to AD Prediction within Each PC Cluster
2.5. Genes That Predict AD Are Also Predictive of Neurodegenerative Diseases
3. Discussion
4. Materials and Methods
4.1. Identification of Publicly Available Transcriptomic Datasets
4.2. Data Processing
4.3. Feature Selection Using Unsupervised Machine Learning Methods and Functional Enrichment Analyses on GSE97760
4.4. Supervised Machine Learning to Identify the Best Predictors of AD
4.5. Feature Selection to Identify the Genes That Contribute to AD Prediction
4.6. Supervised Machine Learning to Identify if AD Gene Biomarkers Are Unique to AD or Generalizable to Other Neurodegenerative Diseases
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Dementia; World Health Organization: Geneva, Switzerland, 2020. [Google Scholar]
- Hansson, O.; Edelmayer, R.M.; Boxer, A.L.; Carrillo, M.C.; Mielke, M.M.; Rabinovici, G.D.; Salloway, S.; Sperling, R.; Zetterberg, H.; Teunissen, C.E. The Alzheimer’s Association appropriate use recommendations for blood biomarkers in Alzheimer’s disease. Alzheimer’s Dement. 2022, 18, 2669–2686. [Google Scholar] [CrossRef] [PubMed]
- Cummings, J.L. The National Institute on Aging—Alzheimer’s Association framework on Alzheimer’s diasease: Application to clinical trials. Alzheimer’s Dement. 2019, 15, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Vernerk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Karikari, T.; Pascoal, T.A.; Ashton, N.J.; Janelidze, S.; Benedet, A.L.; Rodriguez, J.L.; Chamoun, M.; Savard, M.; Kang, M.S.; Therriault, J.; et al. Blood phosphorylated tau 181 as a biomarker for Alzheimer’s disease: A diagnostic performance and prediction modelling study using data from four prospective cohorts. Lancet Neurol. 2020, 19, 422–433. [Google Scholar] [CrossRef] [PubMed]
- Japkowicz, N.; Shaju, S. The class imbalance problem: A systematic study. Intell. Data Anal. 2002, 6, 429–449. [Google Scholar] [CrossRef]
- Guo, X.; Yin, Y.; Dong, C.; Yang, G.; Zhou, G. On the class imbalance problem. In Proceedings of the IEEE Fourth International Conference on Natural Computation, Jinan, China, 18–20 October 2008; Volume 4. [Google Scholar]
- Finney, C.A.; Delerue, F.; Gold, W.A.; Brown, D.A.; Shvetcov, A. Artificial intelligence-driven meta-analysis of brain gene expression identifies novel gene candidates and a role for mitochondria in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2023, 21, 388–400. [Google Scholar] [CrossRef]
- Yao, F.; Zhang, K.; Zhang, Y.; Guo, Y.; Li, A.; Xiao, S.; Liu, Q.; Shen, L.; Ni, J. Identification of blood biomarkers for Alzheimer’s diseae through computational prediction and experimental validation. Front. Neurol. 2019, 9, 1158. [Google Scholar] [CrossRef]
- Yu, H.; Liu, Y.; He, B.; He, T.; Chen, C.; He, J.; Yang, X.; Wang, J.-Z. Platelet biomarkers for a descending cognitive function: A proteomic approach. Aging Cell 2021, 20, e13358. [Google Scholar] [CrossRef]
- Salman, A.; Lapidot, I.; Shufan, E.; Agbaria, A.H.; Katz, B.-S.P.; Mordechai, S. Potential of infrared microscopy to differentiate between dementia with Lewy bodies and Alzheimer’s diseases using peripheral blood samples and machine learning algorithms. J. Biomed. Opt. 2020, 25, 1–15. [Google Scholar] [CrossRef]
- Ludwig, N.; Fehlmann, T.; Kern, F.; Gogol, M.; Maetzler, W.; Deutscher, S.; Gurlit, S.; Schulte, C.; von Thaler, A.-K.; Deuschle, C.; et al. Machine learning to detect Alzheimer’s disease from circulating non-coding RNAs. Genom. Proteom. Bioinform. 2019, 17, 430–444. [Google Scholar] [CrossRef]
- Chiricosta, L.; D’Angiolini, S.; Gugliandolo, A.; Mazzon, E. Artificial intelligence predictor for Alzheimer’s disease trained on blood transcriptome: The role of oxidative stress. Int. J. Mol. Sci. 2022, 23, 5237. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhan, D.; Wang, L. Ribosomal proteins are blood biomarkers and associated with CD4+ T cell activation in Alzheimer’s disease: A study based on machine learning strategies and scRNA-Seq data validation. Am. J. Transl. Res. 2023, 15, 2498–2514. [Google Scholar] [PubMed]
- Huseby, C.J.; Delvaux, E.; Brokaw, D.L.; Coleman, P.D. Blood RNA transcripts reveal similar and differential alterations in fundamental cellular processes in Alzheimer’s disease and other neurodegenerative diseases. Alzheimer’s Dement. 2022, 19, 2618–2632. [Google Scholar] [CrossRef] [PubMed]
- Raudys, S.J.; Jain, A.K. Small sample size effects in statistical pattern recognition: Recommendations for practitioners. IEEE Trans. Pattern Anal. Mach. Intell. 1991, 13, 252–264. [Google Scholar] [CrossRef]
- Kanal, L.; Chandrasekaran, B. On dimensionality and sample size in statistical pattern classification. Pattern Recognit. 1971, 3, 225–234. [Google Scholar] [CrossRef]
- Cawley, G.C.; Talbot, N.L.C. On over-fitting in model selection and subsequent selection bias in performance evaluation. J. Mach. Learn. Res. 2010, 11, 2079–2107. [Google Scholar]
- Huseby, C.J.; Delvaux, E.; Brokaw, D.L.; Coleman, P.D. Blood transcript biomarkers selected by machine learning algorithm classify neurodegenerative diseases including Alzheimer’s disease. Biomolecules 2022, 12, 1592. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, R.A.; Lantos, P.L.; Cairns, N.J. Overlap between neurodegenerative disorders. Neuropathology 2005, 25, 111–124. [Google Scholar] [CrossRef]
- Gan, L.; Cookson, M.R.; Petrucelli, L.; La Spada, A.R. Converging pathways in neurodegneration, from genetics to mechanisms. Nat. Neurosci. 2018, 21, 1300–1309. [Google Scholar] [CrossRef]
- Arneson, D.; Zhang, Y.; Yang, X.; Narayanan, M. Shared mechanisms among neurodegenerative diseases: From genetic factors to gene networks. J. Genet. 2018, 97, 795–806. [Google Scholar] [CrossRef]
- Luo, H.; Han, G.; Wang, J.; Zeng, F.; Li, Y.; Shao, S.; Song, F.; Bai, Z.; Peng, X.; Wang, Y.-J.; et al. Common aging signature in the peripheral blood of vascular dementia and Alzheimer’s disease. Mol. Neurobiol. 2015, 53, 3596–3605. [Google Scholar] [CrossRef] [PubMed]
- Nabais, M.F.; Laws, S.M.; Lin, T.; Vallerga, C.L.; Armstrong, N.J.; Blair, I.P.; Kwok, J.B.; Mather, K.A.; Mellick, G.D.; Sachdev, P.S.; et al. Meta-analysis of genome-wide DNA methylation identifies shared associations across neurodegenerative disorders. Genome Biol. 2021, 22, 90. [Google Scholar] [CrossRef] [PubMed]
- Naughton, B.J.; Duncan, F.J.; Murrey, D.A.; Meadows, A.S.; Newsom, D.E.; Stiocea, N.; White, P.; Scharre, D.W.; Mccarty, D.M.; Fu, H. Blood genome-wide transcriptional profiles reflect broad molecular impairments and strong blood-brain links in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 43, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Annika, G.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N.J.; Hye, A.; Rajkumar, A.P.; Leuzy, A.; Snowden, S.; Suarez-Calvet, M.; Karikari, T.; Scholl, M.; La Joie, R.; Rabinovici, G.D.; et al. An update on blood-based biomarkers for non-Alzheimer neurodegenerative disorders. Nat. Rev. Neurol. 2020, 16, 265–284. [Google Scholar] [CrossRef] [PubMed]
- Day, G.S.; Rappai, T.; Sathyan, S.; Morris, J.C. Deciphering the factors that influence participation in studies requiring serial lumbar punctures. Alzheimer’s Dement. 2020, 12, e12003. [Google Scholar] [CrossRef]
- Yang, S.; Rothman, R.E. PCR-based applications for infectious diseases: Uses, limitations, and future applications in acute-care settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Calligaris, R.; Banica, M.; Roncaglia, P.; Robotti, E.; Finaurini, S.; Vlachouli, C.; Antonutti, L.; Iorio, F.; Carissimo, A.; Cattaruzza, T.; et al. Blood transcriptomics of drug-naive sporadic Parkinson’s disease patients. BMC Genom. 2015, 16, 876. [Google Scholar] [CrossRef]
- Tao, Y.; Han, Y.; Yu, L.; Wang, Q.; Leng, S.X.; Zhang, H. The predicted key molecules, functions, and pathways that bridge mild cognitive impairment (MCI) and Alzheimer’s disease (AD). Front. Neurol. 2020, 11, 233. [Google Scholar] [CrossRef]
- Gui, H.; Gong, Q.; Jiang, J.; Liu, M.; Li, H. Identification of the hub genes in Alzheimer’s disease. Comput. Math. Methods Med. 2021, 2021, 6329041. [Google Scholar] [CrossRef]
- Khayer, N.; Mirzaie, M.; Marashi, S.-A.; Jalessi, M. Rps27a might act as a controller of microglia activation in triggering neurodegenerative diseases. PLoS ONE 2020, 15, e0239219. [Google Scholar] [CrossRef] [PubMed]
- Kavanagh, T.; Halder, A.; Drummond, E. Tau interactome and RNA binding proteins in neurodegenerative diseases. Mol. Neurodegener. 2022, 17, 66. [Google Scholar] [CrossRef] [PubMed]
- Mirzaei, M.; Gupta, V.B.; Chick, J.M.; Greco, T.M.; Wu, Y.; Chitranshi, N.; Wall, R.V.; Hone, E.; Deng, L.; Dheer, Y.; et al. Age-related neurodegenerative disease associated pathways identified in retinal and vitreous proteome from human glaucoma eyes. Sci. Rep. 2017, 7, 12685. [Google Scholar] [CrossRef] [PubMed]
- Askenazi, M.; Kavanagh, T.; Pires, G.; Ueberheide, B.; Wisniewski, T.; Drummond, E. Compilation of reported protein changes in the brain in Alzheimer’s disease. Nat. Commun. 2023, 14, 4466. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Lei, P.; Zhang, S.; Li, D.; Zhao, J.; Zhang, B. Integrated microarray analysis provided a new insight of the pathogenesis of Parkinson’s disease. Neurosci. Lett. 2017, 662, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Haify, S.N.; Botta-Orfila, T.; Hukema, R.K.; Tartaglia, G.G. In silico, in vitro, and in vivo approaches to identify molecular players in Fragile X Tremor and Ataxia Syndrome. Front. Mol. Biosci. 2020, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Chinopoulos, C.; Batzios, S.; van den Heuvel, L.P.; Rodenburg, R.; Smeets, R.; Waterham, H.R.; Turkenburg, M.; Ruiter, J.P.; Wanders, R.J.A.; Doczi, J.; et al. Mutated SUCLG1 causes mislocalization of SUCLG2 protein, morphological alterations of mitochondria and an early-onset severe neurometabolic disorder. Mol. Genet. Metab. 2019, 126, 43–52. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Scaglia, F. Mitochondrial DNA depletion syndromes: Review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics 2013, 10, 186–198. [Google Scholar] [CrossRef]
- Swerdlow, R.H. The neurodegenerative mitochondriopathies. J. Alzheimer’s Dis. 2009, 17, 737–751. [Google Scholar] [CrossRef]
- Wilkins, H.M.; Weidling, I.W.; Ji, Y.; Swerdlow, R.H. Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front. Immunol. 2017, 8, 508. [Google Scholar] [CrossRef]
- Aguilar, K.; Comes, G.; Canal, C.; Quintana, A.; Sanz, E.; Hidalgo, J. Microglial response promotes neurodegeneration in the ndufs4 KO mouse model of Leigh syndrome. Glia 2022, 70, 2032–2044. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, M.; Jain, I.H.; Goldberger, O.; Rezoagli, R.; Cheng, K.-H.; Sosnovik, D.E.; Scherrer-Crosbie, M.; Mootha, V.K.; Zapol, W.M. Hypoxia treatment reverses neurodegenerative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, E4241–E4250. [Google Scholar] [CrossRef]
- Hedden, T.; Gabrieli, J.D.E. Insights into the ageing mind: A view from cognitive neuroscience. Nat. Rev. Neurosci. 2004, 5, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Bianco, A.; Antonacci, Y.; Liguori, M. Sex and gender differences in neurodegenerative diseases: Challenges for therapeutic opportunities. Int. J. Mol. Sci. 2023, 24, 6354. [Google Scholar] [CrossRef]
- Vila-Castelar, C.; Chen, Y.; Langella, S.; Lopera, F.; Zetterberg, H.; Hansson, O.; Dage, J.L.; Janelidze, S.; Su, Y.; Chen, K.; et al. Sex differences in blood biomarkers and cognitive performance in individuals with autosomal dominant Alzheimer’s disease. Alzheimer’s Dement. 2023, 19, 4127–4138. [Google Scholar] [CrossRef] [PubMed]
- Kerkela, E.; Lahtela, J.; Larjo, A.; Impola, U.; Maenpaa, L.; Mattila, P. Exploring transcriptomic landscapes in red blood cells, in their extracellular vesicles and on a single-cell level. Int. J. Mol. Sci. 2022, 23, 12897. [Google Scholar] [CrossRef] [PubMed]
- Doss, J.; Corcoran, D.L.; Jima, D.D.; Telen, M.J.; Dave, S.S.; Chi, J.-T. A comprehensive joint analysis of the long and short RNA transcriptomes of human erythrocytes. BMC Genom. 2015, 16, 952. [Google Scholar] [CrossRef]
- Rowley, J.W.; Schwertz, H.; Weyrich, A.S. Platelet mRNA: The meaning behind the message. Curr. Opin. Hematol. 2012, 19, 385–391. [Google Scholar] [CrossRef]
- Mohr, S.; Liew, C.-C. The peripheral-blood transcriptome: New insights into disease and risk assessment. Trends Mol. Med. 2007, 13, 422–432. [Google Scholar] [CrossRef]
- Karaaslan, Z.; Kahraman, O.T.; Sanli, E.; Ergen, H.A.; Ulusoy, C.; Bilgic, B.; Yilmaz, V.; Tuzun, E.; Hanagasi, H.A.; Kucukali, C.I. Inflammation and regulatory T cell genes are differentially expressed in peripheral blood mononuclear cells of Parkinson’s disease patients. Sci. Rep. 2021, 11, 2316. [Google Scholar] [CrossRef]
- Phongpreecha, T.; Fernandez, R.; Mrdjen, D.; Culos, A.; Gajera, C.R.; Wawro, A.M.; Stanley, N.; Gaudilliere, B.; Poston, K.L.; Aghaeepour, N.; et al. Single-cell peripheral immunoprofiling of Alzheimer’s and Parkinson’s disease. Sci. Adv. 2020, 6, eabd5575. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Zaldivar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalan, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular vesicles through the blood-brain barrier: A review. Fluids Barriers CNS 2022, 19, 60. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, L.; Mao, Z.; Wang, Z.; Zhang, Z.; Li, M. Bidirectional communication between the brain and other organs: The role of extracellular vesicles. Cell. Mol. Neurobiol. 2023, 43, 2675–2696. [Google Scholar] [CrossRef] [PubMed]
- Jolliffe, I.T.; Cadima, J. Principal component analysis: A review and recent developments. Philos. Trans. R. Soc. A 2016, 374, 20150202. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.Y.; Ruzzo, W.L. Principal component analysis for clustering gene expression data. Bioinformatics 2001, 17, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed]
- The Gene Ontology Consortium. The Gene Ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Fushiki, T. Estimation of prediction error by using K-fold cross-validation. Stat. Comput. 2011, 21, 137–146. [Google Scholar]
- Trevethan, R. Sensitivity, specificity, and predictive values: Foundations, pliabilities, and pitfalls in research and practice. Front. Public Health 2017, 5, 307. [Google Scholar] [CrossRef]
- Chen, Q.; Meng, Z.; Liu, X.; Jin, Q.; Su, R. Decision variants for the automatic detection of optimal feature subset in RF-RFE. Genes 2018, 9, 301. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GSE97760 | GSE63061 | GSE63060 | ||||
|---|---|---|---|---|---|---|
| AD | Control | AD | Control | AD | Control | |
| Sample size | 9 | 10 | 139 | 134 | 145 | 104 |
| Sex (M:F) | 0:9 | 0:10 | 54:85 | 53:81 | 46:99 | 42:62 |
| Age (average ± SEM) | 79.3 ± 4.1 | 72.1 ± 4.1 | 77.9 ± 0.6 | 75.3 ± 0.5 | 75.4 ± 0.6 | 72.4 ± 0.6 |
| AD diagnostic criteria | NIH diagnostic guidelines for AD | NINCDS-ADRDA and DSM-IV | NINCDS-ADRDA and DSM-IV | |||
| Platform | GPL16699 | GPL10558 | GPL6947 | |||
| k-Means Cluster | Biological Function (GO) | FDR | Cellular Component (GO) | FDR | Number of Central Gene Nodes | Roles of Central Gene Nodes |
|---|---|---|---|---|---|---|
| 1 (Red) | Cellular localization | 3.61 × 10−11 | Endomembrane system | 6.02 × 10−22 | 3 | Vesicle formation; SNARE complex; signal transduction |
| 2 (Yellow) | Metabolic process | 5.29 × 10−25 | Mitochondrion Ribonucleoprotein complex | 2.87 × 10−40 5.93 × 10−12 | 62 | ATP synthase (complex V); mitochondrial ribosomes; mitochondrial respiration |
| 3 (Green) | Gene expression | 3.87 × 10−28 | Nucleus | 1.75 × 10−44 | 39 | RNA regulation; transcription |
| 4 (Blue) | Cellular response to stimulus Protein folding | 1.69 × 10−9 1.19 × 10−16 | Cytosol | 2.8 × 10−9 | 23 | Protein folding, maintenance, stabilization, and degradation |
| k-Means Cluster | Biological Function (GO) | FDR | Cellular Component (GO) | FDR | Number of Central Gene Nodes | Roles of Central Gene Nodes |
|---|---|---|---|---|---|---|
| 1 (Red) | Gene expression | 2.32 × 10−9 | Nucleus Ribonucleoprotein complex | 1.83 × 10−21 0.00024 | 16 | mRNA regulation; mitochondrial ribosomes |
| 2 (Yellow) | Metabolic process | 2.58 × 10−22 | Cytosol Mitochondrion | 1.78 × 10−16 4.40 × 10−6 | 41 | Cytochrome c oxidase (complex IV); mitochondrial ribosomes; NADH dehydrogenase (complex I); ribosomes |
| 3 (Green) | Transport | 4.58 × 10−6 | Cytoplasm | 4.19 × 10−10 | 18 | Vesicular transport; protein trafficking; haemoglobin |
| 4 (Blue) | Regulation of cellular processes | 2.49 × 10−10 | Plasma membrane | 0.00062 | 12 | Protein kinase signalling; estrogen signalling; transcription; protein chaperone |
| AUC | Sensitivity | Specificity | Precision | |||||
|---|---|---|---|---|---|---|---|---|
| Cluster | A | B | A | B | A | B | A | B |
| Cellular localization | 0.53 | 0.54 | 0.48 | 0.64 | 0.39 | 0.26 | 0.31 | 0.67 |
| Metabolic process | 0.55 | 0.79 | 0.54 | 0.72 | 0.46 | 0.81 | 0.51 | 0.89 |
| Gene expression * | 0.70 | 0.72 | 0.65 | 0.69 | 0.66 | 0.61 | 0.65 | 0.88 |
| Cellular response to stimulus/protein folding | 0.64 | 0.74 | 0.58 | 0.68 | 0.59 | 0.69 | 0.55 | 0.89 |
| AUC | Sensitivity | Specificity | Precision | |||||
|---|---|---|---|---|---|---|---|---|
| Cluster | A | B | A | B | A | B | A | B |
| Gene expression | 0.68 | 0.64 | 0.73 | 0.65 | 0.53 | 0.58 | 0.56 | 0.78 |
| Metabolic process * | 0.73 | 0.78 | 0.65 | 0.75 | 0.67 | 0.72 | 0.7 | 0.83 |
| Transport | 0.58 | 0.43 | 0.55 | 0.60 | 0.48 | 0.55 | 0.55 | 0.87 |
| Regulation of cellular process | 0.57 | 0.58 | 0.54 | 0.67 | 0.42 | 0.41 | 0.40 | 0.64 |
| Abbreviation | Name | Function (NCBI) |
|---|---|---|
| PC1 Gene Expression Cluster | ||
| LSM3 | U6 snRNA and mRNA Degradation Associated | Component of the U4/U6-U5 tri-snRNP complex involved in pre-mRNA splicing and spliceosome assembly |
| GFT2A2 | General transcription factor IIA subunit 2 | Component of RNA polymerase II transcription machinery, role in transcriptional activation |
| EBNA1BP2 | EBNA1 binding protein 2 | RNA binding activity |
| EXOSC7 | Exosome component 7 | Enables RNA binding, 3′-5′-exoribonuclease activity, and RNA metabolism |
| CCDC12 | Coiled-coil domain containing 12 | Component of U2-type and post-mRNA release spliceosomal complexes |
| SUCLG1 | Succinate-CoA ligase GDP/ADP-forming subunit alpha | Alpha subunit of the heterodimeric enzyme succinate coenzyme A ligase, catalyses conversion of succinyl CoA and ADP to succinate and ATP or of GDP to GTP |
| CCT8 | Chaperone containing TCP1 subunit 8 | Component of molecular chaperonin-containing T-complex (TRiC), assists in folding of proteins upon ATP hydrolysis |
| GTF2E2 | General transcription factor IIE subunit 2 | Component of RNA polymerase II transcription initiation complex, involved in recruitment of general transcription factor IIH to initiation complex and stimulation of RNA polymerase II C-terminal domain kinase and DNA-dependent ATPase activity |
| PC2 Metabolic Process Cluster | ||
| RPS27A | Ribosomal protein 27A | Component of 40S subunit of the ribosome involved in protein synthesis |
| NDUFB3 | NADH:ubiquinone oxidoreductase subunit B3 | Component of accessory subunit of mitochondrial membrane respiratory chain NADH dehydrogenase (complex I) |
| RPL17 | Ribosomal protein L17 | Component of the ribosomal 60S subunit, involved in protein synthesis |
| RPL26 | Ribosomal protein L26 | Component of the ribosomal 60S subunit, involved in protein synthesis |
| SNRPG | Small nuclear ribonucleoprotein polypeptide G | Component of the U1, U2, U4, and U5 small nuclear ribonucleoprotein complexes, involved in processing of 3′ end of histone transcripts |
| NDUFS4 | NADH:ubiquinone oxidoreductase subunit S4 | Component of nuclear-encoded accessory subunit of mitochondrial membrane respiratory chain NADH dehydrogenase (complex I) |
| MRPL50 | Mitochondrial ribosomal protein L50 | Encodes a putative 39S subunit protein of mitochondrial ribosomes (mitoribosomes) |
| RPL21 | Ribosomal protein L21 | Component of the ribosomal 60S subunit, involved in protein synthesis |
| GSE6613 | GSE72267 | GSE112681 | ||||
|---|---|---|---|---|---|---|
| PD | Control | PD | Control | ALS | Control | |
| Sample size | 50 | 22 | 40 | 20 | 167 | 137 |
| Sex (M:F) | 39:11 | 11:11 | 22:18 | 10:10 | 96:68 | 79:58 |
| Age (average ± SEM) | 69.4 ± 1.2 | 64.4 ± 2.3 | 68.8 ± 1.1 | 60.3 ± 1.3 | Not reported | Not reported |
| Diagnostic criteria | United Kingdom Parkinson’s Disease Society Brain Bank clinical diagnostic criteria | United Kingdom Parkinson’s Disease Society Brain Bank clinical diagnostic criteria | Revised El Escorial criteria | |||
| RNA quality (RIN average +/− SEM) | Not specified | RIN ≥ 8 | RIN ≥ 7 | |||
| Dataset | AUC | Sensitivity | Specificity | Precision |
|---|---|---|---|---|
| Parkinson’s disease (GSE6613) | 0.48 | 0.69 | 0.30 | 0.69 |
| Parkinson’s disease, drug naïve (GSE72267) | 0.54 | 0.72 | 0.39 | 0.65 |
| Amyotrophic lateral sclerosis (GSE112681) | 0.50 | 0.55 | 0.47 | 0.73 |
| Dataset | AUC | Sensitivity | Specificity | Precision |
|---|---|---|---|---|
| Parkinson’s disease (GSE6613) | 0.52 | 0.68 | 0.27 | 0.68 |
| Parkinson’s disease, drug naïve (GSE72267) | 0.61 | 0.71 | 0.45 | 0.83 |
| Amyotrophic lateral sclerosis (GSE112681) | 0.52 | 0.63 | 0.15 | 0.72 |
| Dataset | AUC | Sensitivity | Specificity | Precision |
|---|---|---|---|---|
| Parkinson’s disease (GSE6613) | 0.44 | 0.64 | 0.5 | 0.78 |
| Parkinson’s disease, drug naïve (GSE72267) | 0.50 | 0.67 | N/A | 1.00 |
| Amyotrophic lateral sclerosis (GSE112681) | 0.89 | 0.91 | 0.71 | 0.82 |
| Dataset | AUC | Sensitivity | Specificity | Precision |
|---|---|---|---|---|
| Parkinson’s disease (GSE6613) | 0.86 | 0.86 | 0.67 | 0.80 |
| Parkinson’s disease, drug naïve (GSE72267) | 1.00 | 1.00 | 1.00 | 1.00 |
| Amyotrophic lateral sclerosis (GSE112681) | 0.89 | 0.91 | 0.74 | 0.84 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shvetcov, A.; Thomson, S.; Spathos, J.; Cho, A.-N.; Wilkins, H.M.; Andrews, S.J.; Delerue, F.; Couttas, T.A.; Issar, J.K.; Isik, F.; et al. Blood-Based Transcriptomic Biomarkers Are Predictive of Neurodegeneration Rather Than Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 15011. https://doi.org/10.3390/ijms241915011
Shvetcov A, Thomson S, Spathos J, Cho A-N, Wilkins HM, Andrews SJ, Delerue F, Couttas TA, Issar JK, Isik F, et al. Blood-Based Transcriptomic Biomarkers Are Predictive of Neurodegeneration Rather Than Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(19):15011. https://doi.org/10.3390/ijms241915011
Chicago/Turabian StyleShvetcov, Artur, Shannon Thomson, Jessica Spathos, Ann-Na Cho, Heather M. Wilkins, Shea J. Andrews, Fabien Delerue, Timothy A. Couttas, Jasmeen Kaur Issar, Finula Isik, and et al. 2023. "Blood-Based Transcriptomic Biomarkers Are Predictive of Neurodegeneration Rather Than Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 19: 15011. https://doi.org/10.3390/ijms241915011
APA StyleShvetcov, A., Thomson, S., Spathos, J., Cho, A.-N., Wilkins, H. M., Andrews, S. J., Delerue, F., Couttas, T. A., Issar, J. K., Isik, F., Kaur, S., Drummond, E., Dobson-Stone, C., Duffy, S. L., Rogers, N. M., Catchpoole, D., Gold, W. A., Swerdlow, R. H., Brown, D. A., & Finney, C. A. (2023). Blood-Based Transcriptomic Biomarkers Are Predictive of Neurodegeneration Rather Than Alzheimer’s Disease. International Journal of Molecular Sciences, 24(19), 15011. https://doi.org/10.3390/ijms241915011