
Successful Treatment of Large B-Cell Lymphoma in a Child with Compound Heterozygous Mutation in the ATM Gene

 , , , and
, , , and
Abstract
:1. Introduction
2. Case Presentation
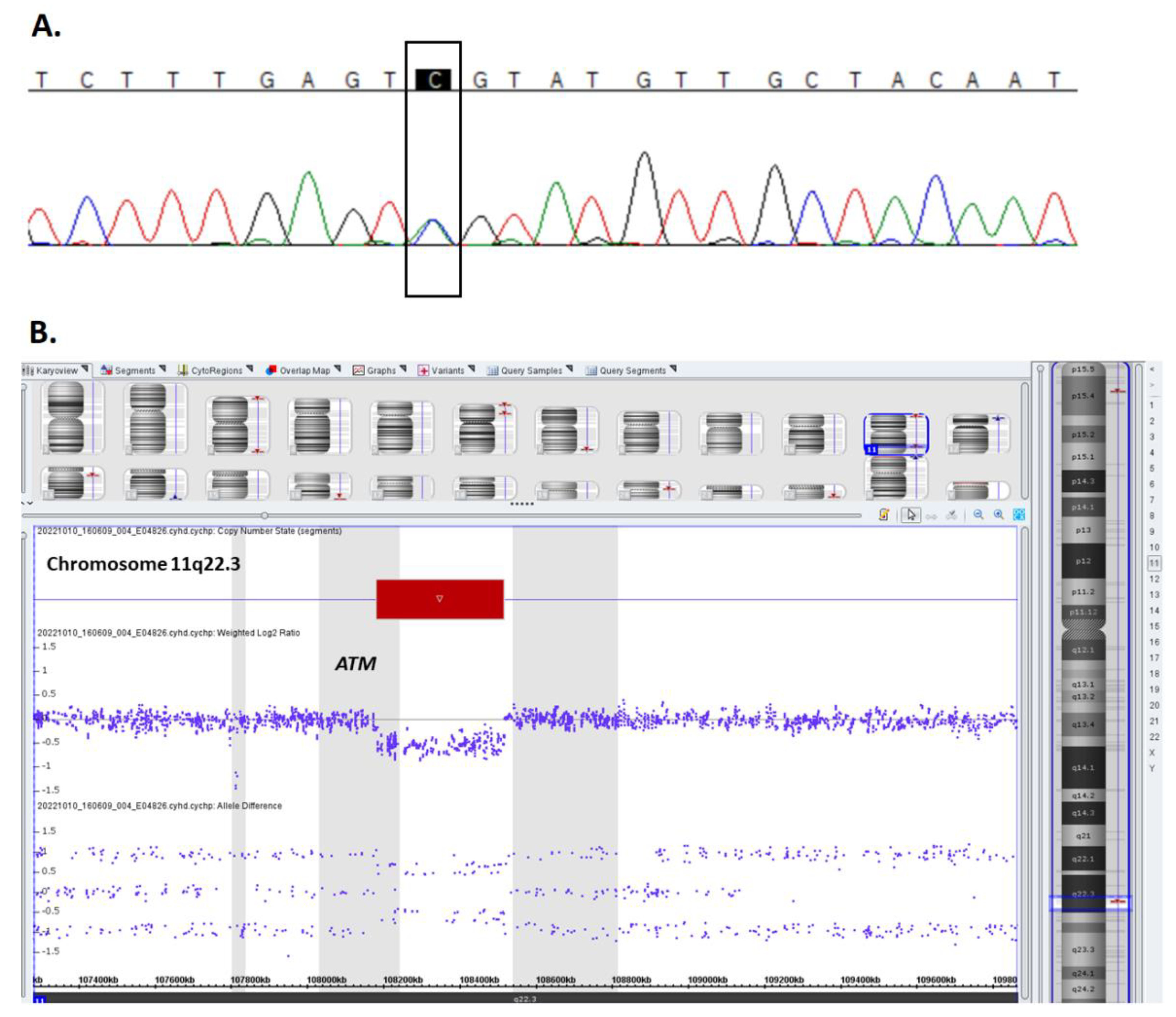
2.1. Ataxia-Telangiectasia Diagnosis and Treatment
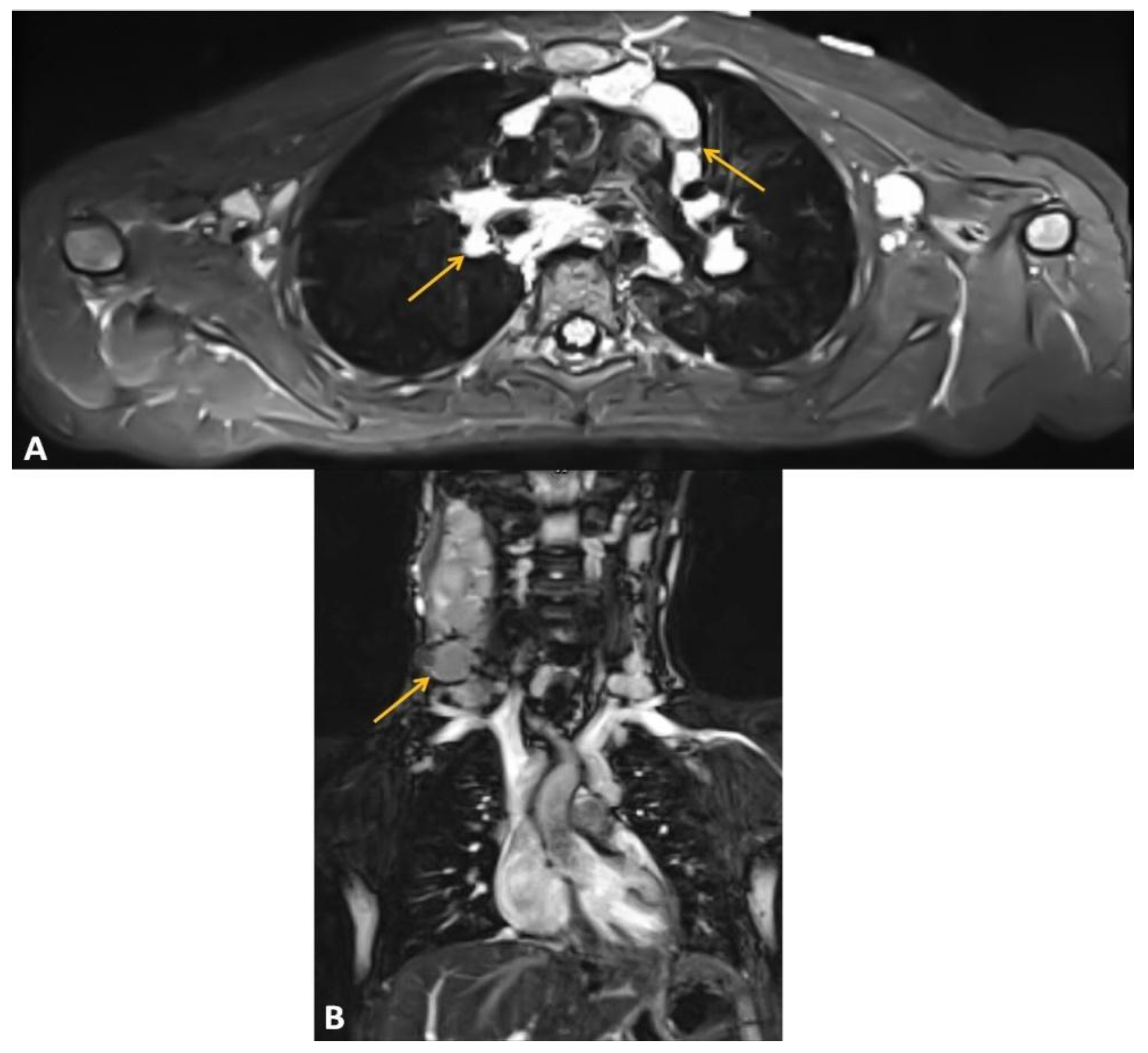
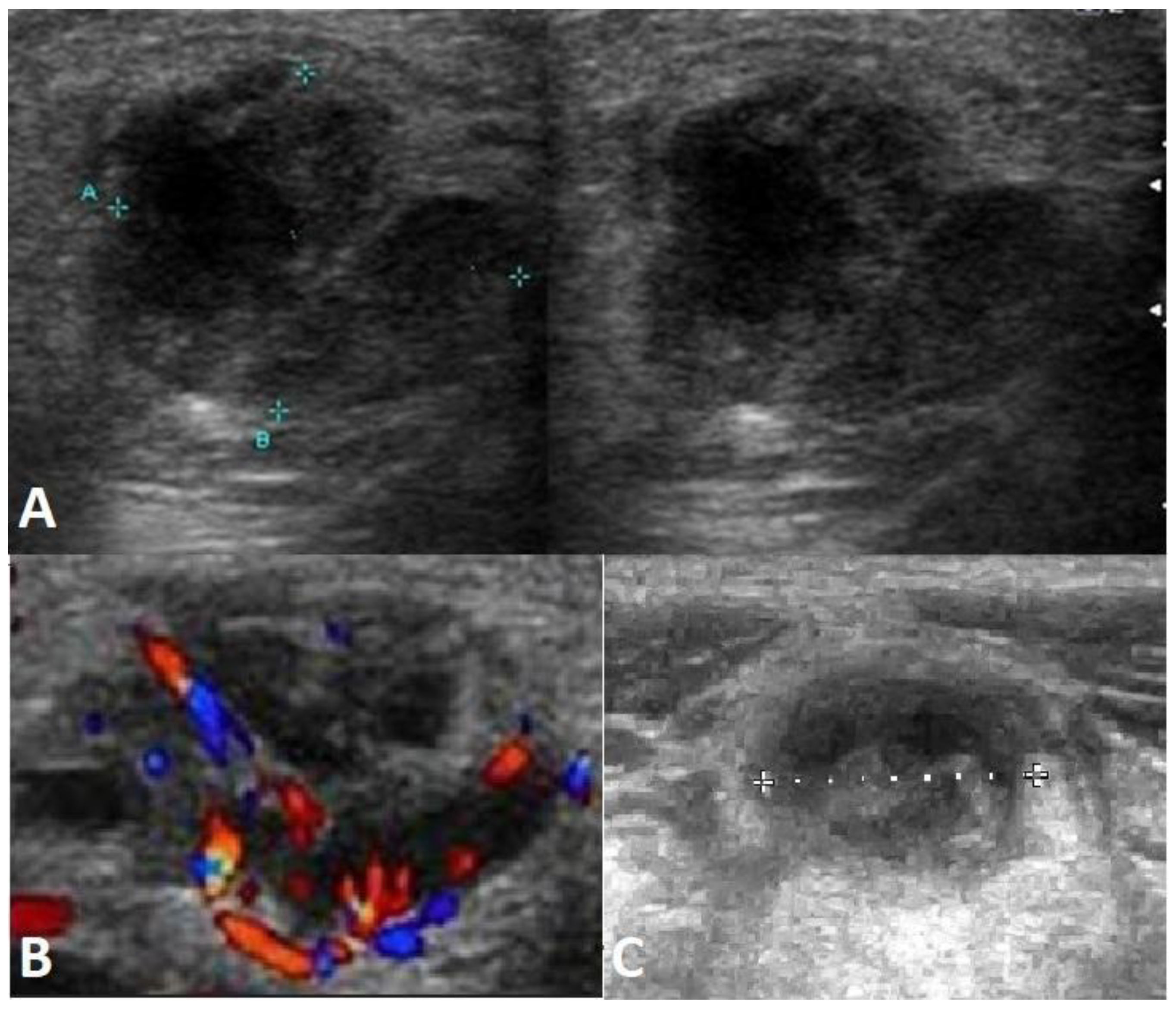
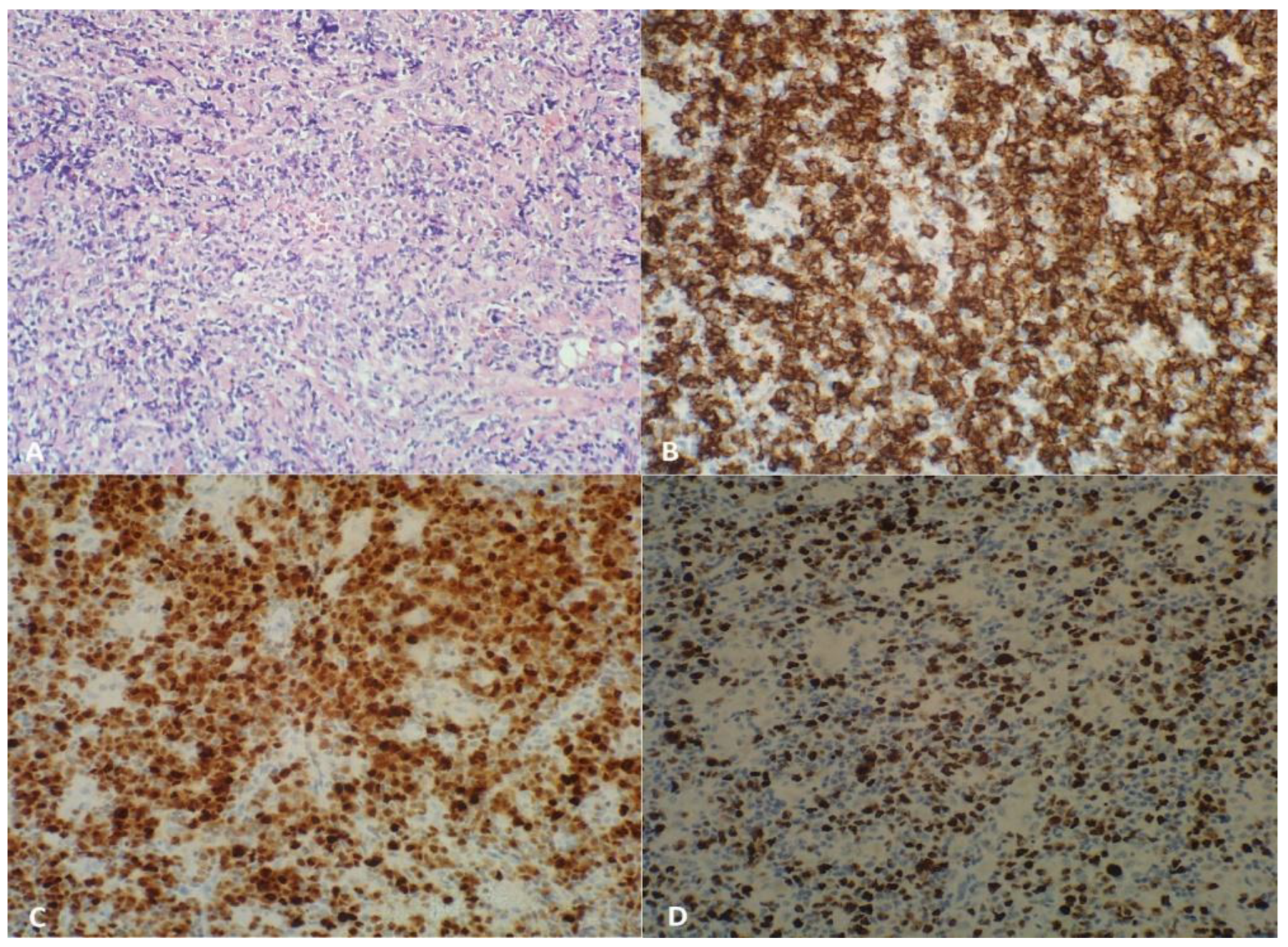
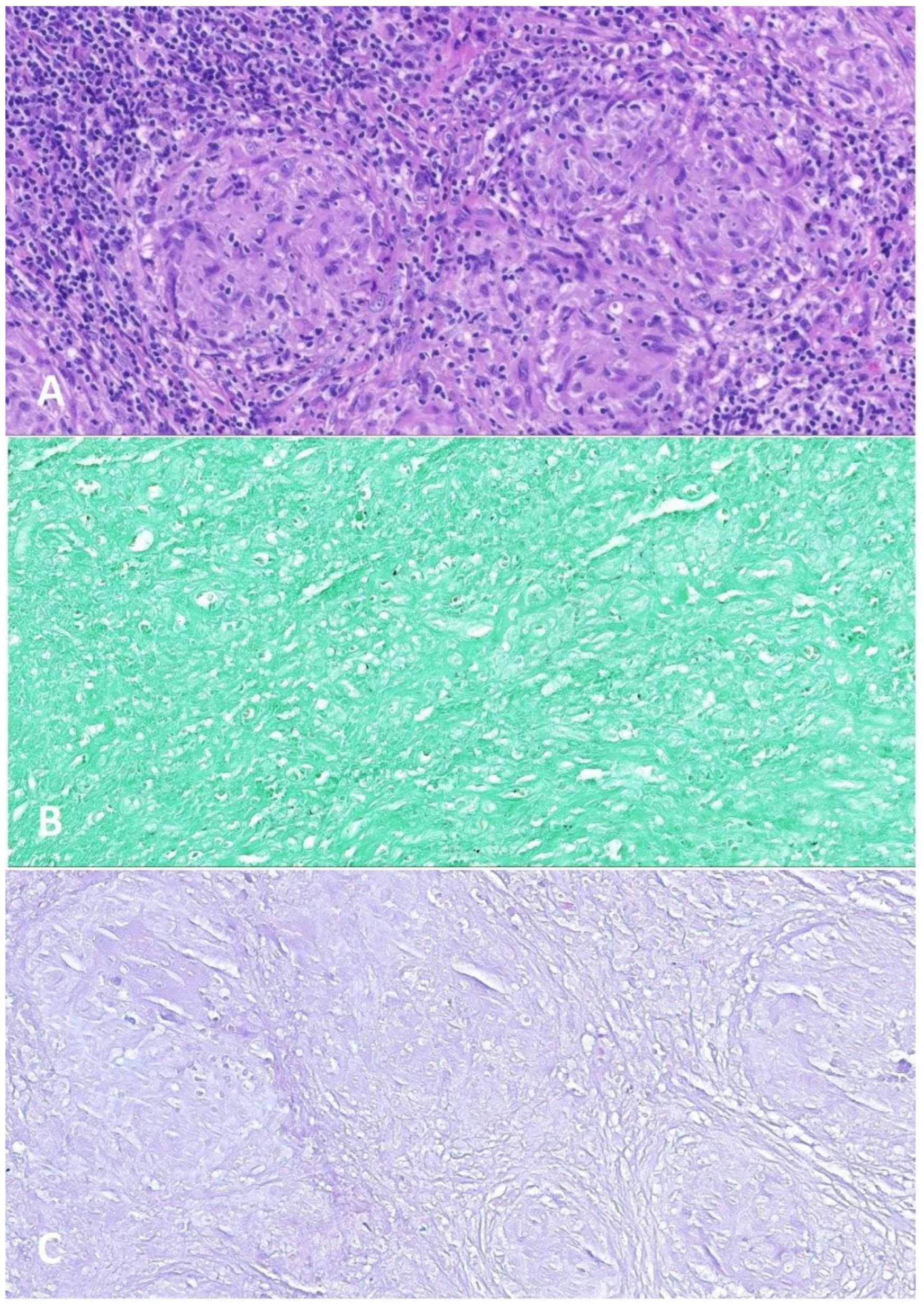
2.2. Large B-Cell Lymphoma Diagnosis
2.3. Large B-Cell Lymphoma Treatment
3. Discussion
3.1. Large B-Cell Lymphoma Biology
3.2. Large B-Cell Lymphoma Treatment in the Presence of Ataxia-Telangiectasia
3.3. Follow-Up of Large B-Cell Lymphoma Treatment in the Light of the Presented Case
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rothblum-Oviatt, C.; Wright, J.; Lefton-Greif, M.A.; McGrath-Morrow, S.A.; Crawford, T.O.; Lederman, H.M. Ataxia Telangiectasia: A Review. Orphanet J. Rare Dis. 2016, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pastorczak, A.; Attarbaschi, A.; Bomken, S.; Borkhardt, A.; van der Werff ten Bosch, J.; Elitzur, S.; Gennery, A.R.; Hlavackova, E.; Kerekes, A.; Křenová, Z.; et al. Consensus Recommendations for the Clinical Management of Hematological Malignancies in Patients with DNA Double Stranded Break Disorders. Cancers 2022, 14, 2000. [Google Scholar] [CrossRef]
- Mitui, M.; Bernatowska, E.; Pietrucha, B.; Piotrowska-Jastrzebska, J.; Eng, L.; Nahas, S.; Teraoka, S.; Sholty, G.; Purayidom, A.; Concannon, P.; et al. ATM Gene Founder Haplotypes and Associated Mutations in Polish Families with Ataxia-Telangiectasia. Ann. Hum. Genet. 2005, 69, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Saviozzi, S. A Late Onset Variant of Ataxia-Telangiectasia with a Compound Heterozygous Genotype, A8030G/7481insA. J. Med. Genet. 2002, 39, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Yanofsky, R.A.; Seshia, S.S.; Dawson, A.J.; Stobart, K.; Greenberg, C.R.; Booth, F.A.; Prasad, C.; Bigio, M.R.D.; Wrogemann, J.J.; Fike, F.; et al. Ataxia-Telangiectasia: Atypical Presentation and Toxicity of Cancer Treatment. Can. J. Neurol. Sci. 2009, 36, 462–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schon, K.; Os, N.; Oscroft, N.; Baxendale, H.; Scoffings, D.; Ray, J.; Suri, M.; Whitehouse, W.P.; Mehta, P.R.; Everett, N.; et al. Genotype, Extrapyramidal Features and Severity of Variant Ataxia-Telangiectasia. Ann. Neurol. 2018, ana.25394. [Google Scholar] [CrossRef]
- Urbańska, Z.; Lejman, M.; Taha, J.; Madzio, J.; Ostrowska, K.; Miarka-Walczyk, K.; Wypyszczak, K.; Styka, B.; Jakubowska, J.; Sędek, Ł.; et al. The Kinetics of Blast Clearance Are Associated with Copy Number Alterations in Childhood B-Cell Acute Lymphoblastic Leukemia. Neoplasia 2023, 35, 100840. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and Accurate Long-Read Alignment with Burrows–Wheeler Transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative Genomics Viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation Prediction for the Deep-Sequencing Age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public Archive of Interpretations of Clinically Relevant Variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- McKusick, V.A. Mendelian Inheritance in Man and Its Online Version, OMIM. Am. J. Hum. Genet. 2007, 80, 588–604. [Google Scholar] [CrossRef] [Green Version]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Shaw, K.; Phillips, A.D.; Cooper, D.N. The Human Gene Mutation Database: Building a Comprehensive Mutation Repository for Clinical and Molecular Genetics, Diagnostic Testing and Personalized Genomic Medicine. Hum. Genet. 2014, 133, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Ramis-Zaldivar, J.E.; Gonzalez-Farré, B.; Balagué, O.; Celis, V.; Nadeu, F.; Salmerón-Villalobos, J.; Andrés, M.; Martin-Guerrero, I.; Garrido-Pontnou, M.; Gaafar, A.; et al. Distinct Molecular Profile of IRF4-Rearranged Large B-Cell Lymphoma. Blood 2020, 135, 274–286. [Google Scholar] [CrossRef]
- Lefebvre, C.; Callet-Bauchu, E.; Chapiro, E.; Nadal, N.; Penther, D.; Antoine-Poirel, H. Cytogenetics in the Management of Lymphomas and Lymphoproliferative Disorders in Adults and Children: An Update by the Groupe Francophone de Cytogénétique Hématologique (GFCH). Ann. De Biol. Clin. 2016, 74, 568–587. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Salaverria, I.; Pittaluga, S.; Jegalian, A.G.; Xi, L.; Siebert, R.; Raffeld, M.; Hewitt, S.M.; Jaffe, E.S. Follicular Lymphomas in Children and Young Adults: A Comparison of the Pediatric Variant with Usual Follicular Lymphoma. Am. J. Surg. Pathol. 2013, 37, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salaverria, I.; Martin-Guerrero, I.; Burkhardt, B.; Kreuz, M.; Zenz, T.; Oschlies, I.; Arnold, N.; Baudis, M.; Bens, S.; García-Orad, A.; et al. High Resolution Copy Number Analysis of IRF4 Translocation-positive Diffuse Large B-cell and Follicular Lymphomas. Genes Chromosom. Cancer 2013, 52, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Al-Kzayer, L.F.Y.; Liu, T.; Kobayashi, N.; Nakazawa, Y.; Koike, K. IFR4/MUM1-Positive Lymphoma in Waldeyer Ring with Co-Expression of CD5 and CD10: Chen et Al. Pediatr. Blood Cancer 2017, 64, 311–314. [Google Scholar] [CrossRef]
- Woessmann, W.; Quintanilla-Martinez, L. Rare Mature B-cell Lymphomas in Children and Adolescents. Hematol. Oncol. 2019, 37, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Au-Yeung, R.K.H.; Arias Padilla, L.; Zimmermann, M.; Oschlies, I.; Siebert, R.; Woessmann, W.; Burkhardt, B.; Klapper, W. Experience with Provisional WHO-entities Large B-cell Lymphoma with IRF4-rearrangement and Burkitt-like Lymphoma with 11q Aberration in Paediatric Patients of the NHL-BFM Group. Br. J. Haematol. 2020, 190, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Ott, G.; Rosenwald, A.; Campo, E. Understanding MYC-Driven Aggressive B-Cell Lymphomas: Pathogenesis and Classification. Hematology 2013, 2013, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Petrich, A.M.; Nabhan, C.; Smith, S.M. MYC-Associated and Double-Hit Lymphomas: A Review of Pathobiology, Prognosis, and Therapeutic Approaches: Double-Hit Lymphomas. Cancer 2014, 120, 3884–3895. [Google Scholar] [CrossRef]
- Sato, D.; Moriya, K.; Nakano, T.; Miyagawa, C.; Katayama, S.; Niizuma, H.; Sasahara, Y.; Kure, S. Refractory T-Cell/Histiocyte-Rich Large B-Cell Lymphoma in a Patient with Ataxia–Telangiectasia Caused by Novel Compound Heterozygous Variants in ATM. Int. J. Hematol. 2021, 114, 735–741. [Google Scholar] [CrossRef]
- Wei, C.; Wei, C.; Alhalabi, O.; Chen, L. T-Cell/Histiocyte-Rich Large B-Cell Lymphoma in a Child: A Case Report and Review of Literature. WJCC 2018, 6, 121–126. [Google Scholar] [CrossRef]
- Kommalapati, A.; Tella, S.H.; Go, R.S.; Nowakowski, G.S.; Goyal, G. T Cell/Histiocyte-rich Large B Cell Lymphoma: Incidence, Demographic Disparities, and Long-term Outcomes. Br. J. Haematol. 2019, 185, 140–142. [Google Scholar] [CrossRef]
- Stankovic, T.; Skowronska, A. The Role of ATM Mutations and 11q Deletions in Disease Progression in Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2014, 55, 1227–1239. [Google Scholar] [CrossRef]
- Mareckova, A.; Malcikova, J.; Tom, N.; Pal, K.; Radova, L.; Salek, D.; Janikova, A.; Moulis, M.; Smardova, J.; Kren, L.; et al. ATM and TP53 Mutations Show Mutual Exclusivity but Distinct Clinical Impact in Mantle Cell Lymphoma Patients. Leuk. Lymphoma 2019, 60, 1420–1428. [Google Scholar] [CrossRef]
- Spiegel, J.Y.; Patel, S.; Muffly, L.; Hossain, N.M.; Oak, J.; Baird, J.H.; Frank, M.J.; Shiraz, P.; Sahaf, B.; Craig, J.; et al. CAR T Cells with Dual Targeting of CD19 and CD22 in Adult Patients with Recurrent or Refractory B Cell Malignancies: A Phase 1 Trial. Nat. Med. 2021, 27, 1419–1431. [Google Scholar] [CrossRef] [PubMed]
- Jończyk-Potoczna, K.; Potoczny, J.; Szczawińska-Popłonyk, A. Imaging in Children with Ataxia-Telangiectasia—The Radiologist’s Approach. Front. Pediatr. 2022, 10, 988645. [Google Scholar] [CrossRef] [PubMed]
- Szczawińska-Popłonyk, A.; Tąpolska-Jóźwiak, K.; Schwartzmann, E.; Pietrucha, B. Infections and Immune Dysregulation in Ataxia-Telangiectasia Children with Hyper-IgM and Non-Hyper-IgM Phenotypes: A Single-Center Experience. Front. Pediatr. 2022, 10, 972952. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PB Lymphocyte Subsets | Results | Reference Values |
|---|---|---|
| WBC | 5600 cc | |
| Lymphocytes CD45+/SSC low | 35.0%, 1960 cc | 29.6–69.2%, 2300–6900 cc |
| B CD19+ | 35.0%, 700 cc | 14.1–28.5%, 400–1700 cc |
| Transitional B CD19+CD38+IgM++ | 4.3%, 30 cc | 3.1–12.3%, 20–200 cc |
| Mature naïve B CD19+CD27-IgD+ | 36,2%, 253 cc | 54.0–88.4%, 280–1330 cc |
| Non-switched memory B (MZL) CD19+CD27+IgD+ | 39.4%, 276 cc | 2.7–19.8%, 20–180 cc |
| Switched memory B CD19+CD27+IgD- | 22.1%, 155 cc | 4.7–21.2%, 20–220 cc |
| Immature B CD19+CD21lo | 44.4%, 311 cc | 4.1–24.4%, 20–230 cc |
| Activated B CD19+CD38loCD21lo | 4,6%, 11 cc | 1.7–5.4%, 10–60 cc |
| Plasmablasts CD19+CD38++IgM- | 0.3%, 2 cc | 0.6–4.0%, 5–10 cc |
| T CD3+ | 17.0%, 340 cc | 52.0–92.0%, 850–4300 cc |
| T helper CD3+CD4+ | 9.0%, 180 cc | 25.0–66.0%, 500–2700 cc |
| T suppressor/cytotoxic CD3+CD8+ | 7.0%, 140 cc | 9.0–49.0%, 200–1800 cc |
| CD4+/CD8+ | 1.29 | 1.5–2.5 |
| CD4+CD45RA+/CD4+CD45RO+ | 0.03 | >1.0 |
| Recent thymic emigrants CD3+CD4+CD45RA+CD31+ | 2.2%, 4 cc | 37.0–100%, 190–2600 cc |
| Naïve T helper CD3+CD4+CD45RA+CD27+ | 1.8%, 3 cc | 52.0–92.0%, 300–2300 cc |
| Central memory T helper CD3+CD4+CD45RA-CD27+ | 97.2%, 167 cc | 15.0–56.0%, 160–660 cc |
| Effector memory T helper CD3+CD4+CD45RA-CD27- | 4.7%, 8 cc | 0.3–9.0%, 3–89 cc |
| Terminally differentiated memory T helper CD3+CD4+CD45RA+CD27- | 0.9%, 2 cc | 0.0–1.2%, 0–16 cc |
| Follicular CXCR5+ T helper CD3+CD4+CD45RO+CD185+ | 48.2%, 84 cc | 6.0–72.0%, 13–170 cc |
| Regulatory T helper CD3+CD4+CD25++CD127- | 1.3%, 2 cc | 3.0–17.0%, 39–150 cc |
| Naïve T suppressor/cytotoxic CD3+CD8+CD27+CD197+ | 12.2%, 17 cc | 19.0–100%, 53–1100 cc |
| Central memory T suppressor/cytotoxic CD3+CD8+CD45RA-CD27+CD197+ | 12.2%, 17 cc | 1.0–9.0%, 4–64 cc |
| Effector memory T suppressor/cytotoxic CD3+CD8+CD45RA-CD27-CD197- | 12.8%, 18 cc | 10.0–55.0%, 24–590 cc |
| Terminally differentiated T suppressor/cytotoxic CD3+CD8+CD45RA+CD27-CD197- | 12.2%, 17 cc | 6.0–83.0%, 25–530 cc |
| NK CD3-CD45+CD16+CD56+ | 41.0%, 820 cc | 2.0–25.0%, 61–510 cc |
| Day | 1 | 2 | 3 | 4 | 5 | 6 = Day 2 of 1st R-COPADM | 7 |
|---|---|---|---|---|---|---|---|
| Vincristine | x | ||||||
| Prednisone | x x | x x | x x | x x | x x | x x | x x |
| Cyclophosphamide | x | ||||||
| IT MTX and HC | x |
| Day | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Rituximab | x | |||||
| Vincristine | x | |||||
| Prednisone | x x | x x | x x | x x | x x | Tail to zero over 3 days |
| Methotrexate | x | |||||
| Folinic acid | xxxx | xxxx | xxxx | |||
| Cyclophosphamide | x x | x x | x x | |||
| Doxorubicin | x | |||||
| IT MTX and HC | x | x |
| Day | 1 | 2 | 3 | 4 | 5 | 6 | 7 |
|---|---|---|---|---|---|---|---|
| Rituximab | x | ||||||
| Methotrexate | x | ||||||
| Folinic acid | xxxx | xxxx | xxxx | ||||
| Cytarabine | x | ||||||
| IT MTX | x | ||||||
| IT HC | x | x | |||||
| IT ARA-C | x | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Czarny, J.; Andrzejewska, M.; Zając-Spychała, O.; Latos-Grażyńska, E.; Pastorczak, A.; Wypyszczak, K.; Szczawińska-Popłonyk, A.; Niewiadomska-Wojnałowicz, I.; Wziątek, A.; Marciniak-Stępak, P.; et al. Successful Treatment of Large B-Cell Lymphoma in a Child with Compound Heterozygous Mutation in the ATM Gene. Int. J. Mol. Sci. 2023, 24, 1099. https://doi.org/10.3390/ijms24021099
Czarny J, Andrzejewska M, Zając-Spychała O, Latos-Grażyńska E, Pastorczak A, Wypyszczak K, Szczawińska-Popłonyk A, Niewiadomska-Wojnałowicz I, Wziątek A, Marciniak-Stępak P, et al. Successful Treatment of Large B-Cell Lymphoma in a Child with Compound Heterozygous Mutation in the ATM Gene. International Journal of Molecular Sciences. 2023; 24(2):1099. https://doi.org/10.3390/ijms24021099
Chicago/Turabian StyleCzarny, Jakub, Marta Andrzejewska, Olga Zając-Spychała, Elżbieta Latos-Grażyńska, Agata Pastorczak, Kamila Wypyszczak, Aleksandra Szczawińska-Popłonyk, Izabela Niewiadomska-Wojnałowicz, Agnieszka Wziątek, Patrycja Marciniak-Stępak, and et al. 2023. "Successful Treatment of Large B-Cell Lymphoma in a Child with Compound Heterozygous Mutation in the ATM Gene" International Journal of Molecular Sciences 24, no. 2: 1099. https://doi.org/10.3390/ijms24021099