
A Mutation in Mouse MT-ATP6 Gene Induces Respiration Defects and Opposed Effects on the Cell Tumorigenic Phenotype
 , ,
, ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
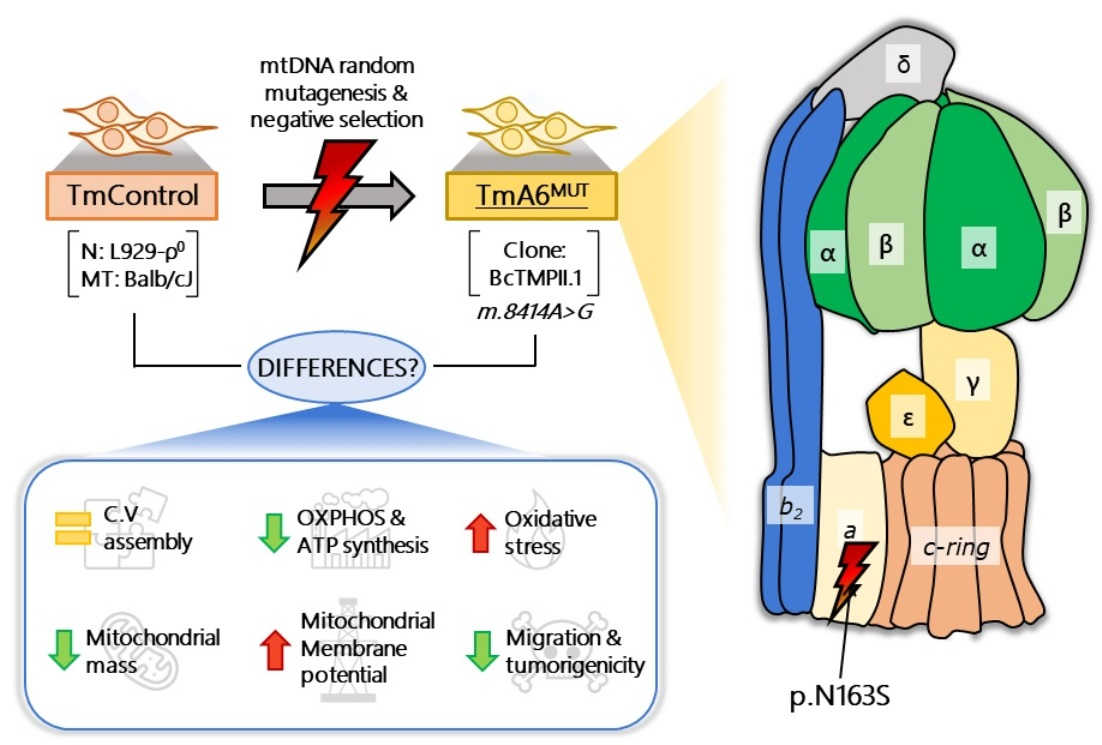
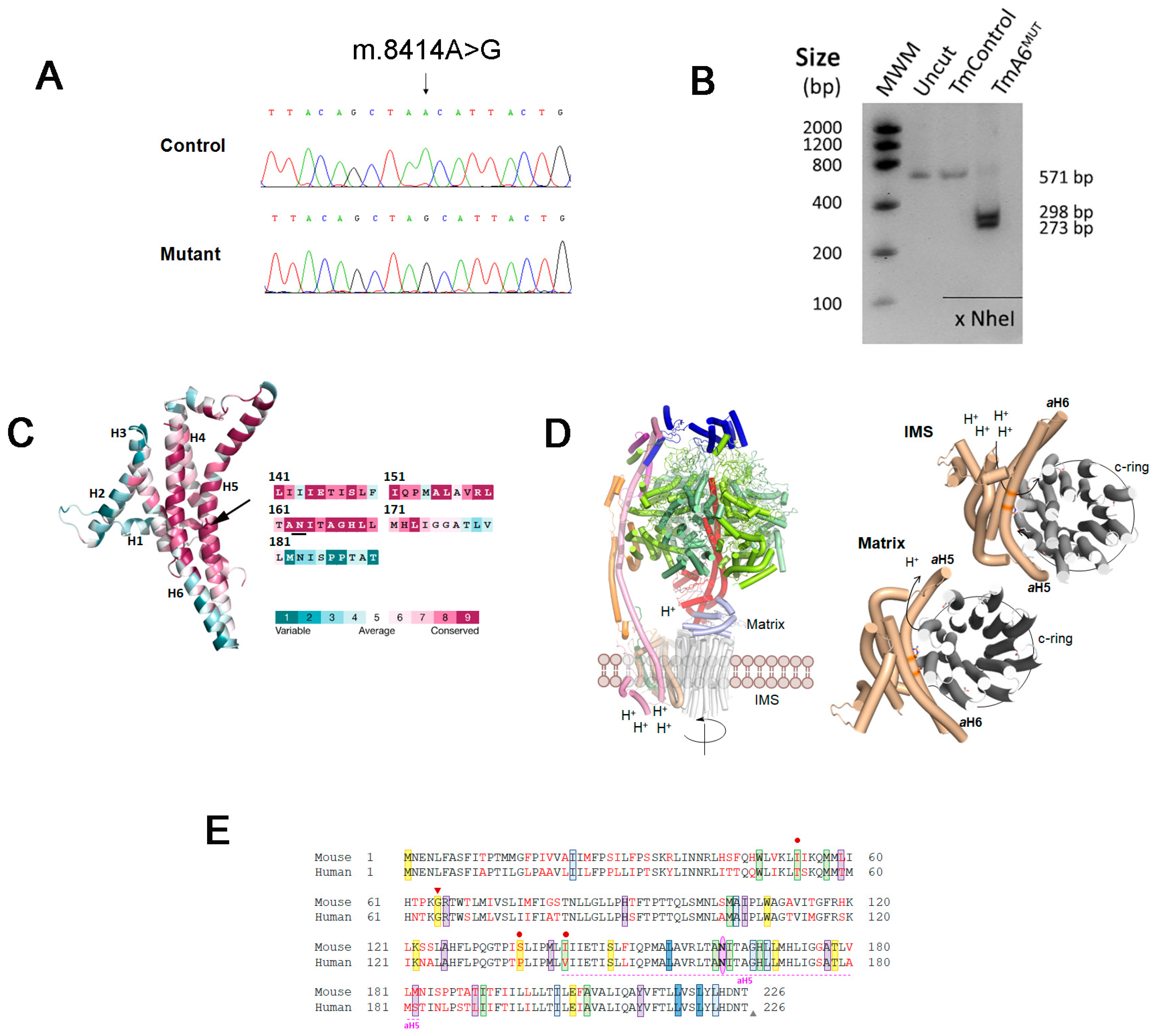
2.1. Isolation of a Mouse Cell Line with an mtDNA Driven Mutation in ATP Synthase
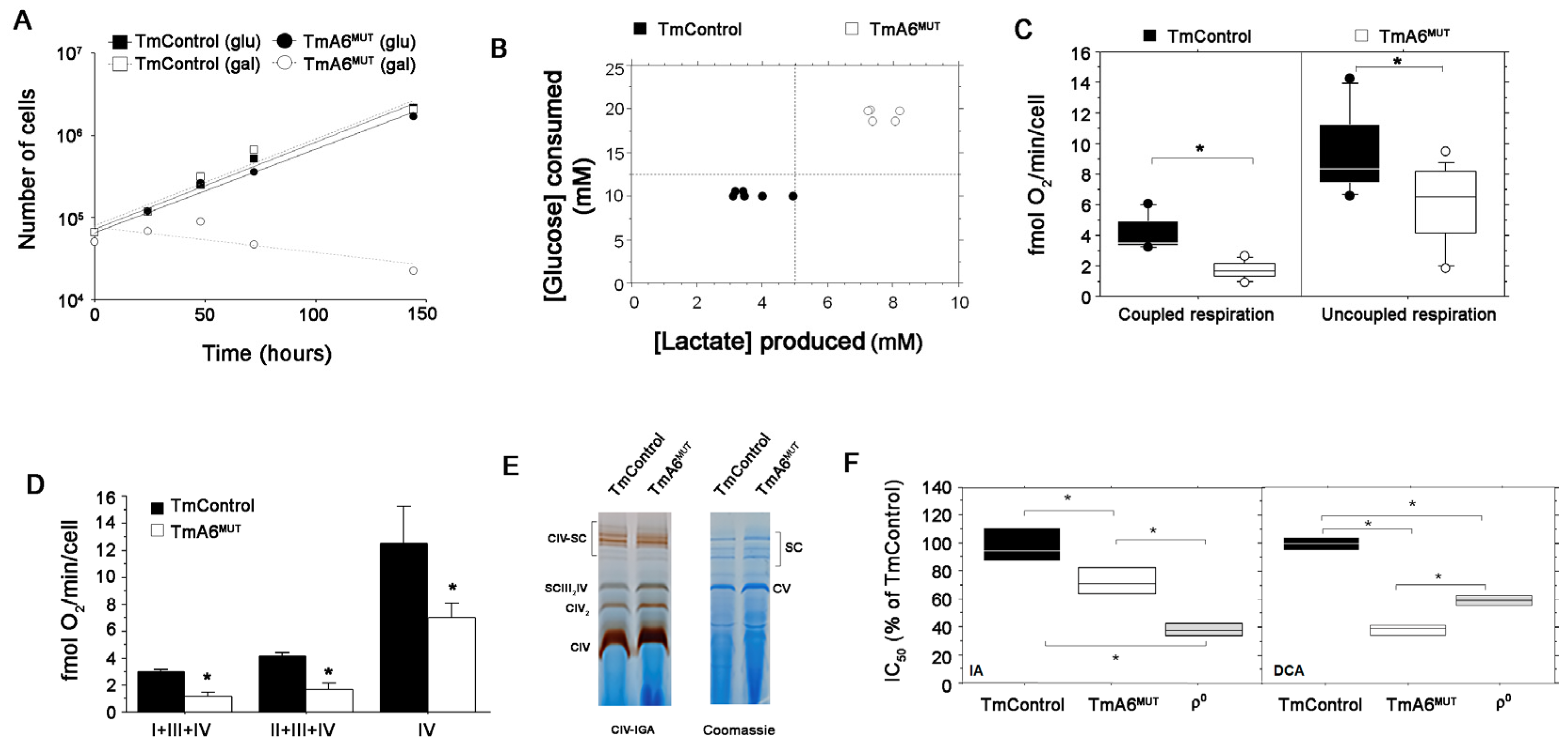
2.2. Cells Carrying the m.8414A>G mtDNA Variant Are Defective in OXPHOS Performance and Dependent on Glycolysis
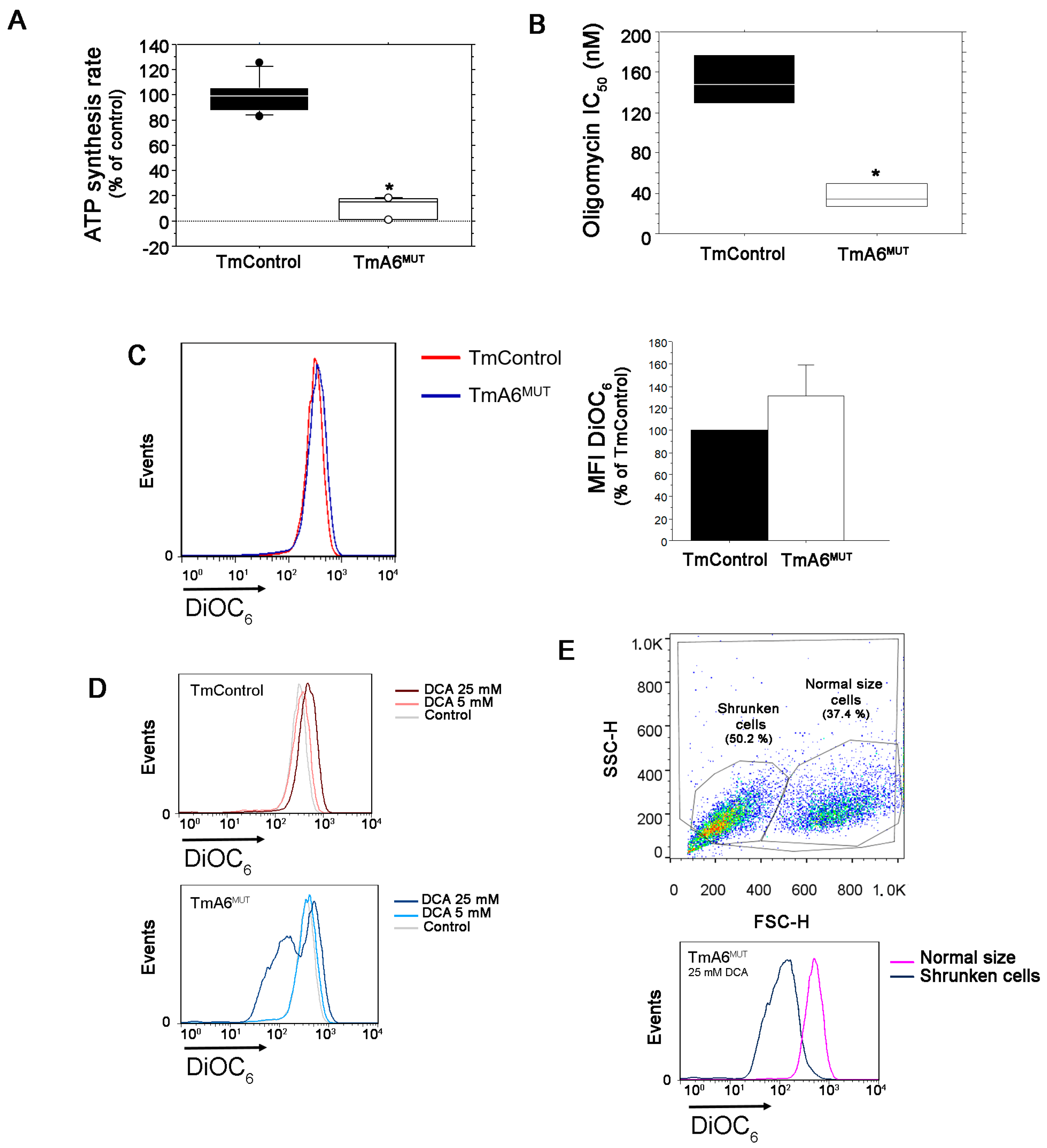
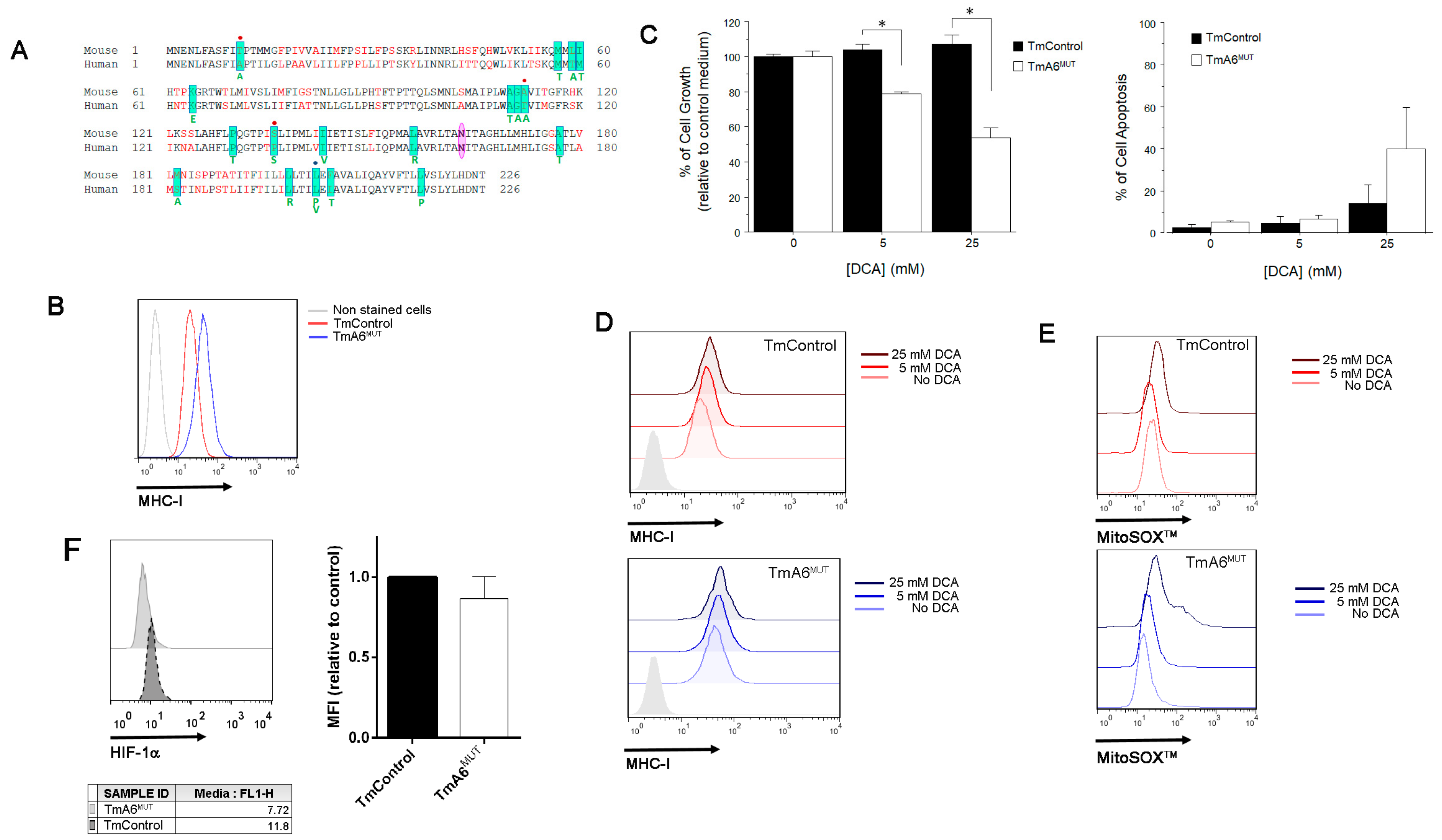
2.3. Mutant Cells Show Impaired ATP Synthesis and Differences in Mitochondrial Membrane Potential
2.4. m.8414A>G Mutation Induces Differences in Mitochondrial Mass and ROS Production
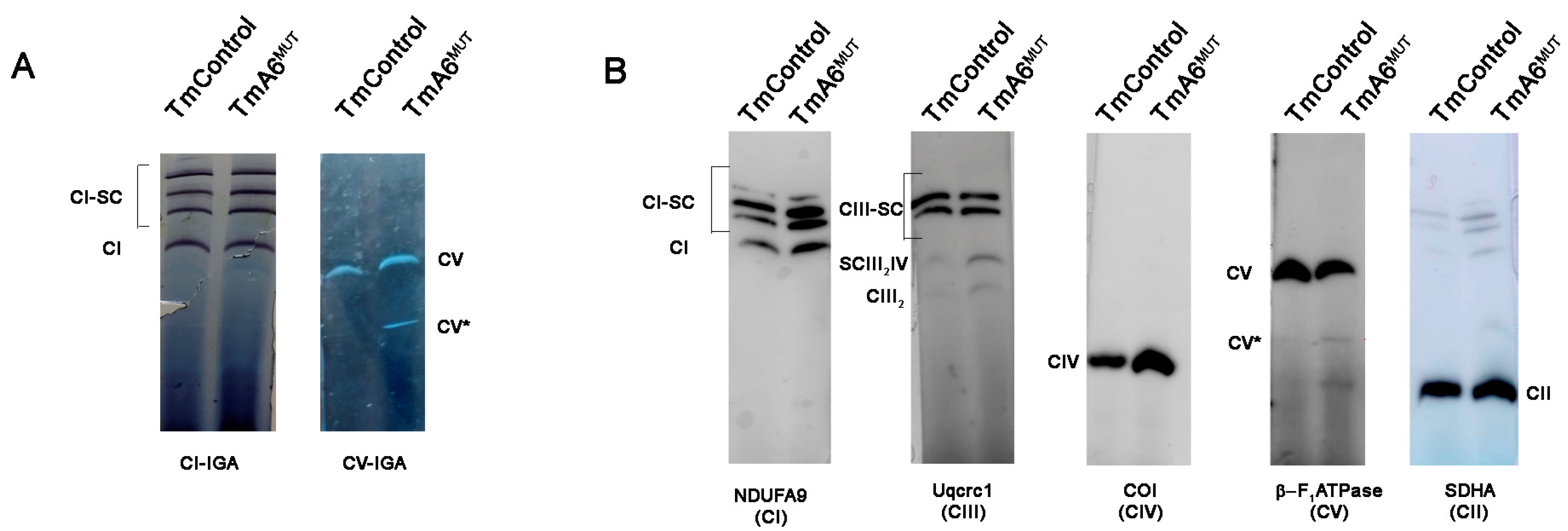
2.5. m.8414A>G Mutation Does Not Affect ATP Synthase Assembly
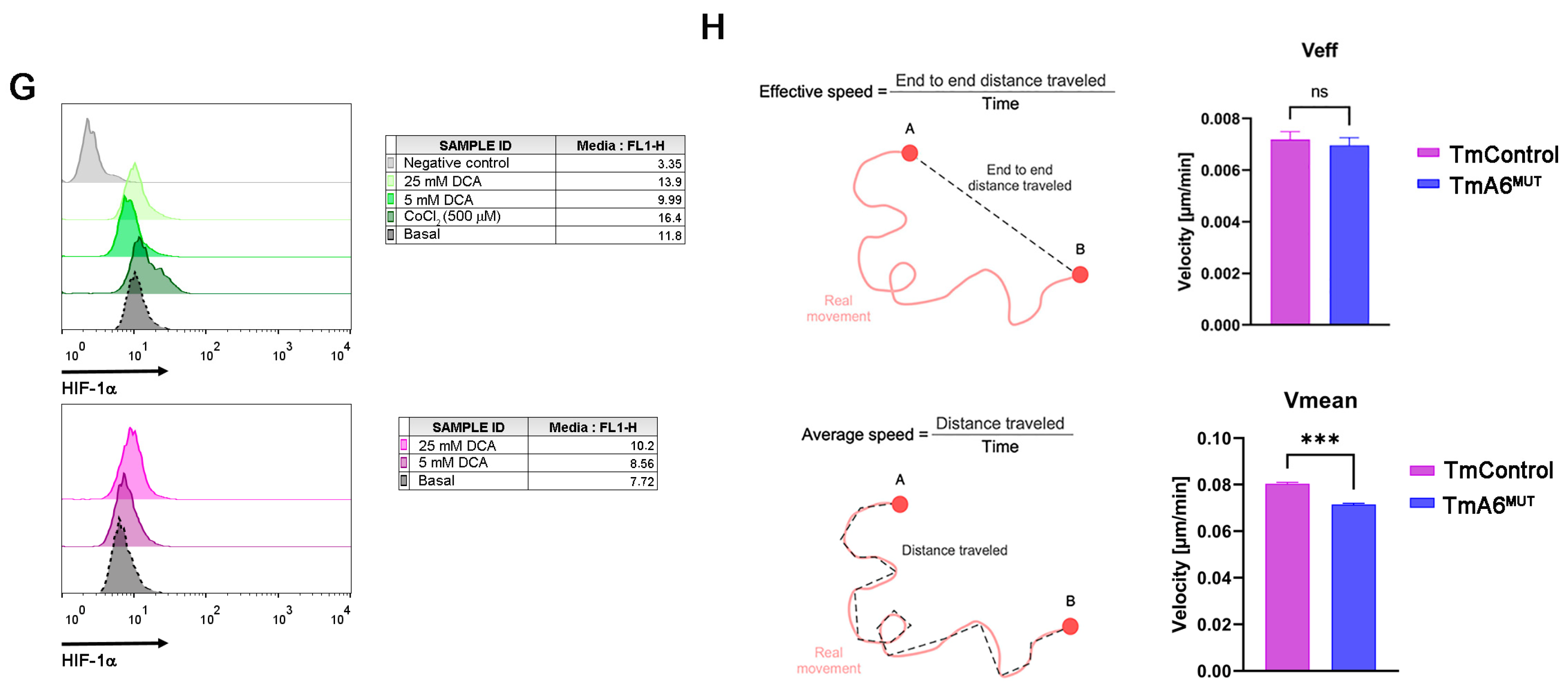
2.6. m.8414A>G Mutation Decreases Migration Ability and Tumorigenic Phenotype in TmA6MUT Cells
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Media
4.2. DNA Analysis
4.3. RFLP Analysis
4.4. Growth Measurements
4.5. Oxygen Consumption Measurements
4.6. Glucose and Lactate Measurements
4.7. Cell Growth Assays
4.8. ATP Synthesis Measurements
4.9. Flow Cytometry Analysis
4.10. mtDNA Copy Number Measurement
4.11. Blue Native Polyacrylamide Electrophoresis
4.12. Respiratory Complex and Supercomplex Analysis
4.13. HIF-1α Expression Evaluation
4.14. Cell Migration Analysis
4.15. Bioinformatic Tools
4.16. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jonckheere, A.I.; Smeitink, J.A.; Rodenburg, R.J. Mitochondrial ATP synthase: Architecture, function and pathology. J. Inherit. Metab. Dis. 2012, 35, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Dominguez-Zorita, S.; Romero-Carraminana, I.; Cuezva, J.M.; Esparza-Molto, P.B. The ATPase Inhibitory Factor 1 is a Tissue-Specific Physiological Regulator of the Structure and Function of Mitochondrial ATP Synthase: A Closer Look Into Neuronal Function. Front. Physiol. 2022, 13, 868820. [Google Scholar] [CrossRef]
- Eydt, K.; Davies, K.M.; Behrendt, C.; Wittig, I.; Reichert, A.S. Cristae architecture is determined by an interplay of the MICOS complex and the F1FO ATP synthase via Mic27 and Mic10. Microb. Cell 2017, 4, 259–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhlbrandt, W. Structure and Mechanisms of F-Type ATP Synthases. Annu. Rev. Biochem. 2019, 88, 515–549. [Google Scholar] [CrossRef] [PubMed]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vazquez, J.; Enriquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef] [PubMed]
- Carraro, M.; Jones, K.; Sartori, G.; Schiavone, M.; Antonucci, S.; Kucharczyk, R.; di Rago, J.P.; Franchin, C.; Arrigoni, G.; Forte, M.; et al. The Unique Cysteine of F-ATP Synthase OSCP Subunit Participates in Modulation of the Permeability Transition Pore. Cell Rep. 2020, 32, 108095. [Google Scholar] [CrossRef]
- Mnatsakanyan, N.; Jonas, E.A. ATP synthase c-subunit ring as the channel of mitochondrial permeability transition: Regulator of metabolism in development and degeneration. J. Mol. Cell. Cardiol. 2020, 144, 109–118. [Google Scholar] [CrossRef]
- Esparza-Molto, P.B.; Nuevo-Tapioles, C.; Cuezva, J.M. Regulation of the H+-ATP synthase by IF1: A role in mitohormesis. Cell. Mol. Life Sci. 2017, 74, 2151–2166. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Reyes, I.; Cuezva, J.M. The H+-ATP synthase: A gate to ROS-mediated cell death or cell survival. Biochim. Biophys. Acta 2014, 1837, 1099–1112. [Google Scholar] [CrossRef] [Green Version]
- Mnatsakanyan, N.; Jonas, E.A. The new role of F1Fo ATP synthase in mitochondria-mediated neurodegeneration and neuroprotection. Exp. Neurol. 2020, 332, 113400. [Google Scholar] [CrossRef]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Capiau, S.; Smet, J.; De Paepe, B.; Yildiz, Y.; Arslan, M.; Stevens, O.; Verschoore, M.; Stepman, H.; Seneca, S.; Vanlander, A. Clinical Heterogeneity in MT-ATP6 Pathogenic Variants: Same Genotype-Different Onset. Cells 2022, 11, 489. [Google Scholar] [CrossRef]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2021, 595, 1062–1106. [Google Scholar] [CrossRef] [PubMed]
- Kucharczyk, R.; Ezkurdia, N.; Couplan, E.; Procaccio, V.; Ackerman, S.H.; Blondel, M.; di Rago, J.P. Consequences of the pathogenic T9176C mutation of human mitochondrial DNA on yeast mitochondrial ATP synthase. Biochim. Biophys. Acta 2010, 1797, 1105–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sgarbi, G.; Baracca, A.; Lenaz, G.; Valentino, L.M.; Carelli, V.; Solaini, G. Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA. Biochem. J. 2006, 395, 493–500. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Schagger, H. Structural organization of mitochondrial ATP synthase. Biochim. Biophys. Acta 2008, 1777, 592–598. [Google Scholar] [CrossRef] [Green Version]
- Wittig, I.; Velours, J.; Stuart, R.; Schagger, H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Mol. Cell. Proteom. 2008, 7, 995–1004. [Google Scholar] [CrossRef] [Green Version]
- Baracca, A.; Sgarbi, G.; Mattiazzi, M.; Casalena, G.; Pagnotta, E.; Valentino, M.L.; Moggio, M.; Lenaz, G.; Carelli, V.; Solaini, G. Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993. Biochim. Biophys. Acta 2007, 1767, 913–919. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Hernandez, P.; Vazquez-Memije, M.E.; Garcia, J.J. ATP6 homoplasmic mutations inhibit and destabilize the human F1F0-ATP synthase without preventing enzyme assembly and oligomerization. J. Biol. Chem. 2007, 282, 1051–1058. [Google Scholar] [CrossRef] [Green Version]
- Houstĕk, J.; Klement, P.; Hermanská, J.; Houstková, H.; Hansíková, H.; Van den Bogert, C.; Zeman, J. Altered properties of mitochondrial ATP-synthase in patients with a T→G mutation in the ATPase 6 (subunit a) gene at position 8993 of mtDNA. Biochim. Biophys. Acta 1995, 1271, 349–357. [Google Scholar] [CrossRef]
- Mattiazzi, M.; Vijayvergiya, C.; Gajewski, C.D.; DeVivo, D.C.; Lenaz, G.; Wiedmann, M.; Manfredi, G. The mtDNA T8993G (NARP) mutation results in an impairment of oxidative phosphorylation that can be improved by antioxidants. Hum. Mol. Genet. 2004, 13, 869–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morava, E.; Rodenburg, R.J.; Hol, F.; de Vries, M.; Janssen, A.; van den Heuvel, L.; Nijtmans, L.; Smeitink, J. Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations. Am. J. Med. Genet. A 2006, 140, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Nijtmans, L.G.; Henderson, N.S.; Attardi, G.; Holt, I.J. Impaired ATP synthase assembly associated with a mutation in the human ATP synthase subunit 6 gene. J. Biol. Chem. 2001, 276, 6755–6762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trounce, I.; Neill, S.; Wallace, D.C. Cytoplasmic transfer of the mtDNA nt 8993 T→G (ATP6) point mutation associated with Leigh syndrome into mtDNA-less cells demonstrates cosegregation with a decrease in state III respiration and ADP/O ratio. Proc. Natl. Acad. Sci. USA 1994, 91, 8334–8338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vazquez-Memije, M.E.; Shanske, S.; Santorelli, F.M.; Kranz-Eble, P.; DeVivo, D.C.; DiMauro, S. Comparative biochemical studies of ATPases in cells from patients with the T8993G or T8993C mitochondrial DNA mutations. J. Inherit. Metab. Dis. 1998, 21, 829–836. [Google Scholar] [CrossRef]
- Aure, K.; Dubourg, O.; Jardel, C.; Clarysse, L.; Sternberg, D.; Fournier, E.; Laforet, P.; Streichenberger, N.; Petiot, P.; Gervais-Bernard, H.; et al. Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations. Neurology 2013, 81, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Craig, K.; Elliott, H.R.; Keers, S.M.; Lambert, C.; Pyle, A.; Graves, T.D.; Woodward, C.; Sweeney, M.G.; Davis, M.B.; Hanna, M.G.; et al. Episodic ataxia and hemiplegia caused by the 8993T->C mitochondrial DNA mutation. J. Med. Genet. 2007, 44, 797–799. [Google Scholar] [CrossRef] [Green Version]
- Kucharczyk, R.; Rak, M.; di Rago, J.P. Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients. Biochim. Biophys. Acta 2009, 1793, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Vilarinho, L.; Barbot, C.; Carrozzo, R.; Calado, E.; Tessa, A.; Dionisi-Vici, C.; Guimaraes, A.; Santorelli, F.M. Clinical and molecular findings in four new patients harbouring the mtDNA 8993T>C mutation. J. Inherit. Metab. Dis. 2001, 24, 883–884. [Google Scholar] [CrossRef]
- Dautant, A.; Meier, T.; Hahn, A.; Tribouillard-Tanvier, D.; di Rago, J.P.; Kucharczyk, R. ATP Synthase Diseases of Mitochondrial Genetic Origin. Front. Physiol. 2018, 9, 329. [Google Scholar] [CrossRef]
- Castagna, A.E.; Addis, J.; McInnes, R.R.; Clarke, J.T.; Ashby, P.; Blaser, S.; Robinson, B.H. Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA. Am. J. Med. Genet. A 2007, 143A, 808–816. [Google Scholar] [CrossRef] [PubMed]
- Childs, A.M.; Hutchin, T.; Pysden, K.; Highet, L.; Bamford, J.; Livingston, J.; Crow, Y.J. Variable phenotype including Leigh syndrome with a 9185T>C mutation in the MTATP6 gene. Neuropediatrics 2007, 38, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Galber, C.; Acosta, M.J.; Minervini, G.; Giorgio, V. The role of mitochondrial ATP synthase in cancer. Biol. Chem. 2020, 401, 1199–1214. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Shidara, Y.; Yamagata, K.; Kanamori, T.; Nakano, K.; Kwong, J.Q.; Manfredi, G.; Oda, H.; Ohta, S. Positive contribution of pathogenic mutations in the mitochondrial genome to the promotion of cancer by prevention from apoptosis. Cancer Res. 2005, 65, 1655–1663. [Google Scholar] [CrossRef] [Green Version]
- Cuezva, J.M.; Krajewska, M.; de Heredia, M.L.; Krajewski, S.; Santamaria, G.; Kim, H.; Zapata, J.M.; Marusawa, H.; Chamorro, M.; Reed, J.C. The bioenergetic signature of cancer: A marker of tumor progression. Cancer Res. 2002, 62, 6674–6681. [Google Scholar]
- Isidoro, A.; Martinez, M.; Fernandez, P.L.; Ortega, A.D.; Santamaria, G.; Chamorro, M.; Reed, J.C.; Cuezva, J.M. Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochem. J. 2004, 378, 17–20. [Google Scholar] [CrossRef] [Green Version]
- Esparza-Molto, P.B.; Cuezva, J.M. The Role of Mitochondrial H+-ATP Synthase in Cancer. Front. Oncol. 2018, 8, 53. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Cuezva, J.M. The ATPase Inhibitory Factor 1 (IF1): A master regulator of energy metabolism and of cell survival. Biochim. Biophys. Acta 2016, 1857, 1167–1182. [Google Scholar] [CrossRef]
- Fiorillo, M.; Ozsvari, B.; Sotgia, F.; Lisanti, M.P. High ATP Production Fuels Cancer Drug Resistance and Metastasis: Implications for Mitochondrial ATP Depletion Therapy. Front. Oncol. 2021, 11, 740720. [Google Scholar] [CrossRef]
- Fiorillo, M.; Scatena, C.; Naccarato, A.G.; Sotgia, F.; Lisanti, M.P. Bedaquiline, an FDA-approved drug, inhibits mitochondrial ATP production and metastasis in vivo, by targeting the gamma subunit (ATP5F1C) of the ATP synthase. Cell Death Differ. 2021, 28, 2797–2817. [Google Scholar] [CrossRef] [PubMed]
- Povalko, N.; Zakharova, E.; Rudenskaia, G.; Akita, Y.; Hirata, K.; Toyojiro, M.; Koga, Y. A new sequence variant in mitochondrial DNA associated with high penetrance of Russian Leber hereditary optic neuropathy. Mitochondrion 2005, 5, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Shidara, K.; Wakakura, M. Leber’s hereditary optic neuropathy with the 3434, 9011 mitochondrial DNA point mutation. Jpn. J. Ophthalmol. 2012, 56, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Bafaluy, M.P.; Movilla, N.; Perez-Martos, A.; Fernandez-Silva, P.; Enriquez, J.A. Functional genetic analysis of the mammalian mitochondrial DNA encoded peptides: A mutagenesis approach. Methods Mol. Biol. 2008, 457, 379–390. [Google Scholar] [PubMed]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Bueno, M.; Machicado, C.; Fernandez-Silva, P.; Perez-Martos, A.; Montoya, J.; Lopez-Perez, M.J.; Sancho, J.; Enriquez, J.A. An intragenic suppressor in the cytochrome c oxidase I gene of mouse mitochondrial DNA. Hum. Mol. Genet. 2003, 12, 329–339. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Bayona-Bafaluy, M.P.; Fernandez-Silva, P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Bruno, C.; Moraes, C.T.; Enriquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell 2004, 13, 805–815. [Google Scholar] [CrossRef]
- D’Aurelio, M.; Vives-Bauza, C.; Davidson, M.M.; Manfredi, G. Mitochondrial DNA background modifies the bioenergetics of NARP/MILS ATP6 mutant cells. Hum. Mol. Genet. 2010, 19, 374–386. [Google Scholar] [CrossRef] [Green Version]
- Sabri, M.I.; Ochs, S. Inhibition of glyceraldehyde-3-phosphate dehydrogenase in mammalian nerve by iodoacetic acid. J. Neurochem. 1971, 18, 1509–1514. [Google Scholar] [CrossRef]
- Manfredi, G.; Gupta, N.; Vazquez-Memije, M.E.; Sadlock, J.E.; Spinazzola, A.; De Vivo, D.C.; Schon, E.A. Oligomycin induces a decrease in the cellular content of a pathogenic mutation in the human mitochondrial ATPase 6 gene. J. Biol. Chem. 1999, 274, 9386–9391. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Gallardo, E.; Emperador, S.; Solano, A.; Llobet, L.; Martin-Navarro, A.; Lopez-Perez, M.J.; Briones, P.; Pineda, M.; Artuch, R.; Barraquer, E.; et al. Expanding the clinical phenotypes of MT-ATP6 mutations. Hum. Mol. Genet. 2014, 23, 6191–6200. [Google Scholar] [CrossRef] [Green Version]
- Sikorska, M.; Sandhu, J.K.; Simon, D.K.; Pathiraja, V.; Sodja, C.; Li, Y.; Ribecco-Lutkiewicz, M.; Lanthier, P.; Borowy-Borowski, H.; Upton, A.; et al. Identification of ataxia-associated mtDNA mutations (m.4052T>C and m.9035T>C) and evaluation of their pathogenicity in transmitochondrial cybrids. Muscle Nerve 2009, 40, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Acin-Perez, R.; Fernandez-Silva, P.; Movilla, N.; Perez-Martos, A.; de Cordoba, S.R.; Gallardo, M.E.; Enriquez, J.A. Differences in reactive oxygen species production explain the phenotypes associated with common mouse mitochondrial DNA variants. Nat. Genet. 2006, 38, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Loshuertos, R.; Ferrín, G.; Acín-Pérez, R.; Gallardo, M.E.; Viscomi, C.; Pérez-Martos, A.; Zeviani, M.; Fernández-Silva, P.; Enríquez, J.A. Evolution Meets Disease: Penetrance and Functional Epistasis of Mitochondrial tRNA Mutations. PLoS Genet. 2011, 7, e1001379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bugiardini, E.; Bottani, E.; Marchet, S.; Poole, O.V.; Beninca, C.; Horga, A.; Woodward, C.; Lam, A.; Hargreaves, I.; Chalasani, A.; et al. Expanding the molecular and phenotypic spectrum of truncating MT-ATP6 mutations. Neurol. Genet. 2020, 6, e381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kenvin, S.; Torregrosa-Muñumer, R.; Reidelbach, M.; Pennonen, J.; Turkia, J.J.; Rannila, E.; Kvist, J.; Sainio, M.T.; Huber, N.; Herukka, S.K.; et al. Threshold of heteroplasmic truncating MT-ATP6 mutation in reprogramming, Notch hyperactivation and motor neuron metabolism. Hum. Mol. Genet. 2022, 31, 958–974. [Google Scholar] [CrossRef]
- Gatto, C.; Grandi, M.; Solaini, G.; Baracca, A.; Giorgio, V. The F1Fo-ATPase inhibitor protein IF1 in pathophysiology. Front. Physiol. 2022, 13, 917203. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [Green Version]
- Grzybowska-Szatkowska, L.; Slaska, B.; Rzymowska, J.; Brzozowska, A.; Florianczyk, B. Novel mitochondrial mutations in the ATP6 and ATP8 genes in patients with breast cancer. Mol. Med. Rep. 2014, 10, 1772–1778. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Morales, S.; Perez-Amado, C.J.; Langley, E.; Hidalgo-Miranda, A. Overview of mitochondrial germline variants and mutations in human disease: Focus on breast cancer (Review). Int. J. Oncol. 2018, 53, 923–936. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Akhtari, M.; Alachkar, H. Characterization of Mutations in the Mitochondrial Encoded Electron Transport Chain Complexes in Acute Myeloid Leukemia. Sci. Rep. 2018, 8, 13301. [Google Scholar] [CrossRef] [Green Version]
- Marco-Brualla, J.; Al-Wasaby, S.; Soler, R.; Romanos, E.; Conde, B.; Justo-Mendez, R.; Enriquez, J.A.; Fernandez-Silva, P.; Martinez-Lostao, L.; Villalba, M.; et al. Mutations in the ND2 Subunit of Mitochondrial Complex I Are Sufficient to Confer Increased Tumorigenic and Metastatic Potential to Cancer Cells. Cancers 2019, 11, 1027. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1: Upstream and downstream of cancer metabolism. Curr. Opin. Genet. Dev. 2010, 20, 51–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, O.M.; Gorman, G.S.; Lightowlers, R.N.; Turnbull, D.M. Mitochondrial Diseases: Hope for the Future. Cell 2020, 181, 168–188. [Google Scholar] [CrossRef] [PubMed]
- Vinothkumar, K.R.; Montgomery, M.G.; Liu, S.; Walker, J.E. Structure of the mitochondrial ATP synthase from Pichia angusta determined by electron cryo-microscopy. Proc. Natl. Acad. Sci. USA 2016, 113, 12709–12714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spikes, T.E.; Montgomery, M.G.; Walker, J.E. Structure of the dimeric ATP synthase from bovine mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 23519–23526. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Zidek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Campanella, M.; Casswell, E.; Chong, S.; Farah, Z.; Wieckowski, M.R.; Abramov, A.Y.; Tinker, A.; Duchen, M.R. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008, 8, 13–25. [Google Scholar] [CrossRef] [Green Version]
- Faccenda, D.; Tan, C.H.; Seraphim, A.; Duchen, M.R.; Campanella, M. IF1 limits the apoptotic-signalling cascade by preventing mitochondrial remodelling. Cell Death Differ. 2013, 20, 686–697. [Google Scholar] [CrossRef] [Green Version]
- Barbato, S.; Sgarbi, G.; Gorini, G.; Baracca, A.; Solaini, G. The inhibitor protein (IF1) of the F1F0-ATPase modulates human osteosarcoma cell bioenergetics. J. Biol. Chem. 2015, 290, 6338–6348. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Bermudez, J.; Sanchez-Arago, M.; Soldevilla, B.; Del Arco, A.; Nuevo-Tapioles, C.; Cuezva, J.M. PKA Phosphorylates the ATPase Inhibitory Factor 1 and Inactivates Its Capacity to Bind and Inhibit the Mitochondrial H+-ATP Synthase. Cell Rep. 2015, 12, 2143–2155. [Google Scholar] [CrossRef] [PubMed]
- Chomyn, A.; Lai, S.T.; Shakeley, R.; Bresolin, N.; Scarlato, G.; Attardi, G. Platelet-mediated transformation of mtDNA-less human cells: Analysis of phenotypic variability among clones from normal individuals—And complementation behavior of the tRNALys mutation causing myoclonic epilepsy and ragged red fibers. Am. J. Hum. Genet. 1994, 54, 966–974. [Google Scholar] [PubMed]
- Gallardo, M.E.; Moreno-Loshuertos, R.; Lopez, C.; Casqueiro, M.; Silva, J.; Bonilla, F.; Rodriguez de Cordoba, S.; Enriquez, J.A. m.6267G>A: A recurrent mutation in the human mitochondrial DNA that reduces cytochrome c oxidase activity and is associated with tumors. Hum. Mutat. 2006, 27, 575–582. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Bafaluy, M.P.; Acin-Perez, R.; Mullikin, J.C.; Park, J.S.; Moreno-Loshuertos, R.; Hu, P.; Perez-Martos, A.; Fernandez-Silva, P.; Bai, Y.; Enriquez, J.A. Revisiting the mouse mitochondrial DNA sequence. Nucleic Acids Res. 2003, 31, 5349–5355. [Google Scholar] [CrossRef] [Green Version]
- Hofhaus, G.; Shakeley, R.M.; Attardi, G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996, 264, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Yang, L.; Manfredi, G. Assay of mitochondrial ATP synthesis in animal cells and tissues. Methods Cell Biol. 2007, 80, 155–171. [Google Scholar] [CrossRef]
- Schagger, H. Native electrophoresis for isolation of mitochondrial oxidative phosphorylation protein complexes. Methods Enzymol. 1995, 260, 190–202. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Fernandez-Silva, P.; Peleato, M.L.; Perez-Martos, A.; Enriquez, J.A. Respiratory active mitochondrial supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Perales-Clemente, E.; Fernandez-Vizarra, E.; Acin-Perez, R.; Movilla, N.; Bayona-Bafaluy, M.P.; Moreno-Loshuertos, R.; Perez-Martos, A.; Fernandez-Silva, P.; Enriquez, J.A. Five entry points of the mitochondrially encoded subunits in mammalian complex I assembly. Mol. Cell. Biol. 2010, 30, 3038–3047. [Google Scholar] [CrossRef] [Green Version]
- Van Coster, R.; Smet, J.; George, E.; De Meirleir, L.; Seneca, S.; Van Hove, J.; Sebire, G.; Verhelst, H.; De Bleecker, J.; Van Vlem, B.; et al. Blue native polyacrylamide gel electrophoresis: A powerful tool in diagnosis of oxidative phosphorylation defects. Pediatr. Res. 2001, 50, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Shin, Y.; Han, S.; Jeon, J.S.; Yamamoto, K.; Zervantonakis, I.K.; Sudo, R.; Kamm, R.D.; Chung, S. Microfluidic assay for simultaneous culture of multiple cell types on surfaces or within hydrogels. Nat. Protoc. 2012, 7, 1247–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Movilla, N.; Borau, C.; Valero, C.; Garcia-Aznar, J.M. Degradation of extracellular matrix regulates osteoblast migration: A microfluidic-based study. Bone 2018, 107, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Arotzena, O.; Borau, C.; Movilla, N.; Vicente-Manzanares, M.; Garcia-Aznar, J.M. Fibroblast Migration in 3D is Controlled by Haptotaxis in a Non-muscle Myosin II-Dependent Manner. Ann. Biomed. Eng. 2015, 43, 3025–3039. [Google Scholar] [CrossRef] [Green Version]
- De Lano, W.L. PyMOL: An open-source molecular graphics tool. CCP4 725 Newsl. Protein Crystallogr. 2002, 40, 82–92. [Google Scholar]
- Ashkenazy, H.; Abadi, S.; Martz, E.; Chay, O.; Mayrose, I.; Pupko, T.; Ben-Tal, N. ConSurf 2016: An improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res. 2016, 44, W344–W350. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moreno-Loshuertos, R.; Movilla, N.; Marco-Brualla, J.; Soler-Agesta, R.; Ferreira, P.; Enríquez, J.A.; Fernández-Silva, P. A Mutation in Mouse MT-ATP6 Gene Induces Respiration Defects and Opposed Effects on the Cell Tumorigenic Phenotype. Int. J. Mol. Sci. 2023, 24, 1300. https://doi.org/10.3390/ijms24021300
Moreno-Loshuertos R, Movilla N, Marco-Brualla J, Soler-Agesta R, Ferreira P, Enríquez JA, Fernández-Silva P. A Mutation in Mouse MT-ATP6 Gene Induces Respiration Defects and Opposed Effects on the Cell Tumorigenic Phenotype. International Journal of Molecular Sciences. 2023; 24(2):1300. https://doi.org/10.3390/ijms24021300
Chicago/Turabian StyleMoreno-Loshuertos, Raquel, Nieves Movilla, Joaquín Marco-Brualla, Ruth Soler-Agesta, Patricia Ferreira, José Antonio Enríquez, and Patricio Fernández-Silva. 2023. "A Mutation in Mouse MT-ATP6 Gene Induces Respiration Defects and Opposed Effects on the Cell Tumorigenic Phenotype" International Journal of Molecular Sciences 24, no. 2: 1300. https://doi.org/10.3390/ijms24021300