
Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers

 ,
, 
Abstract
:1. Introduction
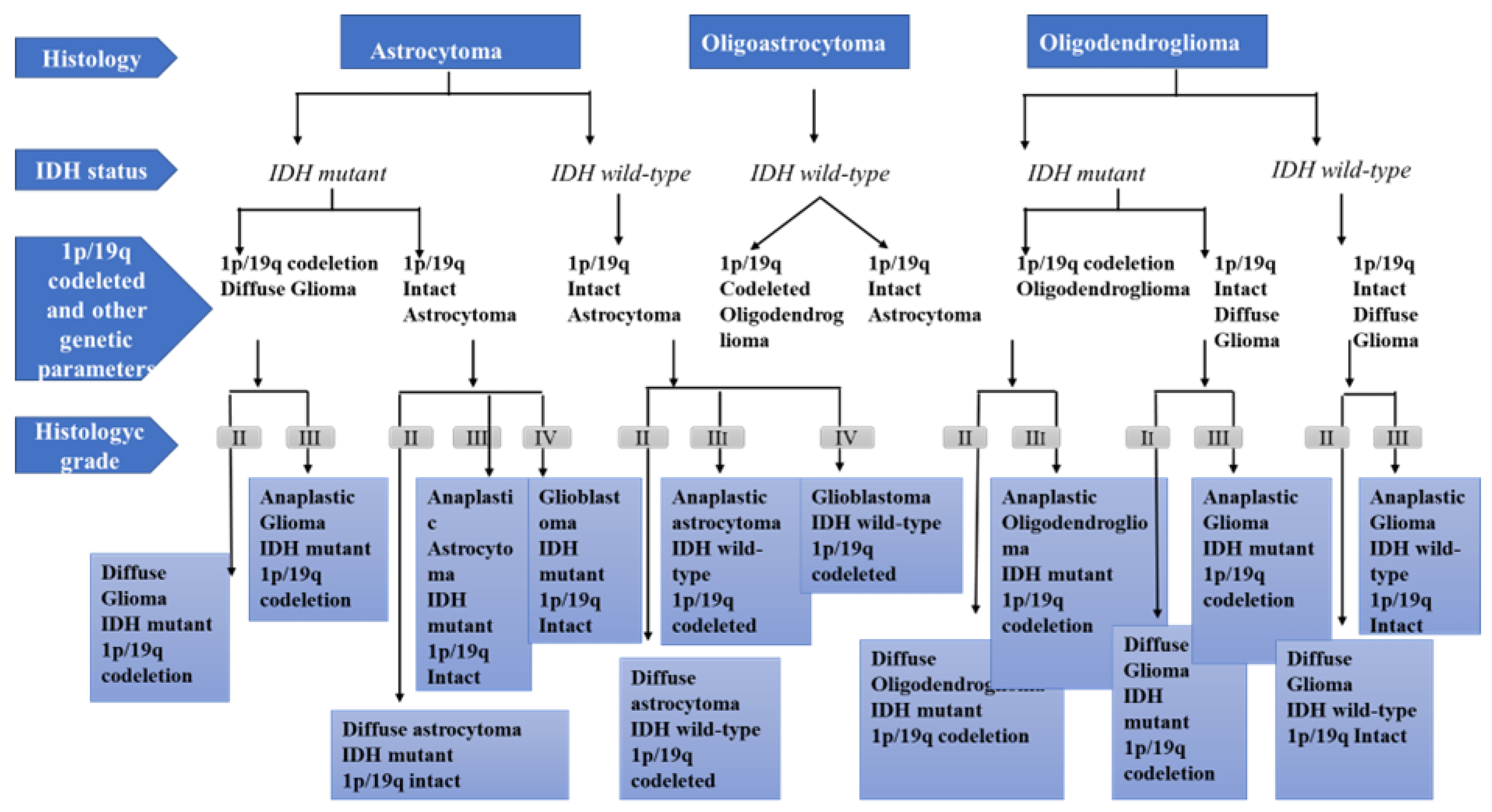
2. Physiopathology of Glioma Development
- Grade I gliomas are low-grade benign lesions, such as pilocytic astrocytomas, that have limited proliferation and are frequent in children; however, they might acquire higher levels of malignancy. Surgical resection is a good therapeutic option in most cases.
- Grade II gliomas have low-grade but infiltrative lesions that tend to show higher recurrence after surgical resection.
- Grade III gliomas have intermediate- to high-grade lesions with atypia, higher mitotic activity, and evidence of malignancy. Patients with these tumors usually are prescribed additional radiation as well as chemotherapy.
- Grade IV tumors are high-grade malignant lesions with higher mitotic activity, microvascular proliferation, high necrosis, and the worst prognosis.
3. Mebendazole: Chemical Structure and Pharmacokinetic Properties
4. Preclinical Evidence of Mebendazole in Brain Cancer—In Vitro and In Vivo
5. Clinical Evidence of Antitumoral Properties of Mebendazole
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xia, C.; Dong, X.; Li, H.; Cao, M.; Sun, D.; He, S.; Yang, F.; Yan, X.; Zhang, S.; Li, N.; et al. Cancer statistics in China and United States, 2022: Profiles, trends, and determinants. Chin. Med. J. 2022, 9, 584–590. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Global Health Estimates 2020: Deaths by Cause, Age, Sex, by Country and by Region, 2000–2019. Available online: https://www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-leading-causes-of-death (accessed on 11 December 2020).
- Sertkaya, A.; Wong, H.H.; Jessup, A.; Beleche, T. Key cost drivers of pharmaceutical clinical trials in the United States. Clin. Trials. 2016, 13, 117–126. [Google Scholar] [CrossRef]
- Yeu, Y.; Yoon, Y.; Park, S. Protein localization vector propagation: A method for improving the accuracy of drug repositioning. Mol. Biosyst. 2015, 11, 2096–2102. [Google Scholar] [CrossRef] [PubMed]
- Pammolli, F.; Magazzini, L.; Riccaboni, M. The productivity crisis in pharmaceutical R&D. Nat. Rev. Drug Discov. 2011, 10, 428–438. [Google Scholar]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug. Discov. 2012, 11, 191–200. [Google Scholar]
- Collier, R. Rapidly rising clinical trial costs worry researchers. CMAJ 2009, 180, 277–278. [Google Scholar] [CrossRef] [Green Version]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Rev. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug repositioning: Identifying and developing new uses for existing drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef]
- Wurth, R.; Thellung, S.; Bajetto, A.; Mazzanti, M.; Florio, T.; Barbieri, F. Drug-repositioning opportunities for cancer therapy: Novel molecular targets for known compounds. Drug Discov. Today 2016, 21, 190–199. [Google Scholar] [CrossRef]
- Schcolnik-Cabrera, A.; Juárez-López, D.; Duenas-Gonzalez, A. Perspectives on Drug Repurposing. Curr. Med. Chem. 2021, 28, 2085–2099. [Google Scholar] [CrossRef]
- Smith, R.B. Repositioned drugs: Integrating intellectual property and regulatory strategies. Drug Discov. Today Ther. Strateg. 2011, 8, 131–137. [Google Scholar] [CrossRef]
- Breckenridge, A.; Jacob, R. Overcoming the legal and regulatory barriers to drug repurposing. Nat. Rev. Drug Discov. 2018, 18, 1–2. [Google Scholar] [CrossRef]
- Nosengo, N. Can you teach old drugs new tricks? Nature 2016, 534, 314–316. [Google Scholar] [CrossRef] [Green Version]
- Bertolini, F.; Sukhatme, V.P.; Bouche, G. Drug repurposing in oncology--patient and health systems opportunities. Nat. Rev. Clin. Oncol. 2015, 12, 732–742. [Google Scholar] [CrossRef]
- Munj, S.A.; Taz, T.A.; Arslanturk, S.; Heath, E.I. Biomarker-driven drug repurposing on biologically similar cancers with DNA-repair deficiencies. Front. Genet. 2022, 13, 1015531. [Google Scholar] [CrossRef]
- Kato, S.; Moulder, S.L.; Ueno, N.T.; Wheler, J.J.; Meric-Bernstam, F.; Kurzrock, R.; Janku, F. Challenges and perspective of drug repurposing strategies in early phase clinical trials. Oncoscience 2015, 2, 576–580. [Google Scholar] [CrossRef]
- Gupta, S.C.; Sung, B.; Prasad, S.; Webb, L.J.; Aggarwal, B.B. Cancer drug discovery by repurposing: Teaching new tricks to old dogs. Trends Pharmacol. Sci. 2013, 34, 508–517. [Google Scholar] [CrossRef]
- Bloom, B.E. The trials and tribulations of repurposing metformin and other generic drugs for tuberculosis. Pharm. Pat. Anal. 2016, 5, 101–105. [Google Scholar] [CrossRef]
- Bloom, B.E. Creating new economic incentives for repurposing generic drugs for unsolved diseases using social finance. ASSAY Drug Dev. Technol. 2015, 13, 606–611. [Google Scholar] [CrossRef]
- Verbaanderd, C.; Rooman, I.; Meheus, L.; Huys, I. On-label or off-label? Overcoming regulatory and financial barriers to bring repurposed medicines to cancer patients. Front. Pharmacol. 2020, 10, 1664. [Google Scholar] [CrossRef] [Green Version]
- Chai, J.Y.; Jung, B.K.; Hong, S.J. Albendazole and Mebendazole as Antiparasitic and Anticancer Agents: An Update. Korean J. Parasitol. 2021, 59, 189–225. [Google Scholar] [CrossRef]
- Guerini, A.E.; Triggiani, L.; Maddalo, M.; Bonù, M.L.; Frassine, F.; Baiguini, A.; Alghisi, A.; Tomasini, D.; Borghetti, P.; Pasinetti, N.; et al. Mebendazole as a candidate for drug repurposing in oncology: An extensive review of current literature. Cancers 2019, 11, 1284. [Google Scholar] [CrossRef]
- Son, D.S.; Lee, E.S.; Adunyah, S.E. The antitumor potentials of benzimidazole anthelmintics as repurposing drugs. Immun. Netw. 2020, 20, 29. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K. World Health Organization Classification of Tumours of the Central Nervous System, 4th ed.; International Agency for Research on Cancer: Lyon, France, 2016.
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro-Oncol. 2021, 2, 1231–1251. [Google Scholar] [CrossRef]
- Hartmann, C.; Hentschel, B.; Wick, W.; Capper, D.; Felsberg, J.; Simon, M.; Westphal, M.; Schackert, G.; Meyermann, R.; Pietsch, T.; et al. Patients with IDH1 wild-type anaplastic astrocytomas exhibit worse prognosis than IDH1-mutated glioblastomas, and IDH1 mutation status accounts for the unfavorable prognostic effect of higher age: Implications for classification of gliomas. Acta Neuropathol. 2010, 120, 707–718. [Google Scholar] [CrossRef] [Green Version]
- Giannini, C.; Burger, P.C.; Berkey, B.A.; Cairncross, J.G.; Jenkins, R.B.; Mehta, M.; Curran, W.J.; Aldape, K. Anaplastic oligodendroglial tumors: Refining the correlation among histopathology, 1p 19q deletion and clinical outcome in Intergroup Radiation Therapy Oncology Group Trial 9402. Brain Pathol. 2008, 18, 360–369. [Google Scholar] [CrossRef]
- Perez-Larraya, J.G.; Ducray, F.; Chinot, O.; Catry-Thomas, I.; Taillandier, L.; Guillamo, J.-S.; Campello, C.; Monjour, A.; Cartalat-Carel, S.; Barrie, M.; et al. Temozolomide in elderly patients with newly diagnosed glioblastoma and poor performance status: An ANOCEF phase II trial. J. Clin. Oncol. 2011, 29, 3050–3055. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Burger, P.; Ellison, D.W.; Reifenberger, G.; von Deimling, A.; Aldape, K.; Brat, D.; Collins, V.P.; Eberhart, C.; et al. International Society of Neuropathology-Haarlem consensus guidelines for nervous system tumor classification and grading. Brain Pathol. 2014, 24, 429–435. [Google Scholar] [CrossRef]
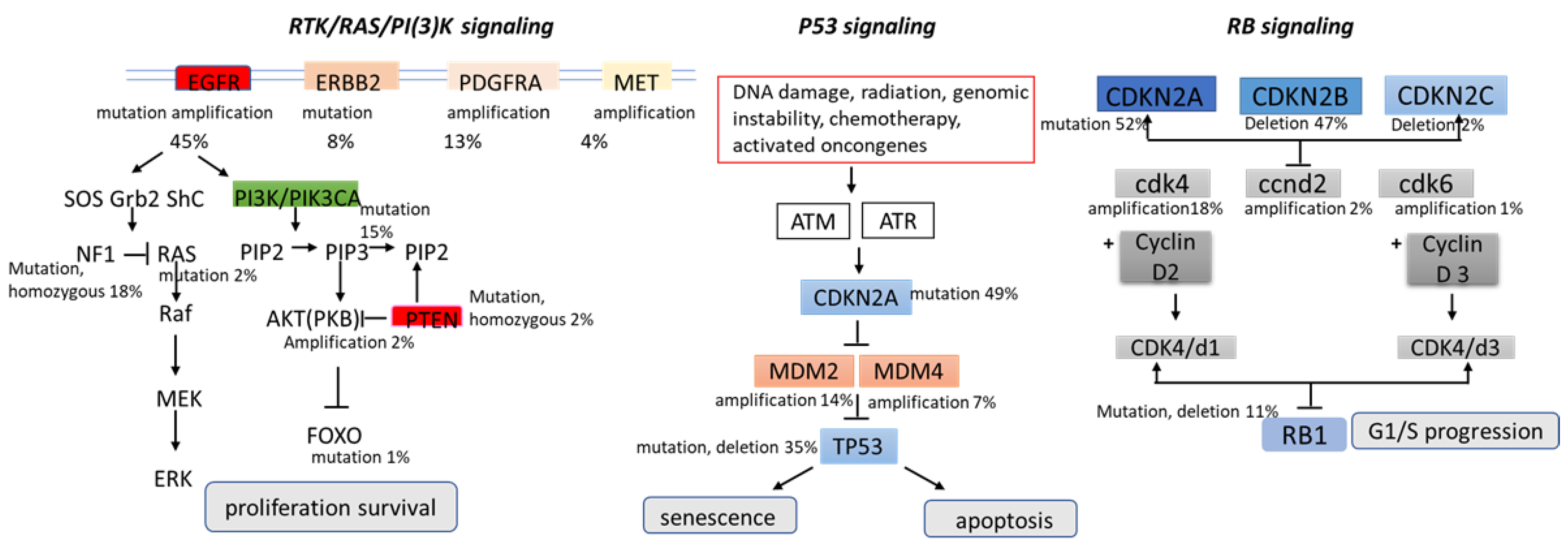
- Wang, H.; Xu, T.; Jiang, Y.; Xu, H.; Yan, Y.; Fu, D.; Chen, J. The challenges and the promise of molecular targeted therapy in malignant gliomas. Neoplasia 2015, 17, 239–255. [Google Scholar] [CrossRef] [Green Version]
- Hatanpa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Nazarenko, I.; Hede, S.M.; He, X.; Hedren, A.; Thompson, J.; Lindstrom, M.S.; Nister, M. PDGF and PDGF receptors in glioma. Upsala J. Med Sci. 2012, 117, 99–112. [Google Scholar] [CrossRef]
- Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 12, 1831. [Google Scholar] [CrossRef]
- Yip, S.; Iafrate, A.J.; Louis, D.N. Molecular diagnostic testing in malignant gliomas: A practical update on predictive markers. J. Neuropathol. Exp. Neurol. 2008, 67, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Sugawa, N.; Ekstrand, A.J.; James, C.D.; Collins, V.P. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc. Natl. Acad. Sci. USA 1990, 87, 8602–8606. [Google Scholar] [CrossRef] [Green Version]
- Mao, H.; LeBrun, D.; Yang, J.; Zhu, V.F.; Li, M. Deregulated signaling pathways in glioblastoma multiforme: Molecular mechanisms and therapeutic targets. Cancer Investig. 2012, 30, 48–56. [Google Scholar] [CrossRef] [Green Version]
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef]
- Karsy, M.; Neil, J.A.; Guan, J.; Mahan, M.A.; Colman, H.; Jensen, R.L. A practical review of prognostic correlations of molecular biomarkers in glioblastoma. Neurosurg. Focus. 2015, 38, E4. [Google Scholar] [CrossRef] [Green Version]
- Huse, J.T.; Holland, E.C. Targeting brain cancer: Advances in the molecular pathology of malignant glioma and medulloblastoma. Nat. Rev. Cancer 2010, 10, 19–331. [Google Scholar] [CrossRef]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462–477. [Google Scholar] [CrossRef]
- Ohgaki, H.; Kleihues, P. Genetic pathways to primary and secondary glioblastoma. Am. J. Pathol. 2007, 170, 1445–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Dick, F.A.; Rubin, S.M. Molecular mechanisms underlying RB protein function. Nat. Rev. Mol. Cell Biol. 2013, 14, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Brugmans, J.P.; Theinpont, D.C.; Wijngaarden, I.V.; Vanparijs, O.F.; Schuermans, V.L.; Lauwers, H.L. Mebendazole in enterobiasis. Radiochemical and pilot clinical study in 1278 subjects. JAMA 1971, 217, 313–316. [Google Scholar] [CrossRef]
- Dawson, M.; Braithwaite, P.A.; Roberts, M.A.; Watson, T.R. The pharmacokinetics and bioavailability of a tracer dose of (3H)-mebendazole in man. Br. J. Clin. Pharmacol. 1985, 19, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Witassek, F.; Burkhardt, B.; Eckhardt, J.; Bircher, J. Chemotherapy of alveolar echinococcosis; comparison of plasma Mebendazole concentrations in animals and man. Eur. J. Clin. Pharmacol. 1981, 20, 427–433. [Google Scholar] [CrossRef]
- Dollery, C.T. Mebendazole. In Therapeutic Drugs, 2nd ed.; Churchill Livingstone: Edinburgh, UK, 1999; Volume 2, pp. M12–M15. [Google Scholar]
- EMEA-European Medicines Evaluation Agency. Mebendazole Summary Report by CVMP; EMEA/MRL/625/99-Final; EMEA: London, UK, 1999. [Google Scholar]
- Edwards, G.; Brechenridge, A.M. Clinical pharmacokinetics of anthelmintic drugs. Clin. Pharmacokinet. 1988, 15, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Pawluk, S.A.; Roles, C.A.; Wilby, K.J.; Ensom, M.H. A review of pharmacokinetic drug-drug interactions with the anthelmintic medications albendazole and mebendazole. Clin. Pharmacokinet. 2015, 54, 371–383. [Google Scholar] [CrossRef]
- Dayan, A.D. Albendazole, mebendazole and praziquantel. Review of non-clinical toxicity and pharmacokinetics. Acta Trop. 2003, 86, 141–159. [Google Scholar] [CrossRef]
- Hamilton, G.; Rath, B. Repurposing of anthelminthics as anticancer drugs. Oncomedicine 2017, 2, 142–149. [Google Scholar] [CrossRef]
- Braithwaite, P.A.; Roberts, M.S.; Allan, R.J.; Watson, T.R. Clinical pharmacokinetics of high dose mebendazole in patients treated for cystic hydatid disease. Eur. J. Clin. Pharmacol. 1982, 22, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Corti, N.; Heck, A.; Rentsch, K.; Zingg, W.; Jetter, A.; Stieger, B.; Pauli-Magnus, C. Effect of ritonavir on the pharmacokinetics of the benzimidazoles albendazole and Mebendazole: An interaction study in healthy volunteers. Eur. J. Clin. Pharmacol. 2009, 65, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Bekhti, A.; Pirotte, J. Cimetidine increases serum mebendazole concentrations. Implications for treatment of hepatic hydatid cysts. Br. J. Clin. Pharmacol. 1987, 24, 390–392. [Google Scholar] [PubMed] [Green Version]
- Luder, P.J.; Siffert, B.; Witassek, F.; Meister, F.; Bircher, J. Treatment of hydatid disease with high oral doses of Mebendazole. Eur. J. Clin. Pharmacol. 1986, 31, 443–448. [Google Scholar] [CrossRef]
- Liu, C.-S.; Zhang, H.-B.; Jiang, B.; Yao, J.-M.; Tao, Y.; Xue, J.; Wen, A.-D. Enhanced bioavailability and cysticidal effect of three mebendazole-oil preparations in mice infected with secondary cysts of Echinococcus granulosus. Parasitol. Res. 2012, 111, 1205–1211. [Google Scholar] [CrossRef]
- Pantziarka, P.; Bouche, G.; Meheus, L.; Sukhatme, V.; Sukhatme, V.P. Repurposing Drugs in Oncology (ReDO)-mebendazole as an anti-cancer agent. Ecancermedicalscience 2014, 8, 443. [Google Scholar] [CrossRef]
- Reuter, S.; Jensen, B.; Buttenschoen, K.; Kratzer, W.; Kern, P. Benzimidazoles in the treatment of alveolar echinococcosis: A comparative study and review of theliterature. J. Antimicrob. Chemother. 2000, 46, 451–456. [Google Scholar] [CrossRef] [Green Version]
- Yangco, B.G.; Klein, T.W.; Deresinski, S.C.; Vickery, A.C.; Craig, C.P. Flubendazole andmebendazole in the treatment of trichuriasis and other helminthiases. Clin. Ther. 1981, 4, 285–290. [Google Scholar]
- Tolomeo, M.; Colomba, C.; Meli, M.; Cascio, A. Hepatotoxicity caused by Mebendazole in a patient with Gilbert’s syndrome. J. Clin. Pharm. Ther. 2019, 44, 985–987. [Google Scholar] [CrossRef]
- Karra, N.; Cohen, R.; Berlin, M.; Dinavitser, N.; Koren, G.; Berkovitch, M. Safety of mebendazole use during lactation: A case series report. Drugs R D 2016, 16, 251–254. [Google Scholar] [CrossRef] [Green Version]
- De Silva, N.; Guyatt, H.; Bundy, D. Anthelmintics. A comparative review of their clinical pharmacology. Drugs 1997, 53, 769–788. [Google Scholar] [CrossRef] [PubMed]
- Göçmen, A.; Toppare, M.F.; Kiper, N. Treatment of hydatid disease in childhood with Mebendazole. Eur. Respir. J. 1993, 6, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.T.; Twu, S.J.; Chang, H.J.; Lin, R.S. Outbreak of Stevens-Johnson syndrome/toxic epidermal necrolysis associated with mebendazole and metronidazole use among Filipino laborers in Taiwan. Am. J. Public Health 2003, 93, 489–492. [Google Scholar] [CrossRef]
- Alavi, S.E.; Ebrahimi Shahmabadi, H. Anthelmintics for drug repurposing: Opportunities and challenges. Saudi Pharm. J. 2021, 29, 434–445. [Google Scholar] [CrossRef]
- De la Torre-Iglesias, P.M.; García-Rodriguez, J.J.; Torrado, G.; Torrado, S.; Torrado-Santiago, S.; Bolás-Fernández, F. Enhanced bioavailability and anthelmintic efficacy of Mebendazole in redispersible microparticles with low-substituted hydroxypropylcellulose. Drug Des. Dev. Ther. 2014, 18, 1467–1479. [Google Scholar]
- Münst, G.J.; Karlaganis, G.; Bircher, J. Plasma concentrations of Mebendazole during treatment of echinococcosis. Eur. J. Clin. Pharmacol. 1980, 17, 375–378. [Google Scholar] [CrossRef]
- Rodriguez-Caabeiro, F.; Criado-Fornelio, A.; Jimenez-Gonzalez, A.; Guzman, L.; Igual, A.; Perez, A.; Pujol, M. Experimental chemotherapy and toxicity in mice of three Mebendazole polymorphic forms. Chemotherapy 1987, 33, 266–271. [Google Scholar] [CrossRef]
- Swanepoel, E.; Liebenberg, W.; de Villiers, M.M. Quality evaluation of generic drugs by dissolution test: Changing the USP dissolution medium to distinguish between active and non-active Mebendazole polymorphs. Eur. J. Pharm. Biopharm. 2003, 55, 345–349. [Google Scholar] [CrossRef]
- Charoenlarp, P.; Waikagul, J.; Muennoo, C.; Srinophakun, S.; Kitayaporn, D. Efficacy of single-dose Mebendazole, polymorphic forms A and C, in the treatment of hookworm and Trichuris infections. Southeast Asian J. Trop. Med. Public Health 1993, 24, 712–716. [Google Scholar] [PubMed]
- Bai, R.Y.; Staedtke, V.; Wanjiku, T.; Rudek, M.A.; Joshi, A.; Gallia, G.L.; Riggins, G.J. Brain Penetration and Efficacy of Different Mebendazole Polymorphs in a Mouse Brain Tumor Model. Clin. Cancer Res. 2015, 21, 3462–3470. [Google Scholar] [CrossRef] [Green Version]
- Armando, R.G.; Mengual Gómez, D.L.; Gomez, D.E. New drugs are not enough drug repositioning in oncology: An update. Int. J. Oncol. 2020, 56, 651–684. [Google Scholar] [CrossRef] [Green Version]
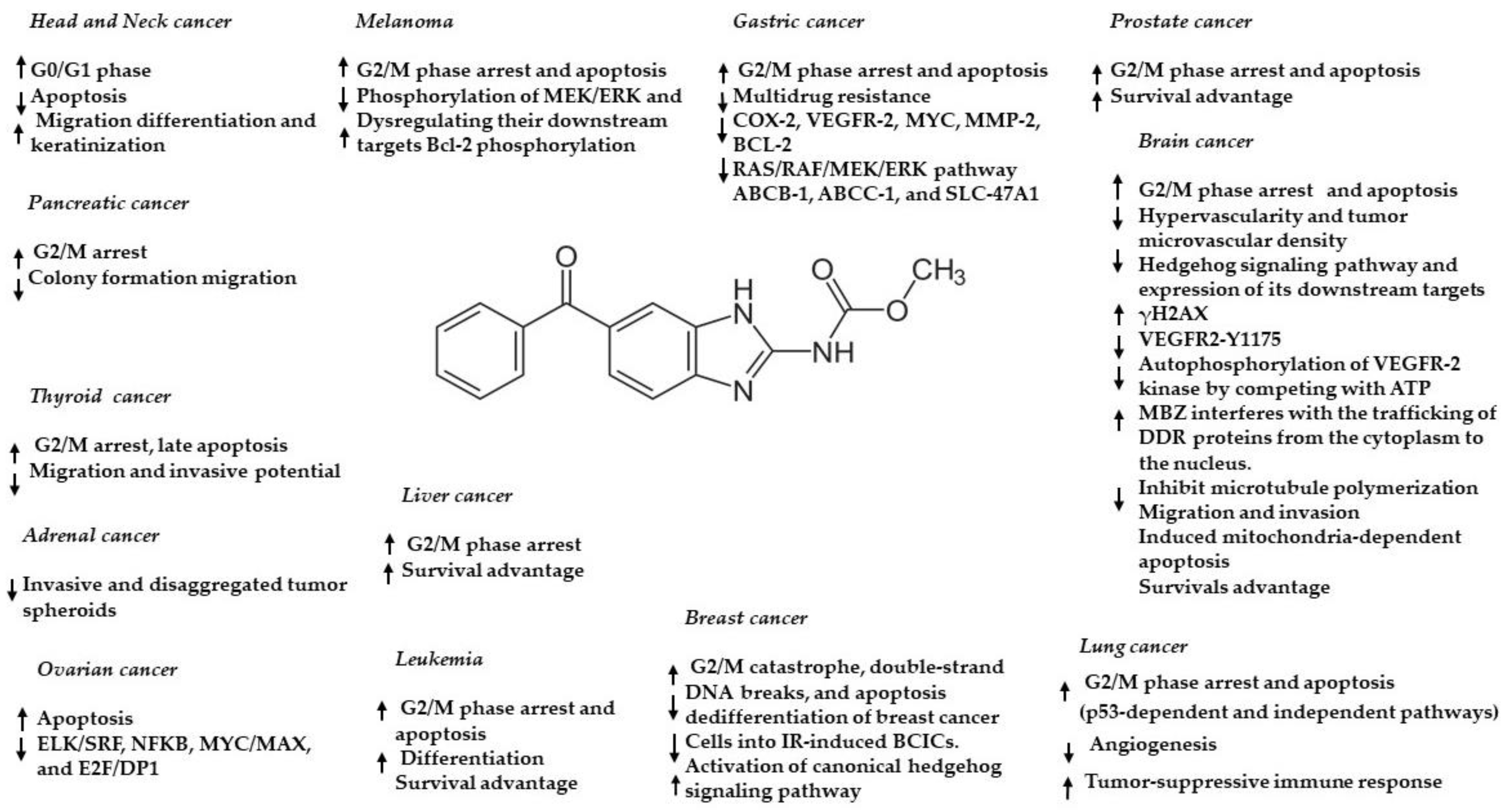
- Nath, J.; Paul, R.; Ghosh, S.K.; Paul, J.; Singha, B.; Debnath, N. Drug repurposing andrelabeling for cancer therapy: Emerging benzimidazole antihelminthics with potent anticancer effects. Life Sci. 2020, 1, 118189. [Google Scholar] [CrossRef] [PubMed]
- Williamson, T.; Mendes, T.B.; Joe, N.; Cerutti, J.M.; Riggins, G.J. Mebendazole inhibits tumor growth and prevents lung metastasis in models of advanced thyroid cancer. Endocr. Relat. Cancer 2020, 27, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Mansoori, S.; Fryknäs, M.; Alvfors, C.; Loskog, A.; Larsson, R.; Nygren, P. A phase 2a clinical study on the safety and efficacy of individualized dosed Mebendazole in patients with advanced gastrointestinal cancer. Sci. Rep. 2021, 26, 8981. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Ko, Y.S.; Jin, H.; Kang, K.M.; Ha, I.B.; Jeong, H.; Song, H.N.; Kim, H.J.; Jeong, B.K. Anticancer Effect of Benzimidazole Derivatives, Especially Mebendazole, on Triple-Negative Breast Cancer (TNBC) and Radiotherapy-Resistant TNBC In Vivo and In Vitro. Molecules 2021, 24, 5118. [Google Scholar] [CrossRef] [PubMed]
- Rushworth, L.K.; Hewit, K.; Munnings-Tomes, S.; Somani, S.; James, D.; Shanks, E.; Dufès, C.; Straube, A.; Patel, R.; Leung, H.Y. Repurposing screen identifies Mebendazole as a clinical candidate to synergise with docetaxel for prostate cancer treatment. Br. J. Cancer 2020, 122, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Williamson, T.; de Abreu, M.C.; Trembath, D.G.; Brayton, C.; Kang, B.; Mendes, T.B.; de Assumpção, P.P.; Cerutti, J.M.; Riggins, G.J. Mebendazole disrupts stromal desmoplasia and tumorigenesis in two models of pancreatic cancer. Oncotarget 2021, 6, 1326–1338. [Google Scholar] [CrossRef]
- Elayapillai, S.; Ramraj, S.; Benbrook, D.M.; Bieniasz, M.; Wang, L.; Pathuri, G.; Isingizwe, Z.R.; Kennedy, A.L.; Zhao, Y.D.; Lightfoot, S.; et al. Potential and mechanism of Mebendazole for treatment and maintenance of ovarian cancer. Gynecol. Oncol. 2021, 160, 302–311. [Google Scholar] [CrossRef]
- Hegazy, S.K.; El-Azab, G.A.; Zakaria, F.; Mostafa, M.F.; El-Ghoneimy, R.A. Mebendazole; from an antiparasitic drug to a promising candidate for drug repurposing in colorectal cancer. Life Sci. 2022, 15, 120536. [Google Scholar] [CrossRef] [PubMed]
- Petersen, J.S.S.M.; Baird, S.K. Treatment of breast and colon cancer cell lines with anti-helmintic benzimidazoles mebendazole or albendazole results in selective apoptotic cell death. J. Cancer Res. Clin. Oncol. 2021, 147, 2945–2953. [Google Scholar] [CrossRef]
- Simbulan-Rosenthal, C.M.; Dakshanamurthy, S.; Gaur, A.; Chen, Y.S.; Fang, H.B.; Abdussamad, M.; Zhou, H.; Zapas, J.; Calvert, V.; Petricoin, E.F.; et al. The repurposed anthelmintic Mebendazole in combination with trametinib suppresses refractory NRASQ61K melanoma. Oncotarget 2017, 21, 12576–12595. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Li, Y.; Zhang, H.; Huang, E.; Gao, L.; Luo, W.; Wei, Q.; Fan, J.; Song, D.; Liao, J.; et al. Anthelmintic mebendazole enhances cisplatin’s effect on suppressing cell proliferation and promotes differentiation of head and neck squamous cell carcinoma (HNSCC). Oncotarget 2017, 21, 12968–12982. [Google Scholar] [CrossRef] [Green Version]
- Freisleben, F.; Modemann, F.; Muschhammer, J.; Stamm, H.; Brauneck, F.; Krispien, A.; Bokemeyer, C.; Kirschner, K.N.; Wellbrock, J.; Fiedler, W. Mebendazole Mediates Proteasomal Degradation of GLI Transcription Factors in Acute Myeloid Leukemia. Int. J. Mol. Sci. 2021, 22, 10670. [Google Scholar] [CrossRef] [PubMed]
- Sawanyawisuth, K.; Williamson, T.; Wongkham, S.; Riggins, G.J. Effect of the antiparasitic drug Menbendaziole on cholangiocarcinoma growth. Southeast Asian J. Trop. Med. Public Health 2014, 45, 1264–1270. [Google Scholar]
- Bai, R.Y.; Staedtke, V.; Aprhys, C.M.; Gallia, G.L.; Riggins, G.J. Antiparasitic Mebendazole shows survival benefit in 2 preclinical models of glioblastoma multiforme. Neuro-Oncol. 2011, 13, 974–982. [Google Scholar] [CrossRef] [Green Version]
- Ren, L.W.; Li, W.; Zheng, X.J.; Liu, J.Y.; Yang, Y.H.; Li, S.; Zhang, S.; Fu, W.Q.; Xiao, B.; Wang, J.H.; et al. Author Correction: Benzimidazoles induce concurrent apoptosis and pyroptosis of human glioblastoma cells via arresting cell cycle. Acta Pharmacol. Sin. 2022, 15, 194–208. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef] [PubMed]
- De Witt, M.; Gamble, A.; Hanson, D.; Markowitz, D.; Powell, C.; Al Dimassi, S.; Atlas, M.; Boockvar, J.; Ruggieri, R.; Symons, M. Repurposing mebendazole as a replacement for vincristine for the treatment of brain tumors. Mol. Med. 2017, 23, 50–56. [Google Scholar] [CrossRef] [Green Version]
- Dakshanamurthy, S.; Issa, N.T.; Assefnia, S.; Seshasayee, A.; Peters, O.J.; Madhavan, S.; Uren, A.; Brown, M.L.; Byers, S.W. Predicting new indications for approved drugs using a proteochemometric method. J. Med. Chem. 2012, 55, 6832–6848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahir, B.K.; Engelhard, H.H.; Lakka, S.S. Tumor Development and Angiogenesis in Adult Brain Tumor: Glioblastoma. Mol. Neurobiol. 2020, 57, 2461–2478. [Google Scholar] [CrossRef] [Green Version]
- Bai, R.Y.; Staedtke, V.; Rudin, C.M.; Bunz, F.; Riggins, G.J. Effective treatment of diverse medulloblastoma models with Mebendazole and its impact on tumor angiogenesis. Neuro-Oncol. 2015, 17, 545–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. Basic Med. Sci. 2018, 20, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, A.R.; Bai, R.-Y.; Chung, J.H.; Borodovsky, A.; Rudin, C.M.; Riggins, G.J.; Bunz, F. Repurposing the antihelmintic Mebendazole as a hedgehog inhibitor. Mol. Cancer Ther. 2015, 14, 3–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodhinayake, I.; Symons, M.; Boockvar, J.A. Repurposing mebendazole for the treatment of medulloblastoma. Neurosurgery 2015, 76, N15–N16. [Google Scholar] [CrossRef] [Green Version]
- Skibinski, C.G.; Williamson, T.; Riggins, G.J. Mebendazole and radiation in combination increase survival through anticancer mechanisms in an intracranial rodent model of malignant meningioma. J. Neurooncol. 2018, 140, 529–538. [Google Scholar] [CrossRef]
- Markowitz, D.; Ha, G.; Ruggieri, R.; Symons, M. Microtubule-targeting agents can sensitize cancer cells to ionizing radiation by an interphase-based mechanism. Onco Targets Ther. 2017, 24, 5633–5642. [Google Scholar] [CrossRef] [Green Version]
- Kipper, F.C.; Silva, A.O.; Marc, A.L.; Confortin, G.; Junqueira, A.V.; Neto, E.P.; Lenz, G. Vinblastine and antihelmintic Mebendazole potentiate temozolomide in resistant gliomas. Investig. New Drugs 2018, 36, 323–331. [Google Scholar] [CrossRef]
- Ariey-Bonnet, J.; Carrasco, K.; Le Grand, M.; Hoffer, L.; Betzi, S.; Feracci, M.; Tsvetkov, P.; Devred, F.; Collette, Y.; Morelli, X.; et al. In silico molecular target prediction unveils mebendazole as a potent MAPK14 inhibitor. Mol. Oncol. 2020, 14, 3083–3099. [Google Scholar] [CrossRef]
- Demuth, T.; Reavie, L.B.; Rennert, J.L.; Nakada, M.; Nakada, S.; Hoelzinger, D.B.; Beaudry, C.E.; Henrichs, A.N.; Anderson, E.M.; Berens, M.E. MAP-ing glioma invasion: Mitogen-activated protein kinase kinase 3 and p38 drive glioma invasion and progression and predict patient survival. Mol. Cancer Ther. 2007, 6, 1212–1222. [Google Scholar] [CrossRef] [Green Version]
- Ben-Hamo, R.; Efroni, S. Gene expression and network-based analysis reveals a novel role for has-miR-9 and drug control over the p38 network in glioblastoma multiforme progression. Genome Med. 2011, 3, 77. [Google Scholar] [CrossRef] [Green Version]
- Dobrosotskaya, I.Y.; Hammer, G.D.; Schteingart, D.E.; Maturen, K.E.; Worden, F.P. Mebendazole monotherapy and long-term disease control in metastatic adrenocortical carcinoma. Endocr. Pract. 2011, 17, 59–62. [Google Scholar] [CrossRef]
- Nygren, P.; Larsson, R. Drug repositioning from bench to bedside: Tumour remission by the antihelmintic drug mebendazole in refractory metastatic colon cancer. Acta Oncol. 2014, 53, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Trisciuzzi, M.T.S.; Riccardi, R.; Piccardi, M.; Iarossi, G.; Buzzonetti, L.; Dickmann, A.; Colosimo, C.; Ruggiero, A.; Di Rocco, C.; Falsini, B. A fast visual evoked potential method for functional assessment and follow-up of childhood optic gliomas. Clin. Neurophysiol. 2004, 115, 217–226. [Google Scholar] [CrossRef]
- Ruggiero, A.; Rizzo, D.; Trombatore, G.; Maurizi, P.; Riccardi, R. The ability of mannitol to decrease cisplatin-induced nephrotoxicity in children: Real or not? Cancer Chemother. Pharmacol. 2016, 77, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.; Brown, J.; Brown, K.; Gregor, A.; Whittle, I.; Grant, R. Practical problems with the collection and interpretation of serial quality of life assessments in patients with malignant glioma. J. Neurooncol. 2003, 63, 179–186. [Google Scholar] [CrossRef]
- Ruggiero, A.; Rizzo, D.; Catalano, M.; Coccia, P.; Triarico, S.; Attiná, G. Acute chemotherapy-induced nausea and vomiting in children with cancer: Still waiting for a common consensus on treatment. J. Int. Med. Res. 2018, 46, 2149–2156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henriksson, R.; Asklund, T.; Poulsen, H.S. Impact of therapy on quality of life, neurocognitive function and their correlates in glioblastoma multiforme: A review. J. Neurooncol. 2011, 104, 639–646. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Mebendazole: | Methyl 5-benzoyl-1H-benzimidazol-2-yl-carbamate |
|---|---|
| Structure |  |
| Chemical formula | C16-H13-N3-O3 |
| Weight | 295.293 g/mol |
| CAS | 60254-95-7 |
| Indication | oral administration |
| Absorption | poor solubility and absorption |
| Distribution | highly bound to plasma protein |
| Metabolism | extensively hepatic first-pass |
| Side effects | gastrointestinal upset, diarrhea, fever, abdominal |
| discomfort, flatulence, hypersensitivity reactions | |
| Excretion | bile and feces and ˂2% urine |
| BBB permeability | Yes |
| GI Absorption | High |
| P-gp Substrate | No |
| T1/2: elimination half-life | 3–6 h |
| Author (Year) | Population | Formulation | Cmax (ng/mL) | AUC (ng.h/mL) | Tmax (h) | T1/2 (h) | MBZ Dose and Coadministration with Other Drugs | Ref | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Brainthwaite (1982) | Patients (n.10) | tablet | 17.2–116.2 | 209.6 | 5.2 | MBZ 10 mg/kg | [55] | |||||
| Chronic treatment | 99.4–500.2 | 649.8 | 2 | MBZ 10 mg/kg | ||||||||
| Dawson (1985) Edwards (1988) | Fasting volunteers (n.3) | oral intravenous | 91, 1–142 | 4.63 ± 1.80 2.27 ± 8.2 | 0.42 ± 0.12 | 0.93 ± 0.25 1.12 ± 0.24 | MBZ 1.18 µg of 3H-mebendazole | [47] | ||||
| Corti (2009) | Healthy volunteers (n.8) Fasting | tablet | 31.0 ± 26.0 | 207.2 ± 157.6 | 2.1 ± 1 | 7.4 ± 2.2 | MBZ 1000 mg/day po on 3 days | [56] | ||||
| tablet | 36.0 ± 22.8 | 228.9 ± 147 | 2.4 ± 1.7 | 9.3 ± 3.5 | MBZ 1000 mg/day po on 3 days + Short-term Ritovaris 200 mg po twice daily × 2 doses | |||||||
| tablet | 11.5 ± 6.2 | 85.9 ± 53.2 | 2.1 ± 0.8 | 10.6 ± 8.6 | MBZ 1000 mg/day po on 3 days + Long-term Ritovaris 200 mg po twice daily × 7 doses | |||||||
| Bekhti (1987) | Patients (n.8) with peptic ulcer and hydatid cysts | tablet | 55.7 ± 30.2 | MBZ 1.5 g three times daily po × 30 days | [57] | |||||||
| 82.3 ± 41.8 | MBZ 1.5 g three times daily po × 30 days + Cimetidine 400 mg three times daily po × 30 days | |||||||||||
| Luder (1986) | Patients (n.10) with Echinococcus multilocularis or granulosus | tablet | HC | MBZ 45–122 mg/kg/day po × 10 weeks + Cimetidine 1 G/day po × 10 weeks | [58] | |||||||
| NC | MBZ 45–122 mg/kg/day po × 10 weeks + Ursodeosycholic acid 500 mg/day | |||||||||||
| Liu (2012) | Mouse | powder | 1.3 ± 0.42 | 11.6 ± 2.0 | 1.3 ± 0.6 | 12.0 ± 5.5 | MBZ-1% tragacanth po 25 mg/kg | [59] | ||||
| 3.3 ± 0.6 | 28.2 ± 2.5 | 1.7 ± 1.6 | 11.5 ± 6.2 | MBZ-Oleic Acid po 25 mg/kg | ||||||||
| 4.8 ± 0.6 | 19.8 ± 2.5 | 1 | 4.2 ± 1.0 | MBZ-Glycerol Trioleate po 25 mg/kg | ||||||||
| 4.4 ± 2.0 | 25.1 ± 4.4 | 1.5 ± 0.9 | 4.9 ± 1.7 | MBZ-Soybean oil po 25 mg/kg | ||||||||
| Ren-Yuan Bai (2015) | MBZ polymorphs in mice (GL261 Glioma model) | tablet | Plasma | Brain | Plasma | Brain | Plasma | Brain | Plasma | Brain | [53] | |
| 379.3 | 3052 | 1 | 3.23 | MBZ Polimorph A | ||||||||
| 2778.3 | 26474 | 6 | 3.18 | MBZ Polimorph B | ||||||||
| 2553.3 | 2016 | 16039 | 13134 | 4 | 4 | 0.90 | 1.64 | MBZ Polimorph C | ||||
| Cancer Type | Cell Lines | Model Used | Molecular Targets | Efficacy | IC50 or Doses | Combination with Other Drugs | Ref |
|---|---|---|---|---|---|---|---|
| Glioblastom | 060919 | In vitro | Inhibit tubulin polymerization | ↓ Cell viability | 0.11 μM | [57] | |
| In vivo orthotopic mouse models | Apoptosis | ↑ Survival: 48 d CT: 65 d MBZ | |||||
| Glioma | GL261 | In vitro | ↓ Cell viability | 0.24 µM | |||
| In vivo orthotopic mouse models | ↑ Survival: 30 d CT: 49 d MBZ ↑ Survival: 29 d CT: 41 d TMZ: 50 d MBZ + TMZ | Temozolomide | |||||
| Glioma | GL261 | In vivo orthotopic mouse models | Inhibit tubulin polymerization | ↑ Survival (MBZ tablets from different suppliers) 29 days CT; 34 days in S2015; 50 days in S2017; 42 days in medley; 44 days in Janssen | 50 mg/kg | [53] | |
| Medulloblastoma | GL261 D425 | In vivo orthotopic mouse models | ↑ Survival with elacridar | Elacridar (P-glycoprotein inhibitor) | |||
| Glioma | GL261 | In vitro | MBZ sensitizes GL261 cells to IR | ↓ Cell viability | 35 nM | IR (ionizing radiation) | [79] |
| Glioblastoma | GBM14 glioma cells | MBZ sensitizes GBM14 cells to IR | ↓ Cell viability | IR (ionizing radiation) | |||
| Glioblastoma | U87-MG | In vitro | Inhibited migration and invasion | ↓ Cell viability | 0.21 μmol/L | [58] | |
| U251-MG | Arrest the cell cycle at the G2/M phase | ↓ Cell viability | 0.25 μmol/L | ||||
| Glioblastoma | GL261 | In vitro | Tubulin disruption | ↓ Cell viability | 160 nM | [71] | |
| In vivo orthotopic mouse models | ↓ Tumor growth ↑ Survival: CT 10 d; 50 MBZ 14 d; 100 MBZ 16.5 d No effect with vincristine | 50 mg/kg 100 mg/kg | |||||
| Medulloblastoma | HUVEC | In vitro | ↓VEGFR2 kinase activity, CD31 | 1–10 µM 4.3 µM | [74] | ||
| D425 | In vivo orthotopic mouse models | ↓ Tumor growth ↑ Survival: CT 21 d; MBZ 48 d | 50 mg/kg | ||||
| PTCH Mutant allograft MB | ↑ Survival: CT 12 d; MBZ 30 d | 50 mg/kg | |||||
| PTCH Mutant D477G allograft MB | ↑ Survival: CT 12 d; MBZ 30 d | 50 mg/kg | |||||
| Medulloblastoma | Daoy | In vitro | Inhibits hedgehog Signaling pathway ↓ GLI1 | ↓ Cell viability and colony formation | 516 nmol/L | [76] | |
| Daoy | In vivo orthotopic mouse models | ↓ GLI1 and PTCH2 | ↓ Tumor growth and hedgehog signaling ↑ Survival: CT 75 d; MBZ 113 d | 50 mg/kg | |||
| Medulloblastoma | D425 SHH SHH- vismodegib-resistant | In vivo orthotopic mouse models | inhibition of VEGFR2 kinase activity | ↑ Survival: CT 21 d; MBZ 48 d | 50 mg/kg | [77] | |
| Meningioma | KT21MG1 IOMMLE AC-1 SF4068 SF6717, SF1335 SF1335 + YAP | In vitro | Apoptosis induction, angiogenesis Inhibitor | ↓ Cell viability and colony formation | 0.39 μM 0.39 μM 0.342 μM 0.42 μM 0.372 μM 0.262 μM | [78] | |
| KT21MG1 | In vivo orthotopic mouse models | ↓ Tumor, Ki67, CD31 ↑ Survival cleaved caspase 3 | ↑ Survival: CT 19 d; MBZ 30 d; radiation 33.5 D; MBZ + R 39 d | 50 mg/kg | Radiation | ||
| Glioblastoma |
U87
A172 U251 U138 C6 (murine) | In vitro | Low expression of FGFR3 and AKT2 was especially sensitive to T + V + M. | ↓ Cell viability | 500 nM | 50 µM TMZ 5 nM VBL | [80] |
| ↓↓↓ Cell viability | T + V + M | ||||||
| Glioblastoma | U87 U87vIII T98G U251 | In vitro | MBZ inhibits ABL1, ERK2/MAPK1, and MAPK14/p38a in vitro, with particularly high potency against MAPK14/p38a | ↓↓↓Cell viability | 2.1 μM 288 nM | [81] |
| Title | Phase | Conditions | Intervention/ Treatment | Institution | Study/Results | Status |
|---|---|---|---|---|---|---|
| Study of Mebendazole in Newly Diagnosed High-Grade Glioma Patients Receiving Temozolomide NCT N. NCT01729260 | Phase 1 | Newly diagnosed high-grade glioma (WHO Grade III or IV) | Mebendazole: 500 mg chewable tablets with meals three times every day on a 28-day cycle | The Johns Hopkins Hospital Baltimore, Maryland, United States, 21287 | No results Available | Study Start Date: 2012 Study Completion Date: 2021 Completed |
| Phase I Study of Mebendazole Therapy for Recurrent/Progressive Pediatric Brain Tumors NCT N. NCT02644291 | Phase 1 | Medulloblastoma Astrocytoma, Grade III Glioblastoma Anaplastic Astrocytoma Brain Stem Neoplasms, Malignant Oligodendroblastoma Anaplastic Oligodendroglioma Malignant Glioma | Mebendazole: 500 mg tablets, three divided doses with meals | Johns Hopkins All Children’s Hospital, Saint Petersburg, Florida, United States, 33701 Johns Hopkins University School of Medicine Baltimore, Maryland, United States, 21231 | No results Available | Study Start Date: 2016 Study Completion Date: June 2022 Completed |
| A Phase I Study of Mebendazole for the Treatment of Pediatric Gliomas Phase I: determine if MBZ is tolerated when used in combination with the current three-drug regimen. Phase II: evaluate the efficacy of this regimen. NCT N. NCT01837862 | Phase 1 Phase 2 | Pilomyxoid Astrocytoma Pilocytic Astrocytoma Glioma, Astrocytic Optic Nerve Glioma Pleomorphic Xanthoastrocytoma Glioblastoma Multforme Anaplastic Astrocytoma Gliosarcoma Diffuse Intrinsic Pontine Glioma (DIPG) Low-grade Glioma Brainstem Glioma | Mebendazole: 50 mg/kg/day, 100 mg/kg/day, or 200 mg/kg/day p.o. and b.d. for 70 weeks for Low-grade Glioma (in combination with vincristine, carboplatin, and temozolomide) and 48 weeks for High-grade Glioma/Pontine Glioma (in combination with bevacizumab and irinotecan) | Cohen Children’s Medical Center of New York Recruiting New Hyde Park, New York, United States, 11040 | No results Available | Study Start Date: 2013 Estimated Primary Completion Date: April 2024 Estimated Study Completion Date: April 2025 Recruiting |
| A Clinical Safety and Efficacy Study of Mebendazole on GI Cancer or Cancer of Unknown Origin NCT N. NCT03628079 | Phase 1 Phase 2 | Cancer of the Gastrointestinal Tract Cancer of Unknown Origin | ReposMBZ Capsules 50 mg, 100 mg, 200 mg Pharmacokinetics analysis | Dept of Oncology, University Hospital, Uppsala, Sweden, 75185 | No Results Available | Study Start Date: 2018 Study Completion Date: 2019 Terminated |
| Clinical Study Evaluating Mebendazole as Adjuvant Therapy in Patients with Colorectal Cancer NCT N. NCT03925662 | Phase 3 | Colorectal Cancer | Folfox with Avastin only Folfox with Avastin with Mebendazole | Sherief Abd-Elsalam, Tanta University | No Results Available | Study Start Date 2019 Study Completion Date: 2028 Recruiting |
| Study of the Safety, Tolerability, and Efficacy of Metabolic Combination Treatments on Cancer (METRICS) NCT N. NCT02201381 | Phase 3 | Cancer Overall Survival | Oral Mebendazole 100 mg p.o. and uid, for the study duration Oral atorvastatin up to 80 mg uid Oral metformin up to 1000 mg uid Oral doxycycline 100 mg uid | Care Oncology Clinic London, United Kingdom, W1G 9PP | Study Start Date: May 2022 Study Completion Date: 2027 Withdrawn (Prospective recruitment not possible) | |
| Mebendazole Monotherapy and Long-Term Disease Control I Metastatic Adrenocortical carcinoma. 2011 | Clinical Study | 48-year-old man with adrenocortical carcinoma | Mebendazole: 100 mg twice daily for 19 months | University of Michigan | Metastases regressed, and the disease remained stable Well tolerated, and the associated adverse effects are minor | |
| Drug repositioning from bench to bedside: Tumor remission by the anthelmintic drug Mebendazole in refractory metastatic colon cancer. 2013 | Clinical Study | 74-year-old man with metastatic colon cancer | Repositioning drugs for use in advanced colon cancer Mebendazole: 100 mg twice a day b.d. for six weeks | Uppsala University, Sweden | Complete remission of the metastases in the lungs and lymph nodes and a good partial remission in the liver No adverse effects from the treatment |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meco, D.; Attinà, G.; Mastrangelo, S.; Navarra, P.; Ruggiero, A. Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. Int. J. Mol. Sci. 2023, 24, 1334. https://doi.org/10.3390/ijms24021334
Meco D, Attinà G, Mastrangelo S, Navarra P, Ruggiero A. Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. International Journal of Molecular Sciences. 2023; 24(2):1334. https://doi.org/10.3390/ijms24021334
Chicago/Turabian StyleMeco, Daniela, Giorgio Attinà, Stefano Mastrangelo, Pierluigi Navarra, and Antonio Ruggiero. 2023. "Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers" International Journal of Molecular Sciences 24, no. 2: 1334. https://doi.org/10.3390/ijms24021334
APA StyleMeco, D., Attinà, G., Mastrangelo, S., Navarra, P., & Ruggiero, A. (2023). Emerging Perspectives on the Antiparasitic Mebendazole as a Repurposed Drug for the Treatment of Brain Cancers. International Journal of Molecular Sciences, 24(2), 1334. https://doi.org/10.3390/ijms24021334