

Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants
 , , ,
, , , 
Abstract
:1. Introduction
2. Results
2.1. Case Descriptions
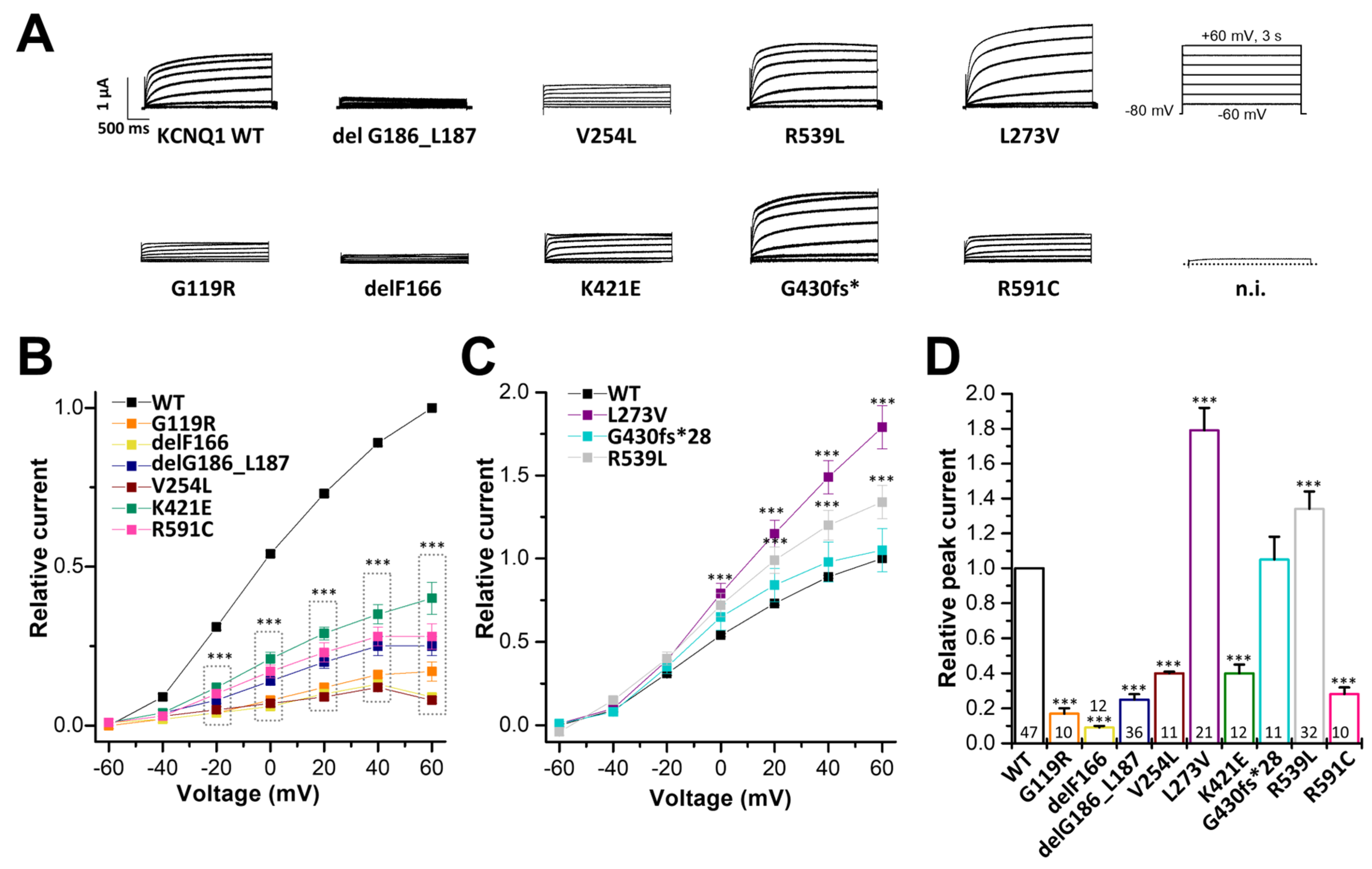
2.2. Electrophysiological Characterization of KCNQ1 Variants
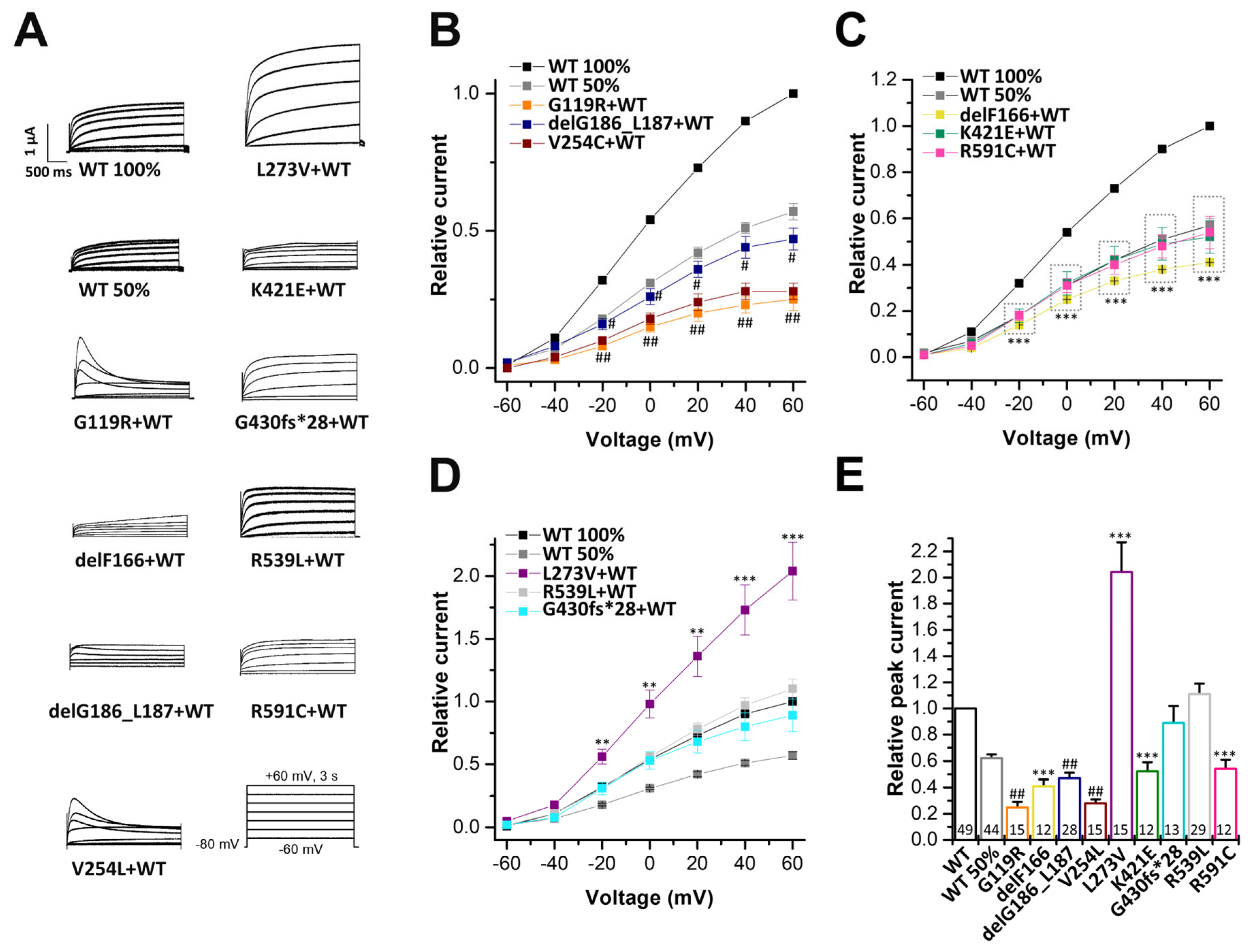
2.3. Electrophysiological Characterization of KCNQ1 Variants Co-Expressed with KCNQ1 Wild-Type Subunits
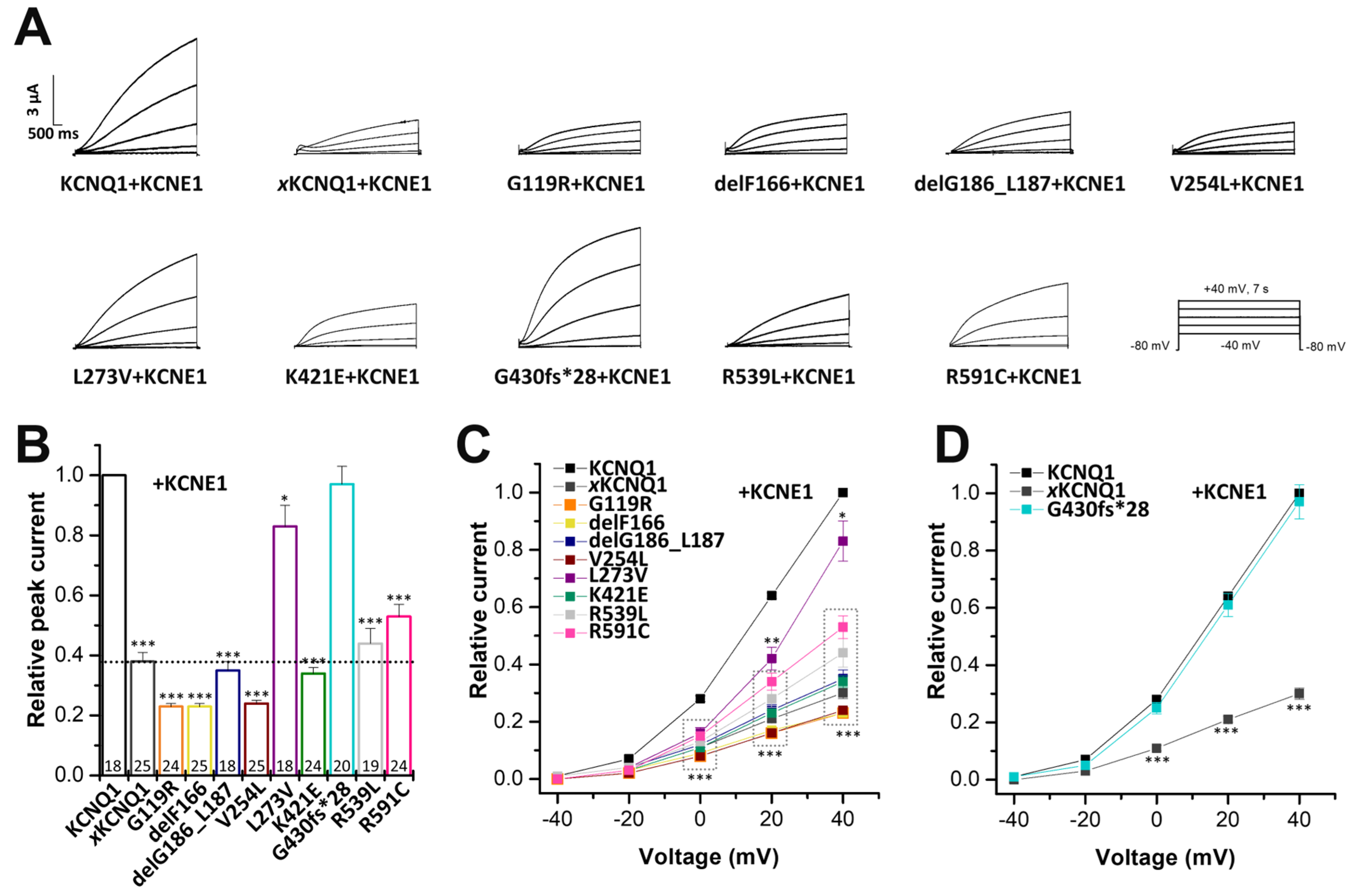
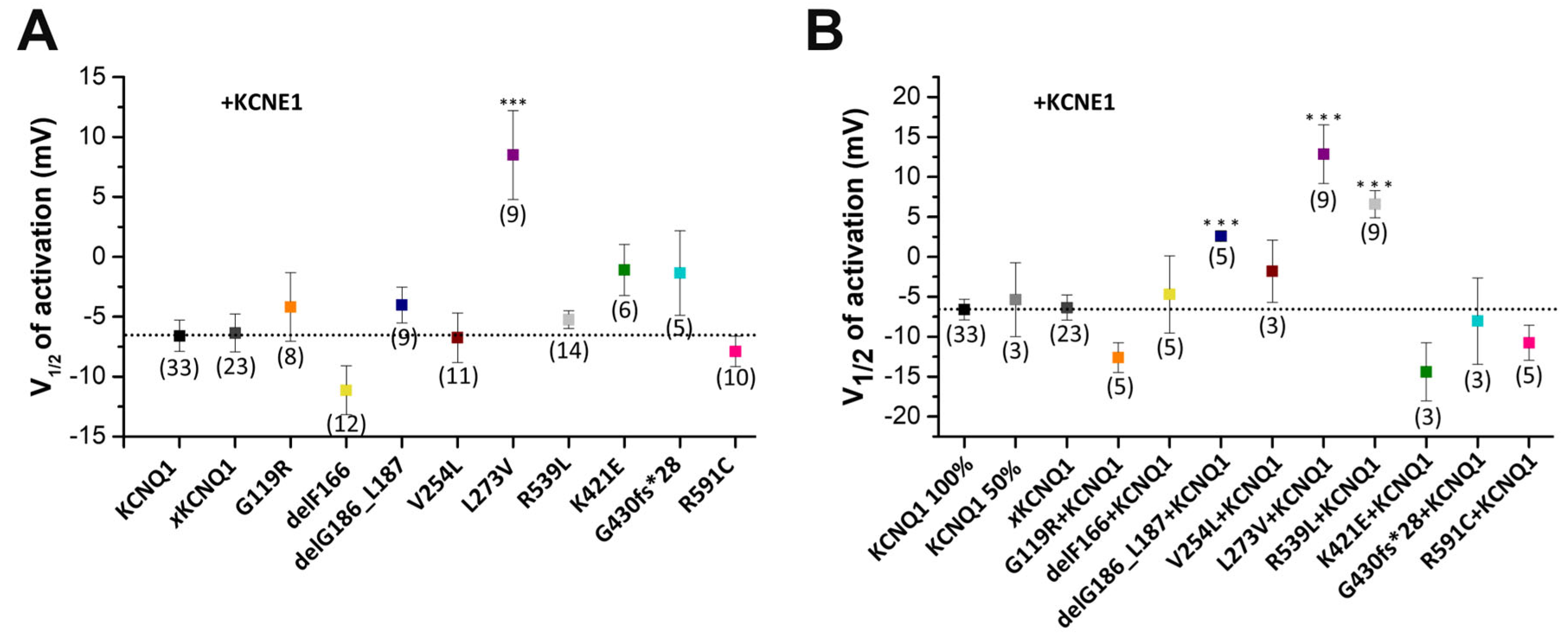
2.4. Electrophysiological Characterization of Homomeric KCNQ1 Variants Co-Expressed with KCNE1
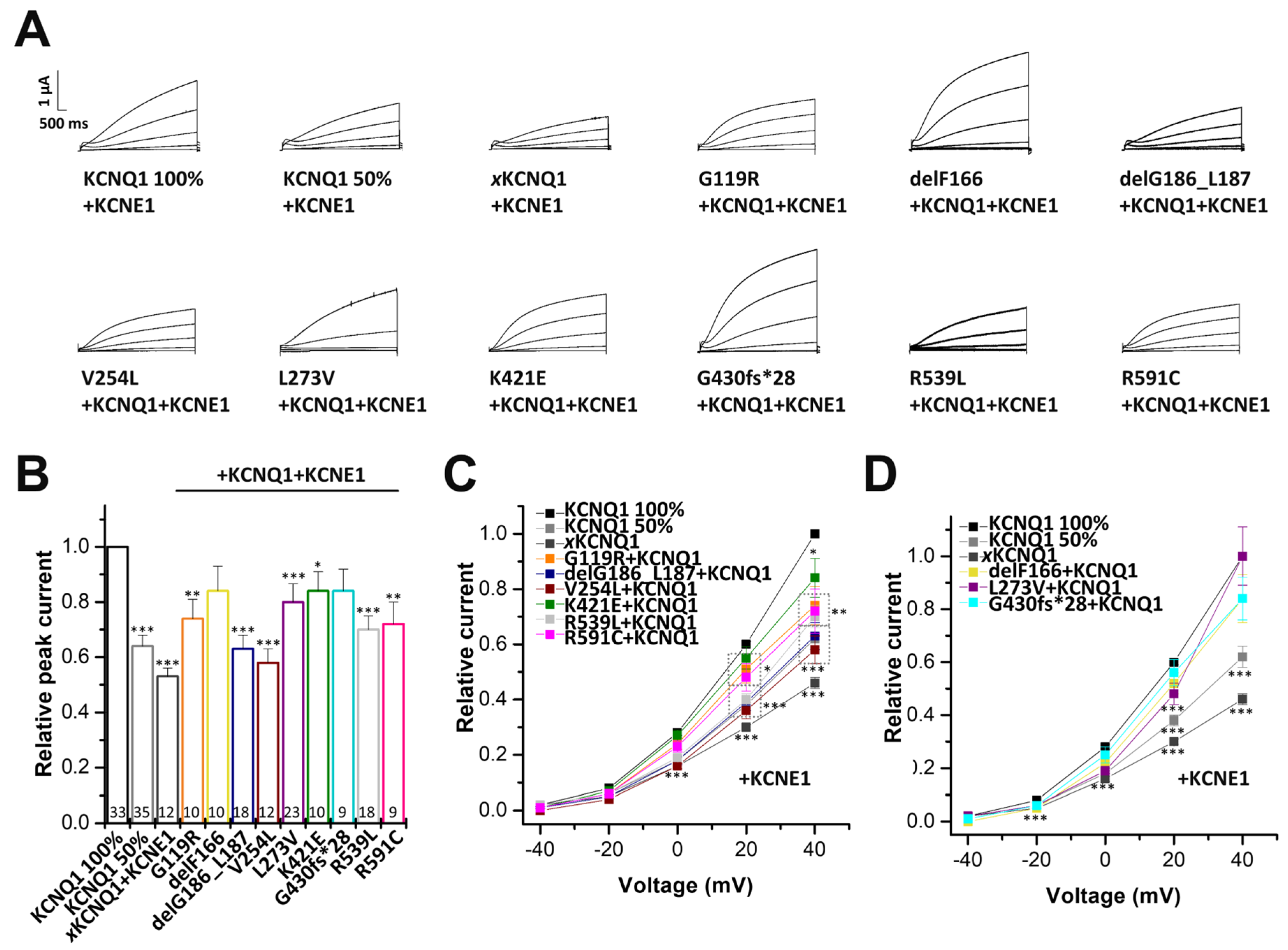
2.5. Electrophysiological Characterization of KCNQ1 Variants Co-Expressed with Wild-Type KCNQ1 and KCNE1
2.6. The delG186_L187, L273V and R539L Variants Alter the Voltage-Dependent Gating of the Heterozygous IKs Channel Complexes
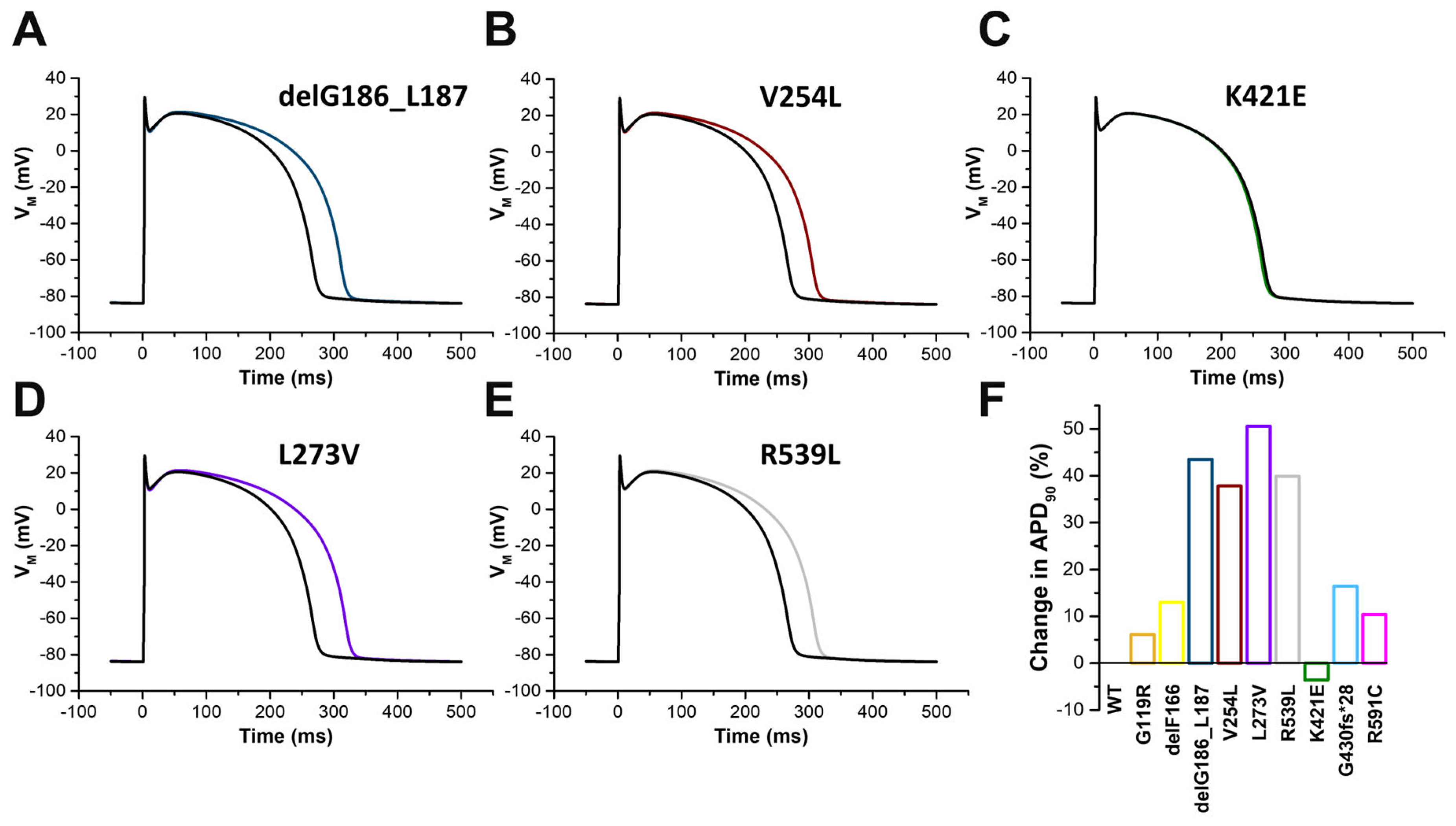
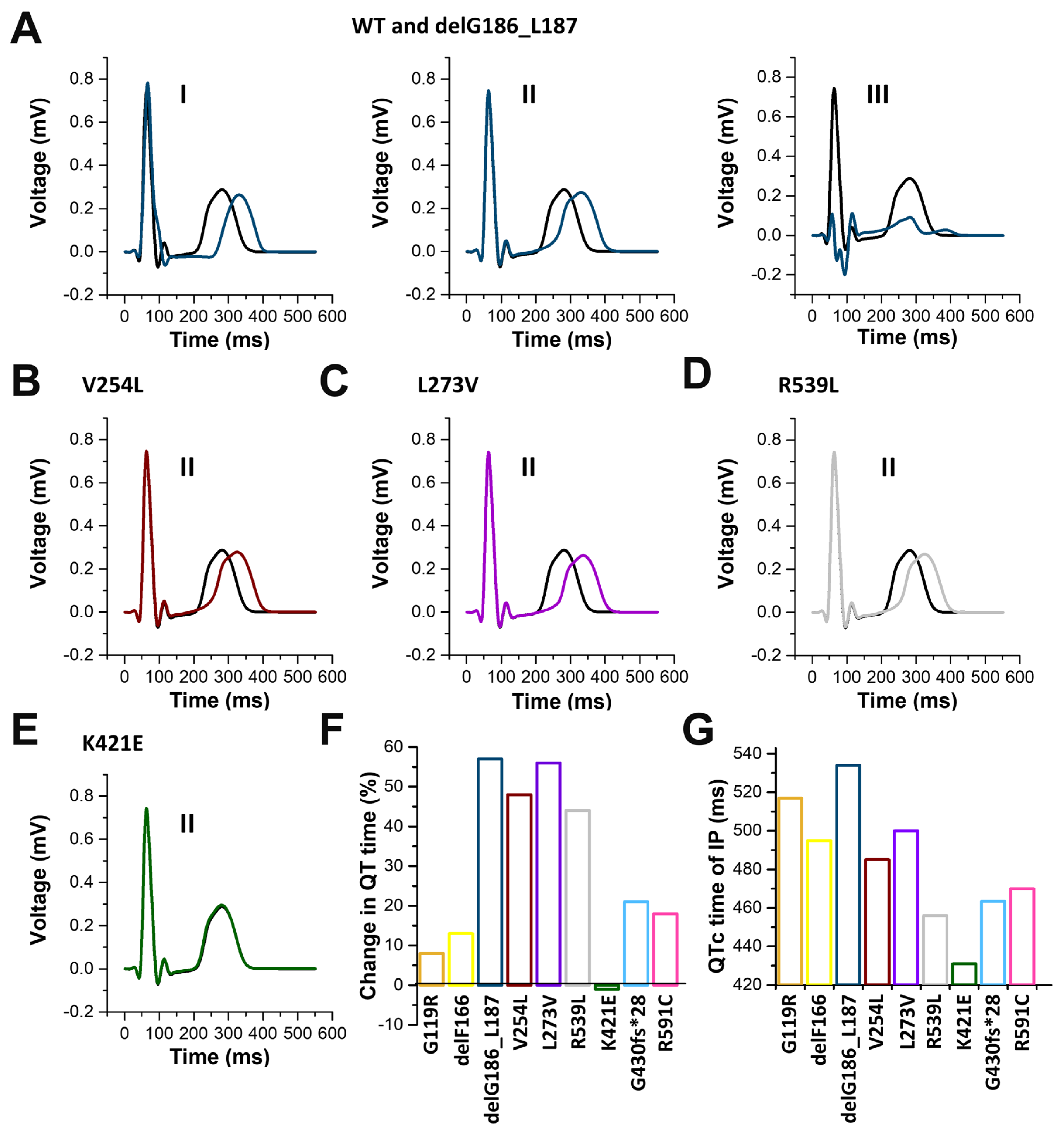
2.7. In Silico Modelling of the Changes to the Human Ventricular Action Potentials for the Different KCNQ1 Variants
3. Discussion
4. Materials and Methods
4.1. Clinical Evaluation
4.2. Classification of Variants
4.3. Molecular Biology
4.4. Electrophysiology
4.5. In Silico Model of Human Ventricular Myocytes
4.6. Data Analysis
4.7. Ethics Statement
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barhanin, J.; Lesage, F.; Guillemare, E.; Fink, M.; Lazdunski, M.; Romey, G. KVLQT1 and lsK (minK) proteins associate to form the IKs cardiac potassium current. Nature 1996, 384, 78–80. [Google Scholar] [CrossRef]
- Sanguinetti, M.C.; Curran, M.E.; Zou, A.; Shen, J.; Spector, P.S.; Atkinson, D.L.; Keating, M.T. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 1996, 384, 80–83. [Google Scholar] [CrossRef]
- Bendahhou, S.; Marionneau, C.; Haurogne, K.; Larroque, M.M.; Derand, R.; Szuts, V.; Escande, D.; Demolombe, S.; Barhanin, J. In vitro molecular interactions and distribution of KCNE family with KCNQ1 in the human heart. Cardiovasc. Res. 2005, 67, 529–538. [Google Scholar] [CrossRef]
- Wrobel, E.; Tapken, D.; Seebohm, G. The KCNE Tango—How KCNE1 Interacts with Kv7.1. Front. Pharmacol. 2012, 3, 142. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.J. Long QT Syndrome. JAMA 2003, 289, 2041–2044. [Google Scholar] [CrossRef]
- Schwartz, P.J.; Stramba-Badiale, M.; Crotti, L.; Pedrazzini, M.; Besana, A.; Bosi, G.; Gabbarini, F.; Goulene, K.; Insolia, R.; Mannarino, S.; et al. Prevalence of the congenital long-QT syndrome. Circulation 2009, 120, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, C.; Priori, S.G.; Schwartz, P.J.; Bloise, R.; Ronchetti, E.; Nastoli, J.; Bottelli, G.; Cerrone, M.; Leonardi, S. Genetic testing in the long QT syndrome: Development and validation of an efficient approach to genotyping in clinical practice. JAMA 2005, 294, 2975–2980. [Google Scholar] [CrossRef] [Green Version]
- Splawski, I.; Shen, J.; Timothy, K.W.; Lehmann, M.H.; Priori, S.; Robinson, J.L.; Moss, A.J.; Schwartz, P.J.; Towbin, J.A.; Vincent, G.M.; et al. Spectrum of mutations in long-QT syndrome genes. KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation 2000, 102, 1178–1185. [Google Scholar] [CrossRef] [Green Version]
- Tester, D.J.; Will, M.L.; Haglund, C.M.; Ackerman, M.J. Compendium of cardiac channel mutations in 541 consecutive unrelated patients referred for long QT syndrome genetic testing. Heart Rhythm 2005, 2, 507–517. [Google Scholar] [CrossRef]
- Jervell, A.; Lange-Nielsen, F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval and sudden death. Am. Heart J. 1957, 54, 59–68. [Google Scholar] [CrossRef]
- Giudicessi, J.R.; Ackerman, M.J. Prevalence and potential genetic determinants of sensorineural deafness in KCNQ1 homozygosity and compound heterozygosity. Circ. Cardiovasc. Genet. 2013, 6, 193–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Splawski, I.; Timothy, K.W.; Vincent, G.M.; Atkinson, D.L.; Keating, M.T. Molecular basis of the long-QT syndrome associated with deafness. N. Engl. J. Med. 1997, 336, 1562–1567. [Google Scholar] [CrossRef] [PubMed]
- Bhuiyan, Z.A.; Wilde, A.A. IKs in heart and hearing, the ear can do with less than the heart. Circ. Cardiovasc. Genet. 2013, 6, 141–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oertli, A.; Rinné, S.; Moss, R.; Kääb, S.; Seemann, G.; Beckmann, B.M.; Decher, N. Molecular Mechanism of Autosomal Recessive Long QT-Syndrome 1 without Deafness. Int. J. Mol. Sci. 2021, 22, 1112. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Curran, M.E.; Splawski, I.; Burn, T.C.; Millholland, J.M.; VanRaay, T.J.; Shen, J.; Timothy, K.W.; Vincent, G.M.; de Jager, T.; et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 1996, 12, 17–23. [Google Scholar] [CrossRef]
- Ackerman, M.J.; Tester, D.J.; Porter, C.J. Swimming, a gene-specific arrhythmogenic trigger for inherited long QT syndrome. Mayo Clin. Proc. 1999, 74, 1088–1094. [Google Scholar] [CrossRef] [PubMed]
- Kapplinger, J.D.; Tester, D.J.; Salisbury, B.A.; Carr, J.L.; Harris-Kerr, C.; Pollevick, G.D.; Wilde, A.A.; Ackerman, M.J. Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart Rhythm 2009, 6, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Seebohm, G.; Westenskow, P.; Lang, F.; Sanguinetti, M.C. Mutation of colocalized residues of the pore helix and transmembrane segments S5 and S6 disrupt deactivation and modify inactivation of KCNQ1 K+ channels. J. Physiol. 2005, 563, 359–368. [Google Scholar] [CrossRef]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, Unit7.20. [Google Scholar] [CrossRef] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucleic Acids Res. 2007, 35, 3823–3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Sims, G.E.; Murphy, S.; Miller, J.R.; Chan, A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE 2012, 7, e46688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckmann, B.M.; Wilde, A.A.; Kaab, S. Dual inheritance of sudden death from cardiovascular causes. N. Engl. J. Med. 2008, 358, 2077–2078. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhang, Q.; Qiu, Y.; Li, Z.; Chen, Z.; Jiang, H.; Li, Y.; Yang, H. Migration of PIP2 lipids on voltage-gated potassium channel surface influences channel deactivation. Sci. Rep. 2015, 5, 15079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seebohm, G.; Scherer, C.R.; Busch, A.E.; Lerche, C. Identification of specific pore residues mediating KCNQ1 inactivation. A novel mechanism for long QT syndrome. J. Biol. Chem. 2001, 276, 13600–13605. [Google Scholar] [CrossRef] [Green Version]
- Kapplinger, J.D.; Tseng, A.S.; Salisbury, B.A.; Tester, D.J.; Callis, T.E.; Alders, M.; Wilde, A.A.; Ackerman, M.J. Enhancing the Predictive Power of Mutations in the C-Terminus of the KCNQ1-Encoded Kv7.1 Voltage-Gated Potassium Channel. J. Cardiovasc. Transl. Res. 2015, 8, 187–197. [Google Scholar] [CrossRef] [Green Version]
- Coyan, F.C.; Abderemane-Ali, F.; Amarouch, M.Y.; Piron, J.; Mordel, J.; Nicolas, C.S.; Steenman, M.; Merot, J.; Marionneau, C.; Thomas, A.; et al. A long QT mutation substitutes cholesterol for phosphatidylinositol-4,5-bisphosphate in KCNQ1 channel regulation. PLoS ONE 2014, 9, e93255. [Google Scholar] [CrossRef] [Green Version]
- Grandi, E. Size matters, proportion too: Coupling of experiments and theory reveals relative roles of K+ channels in action potential stability. J. Physiol. 2017, 595, 2319–2320. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, I.; Zareba, W.; Moss, A.J. Long QT Syndrome. Curr. Probl. Cardiol. 2008, 33, 629–694. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. Off. J. Am. Coll. Med. Genet. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alfoldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Henikoff, S.; Ng, P.C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 2009, 4, 1073–1081. [Google Scholar] [CrossRef]
- Hecht, M.; Bromberg, Y.; Rost, B. Better prediction of functional effects for sequence variants. BMC Genom. 2015, 16 (Suppl. S8), S1. [Google Scholar] [CrossRef] [Green Version]
- Ten Tusscher, K.H.; Panfilov, A.V. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1088–H1100. [Google Scholar] [CrossRef]
- Plank, G.; Loewe, A.; Neic, A.; Augustin, C.; Huang, Y.L.; Gsell, M.A.F.; Karabelas, E.; Nothstein, M.; Prassl, A.J.; Sanchez, J.; et al. The openCARP simulation environment for cardiac electrophysiology. Comput. Methods Programs Biomed. 2021, 208, 106223. [Google Scholar] [CrossRef]
- Bai, W.; Shi, W.; de Marvao, A.; Dawes, T.J.; O’Regan, D.P.; Cook, S.A.; Rueckert, D. A bi-ventricular cardiac atlas built from 1000+ high resolution MR images of healthy subjects and an analysis of shape and motion. Med. Image Anal. 2015, 26, 133–145. [Google Scholar] [CrossRef] [Green Version]
- Schuler, S.; Loewe, A. Biventricular statistical shape model of the human heart adapted for computer simulations [Data set]. Zenodo 2021. [Google Scholar] [CrossRef]
- Schuler, S.; Pilia, N.; Potyagaylo, D.; Loewe, A. Cobiveco: Consistent biventricular coordinates for precise and intuitive description of position in the heart—With MATLAB implementation. Med. Image Anal. 2021, 74, 102247. [Google Scholar] [CrossRef]
- Gillette, K.; Gsell, M.A.F.; Prassl, A.J.; Karabelas, E.; Reiter, U.; Reiter, G.; Grandits, T.; Payer, C.; Stern, D.; Urschler, M.; et al. A Framework for the generation of digital twins of cardiac electrophysiology from clinical 12-leads ECGs. Med. Image Anal. 2021, 71, 102080. [Google Scholar] [CrossRef] [PubMed]
- Keller, D.U.; Weiss, D.L.; Dossel, O.; Seemann, G. Influence of IKs heterogeneities on the genesis of the T-wave: A computational evaluation. IEEE Trans. Bio-Med. Eng. 2012, 59, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.; Strodthoff, N.; Bousseljot, R.D.; Kreiseler, D.; Lunze, F.I.; Samek, W.; Schaeffter, T. PTB-XL, a large publicly available electrocardiography dataset. Sci. Data 2020, 7, 154. [Google Scholar] [CrossRef] [PubMed]
- Salvi, V.; Karnad, D.R.; Kerkar, V.; Panicker, G.K.; Manohar, D.; Natekar, M.; Kothari, S.; Narula, D.; Lokhandwala, Y. Choice of an alternative lead for QT interval measurement in serial ECGs when Lead II is not suitable for analysis. Indian Heart J. 2012, 64, 535–540. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant | PROVEAN (Score) | PolyPhen-2 (Score) | SIFT (Score) | SNAP2 (Score) |
|---|---|---|---|---|
| G119R | deleterious (−6.01) | possible damaging (0.691) | damaging (0.00) | effect (84) |
| F166del | deleterious (−13.9) | |||
| G186_L187del | deleterious (−15.78) | |||
| V254L | deleterious (−2.92) | probably damaging (0.959) | damaging (0.04) | effect (76) |
| L273V | deleterious (−2.92) | probably damaging (0.992) | tolerated (0.22) | neutral (−8) |
| K421E | neutral (−1.8) | possibly damaging (0.827) | tolerated (0.05) | effect (59) |
| R539L | deleterious (−5.73) | probably damaging (0.995) | tolerated (0.05) | effect (70) |
| R591C | deleterious (−5.73) | probably damaging (1) | damaging (0.00) | effect (88) |
| G430fs*28 | deleterious (−4.4) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rinné, S.; Oertli, A.; Nagel, C.; Tomsits, P.; Jenewein, T.; Kääb, S.; Kauferstein, S.; Loewe, A.; Beckmann, B.M.; Decher, N. Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants. Int. J. Mol. Sci. 2023, 24, 1350. https://doi.org/10.3390/ijms24021350
Rinné S, Oertli A, Nagel C, Tomsits P, Jenewein T, Kääb S, Kauferstein S, Loewe A, Beckmann BM, Decher N. Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants. International Journal of Molecular Sciences. 2023; 24(2):1350. https://doi.org/10.3390/ijms24021350
Chicago/Turabian StyleRinné, Susanne, Annemarie Oertli, Claudia Nagel, Philipp Tomsits, Tina Jenewein, Stefan Kääb, Silke Kauferstein, Axel Loewe, Britt Maria Beckmann, and Niels Decher. 2023. "Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants" International Journal of Molecular Sciences 24, no. 2: 1350. https://doi.org/10.3390/ijms24021350