
The polyHIS Tract of Yeast AMPK Coordinates Carbon Metabolism with Iron Availability


Abstract
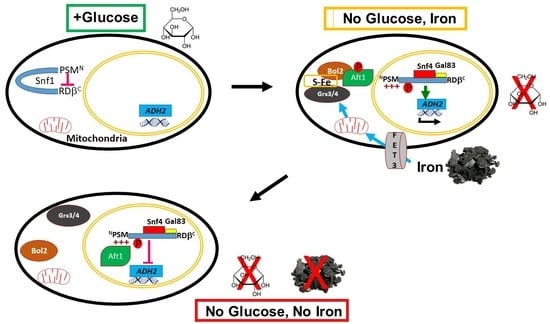
1. Introduction
2. Results
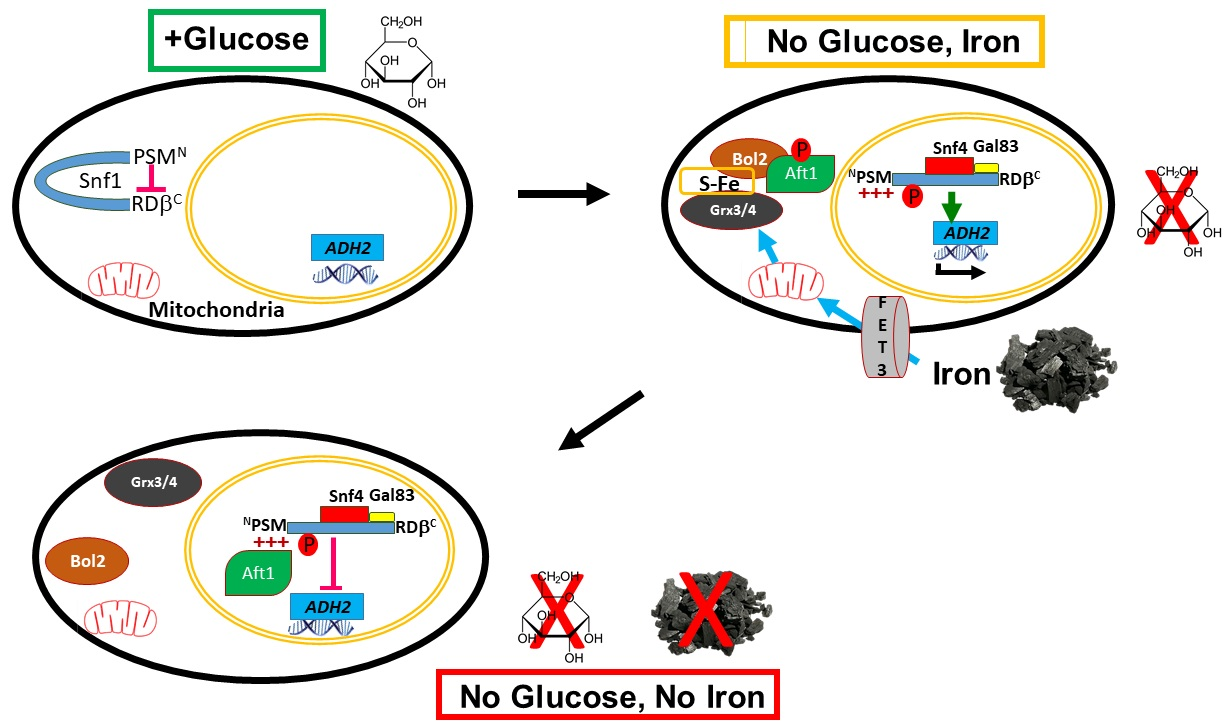
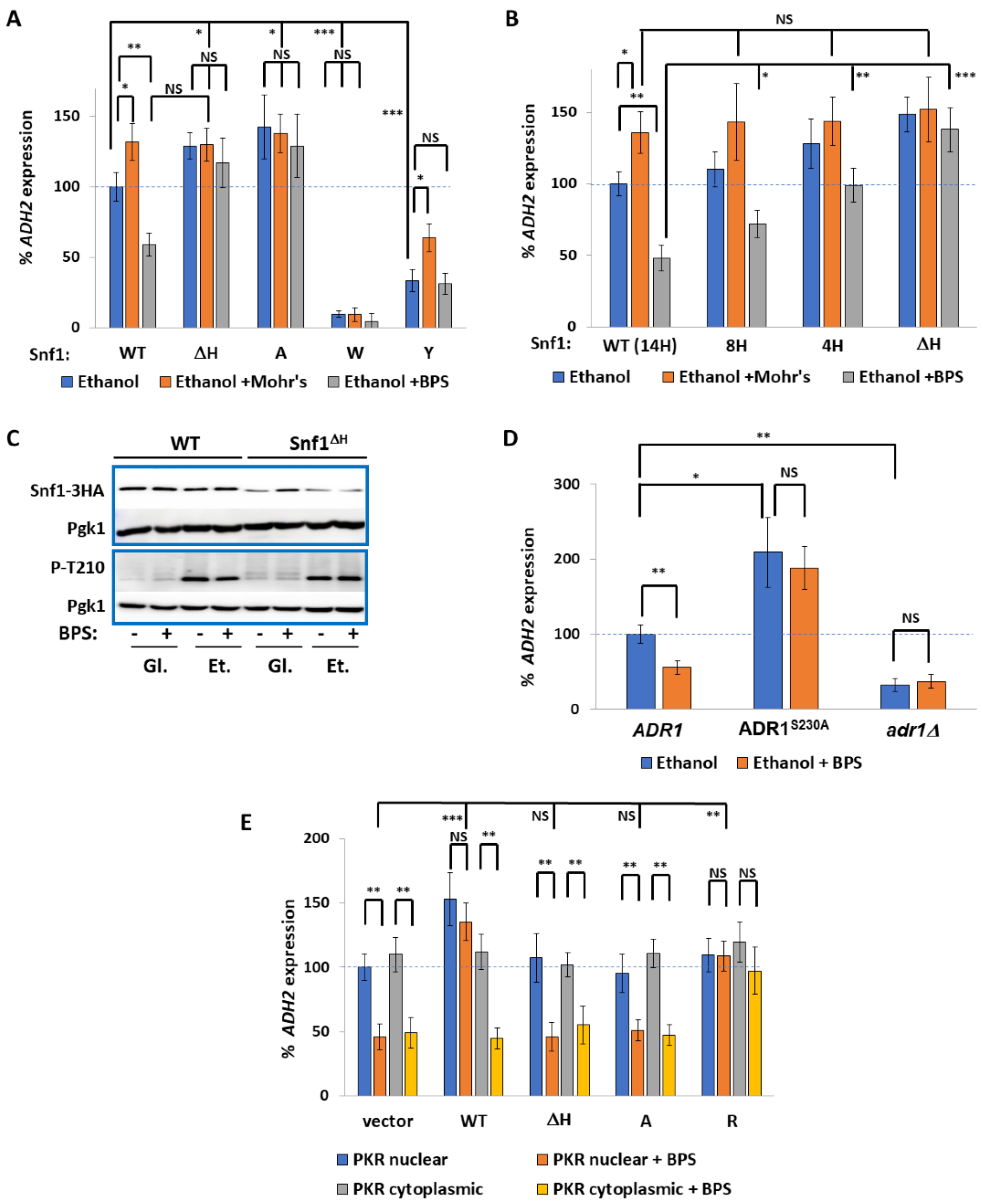
2.1. The PSM Receives a Signal Transmitted by Iron Deprivation
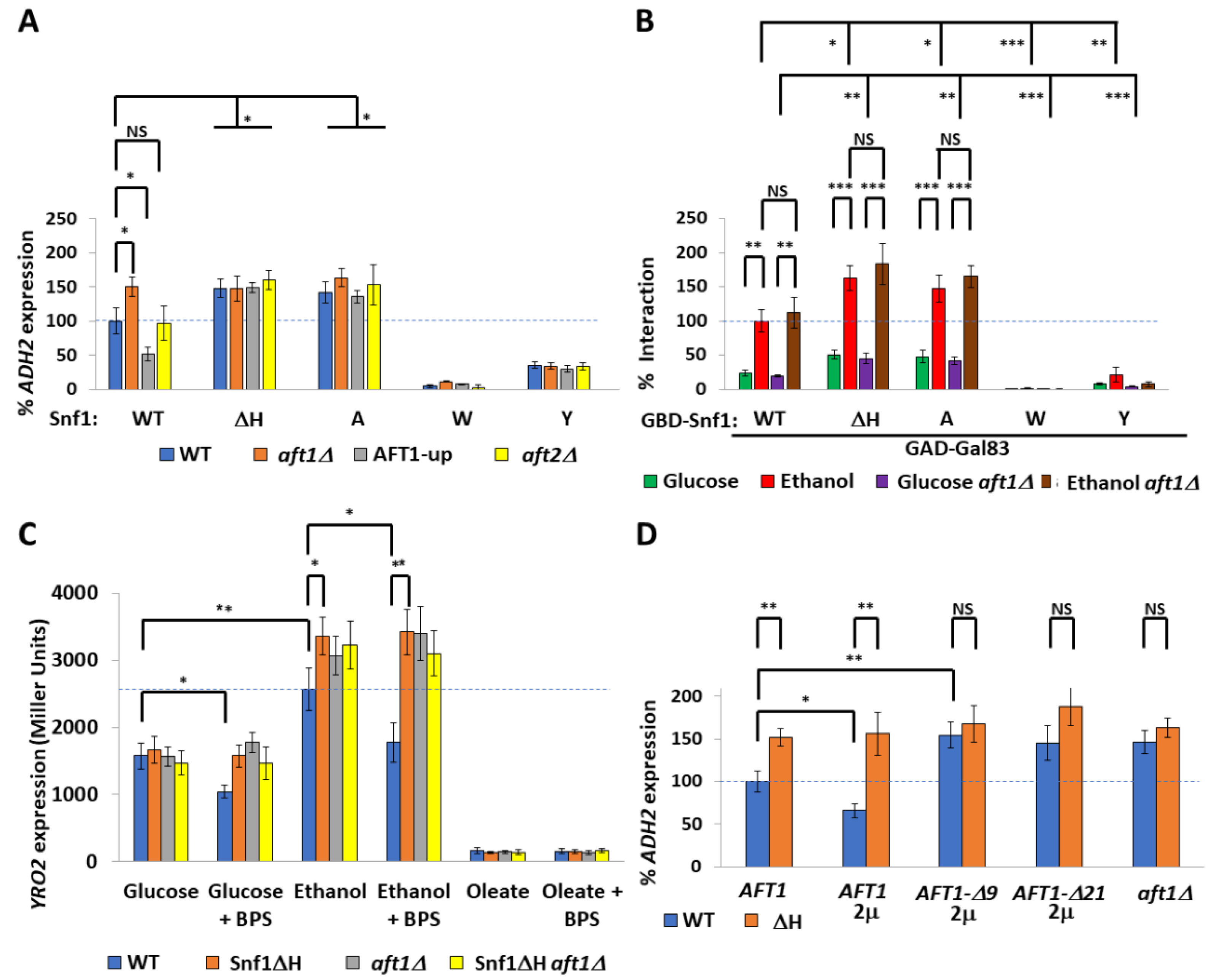
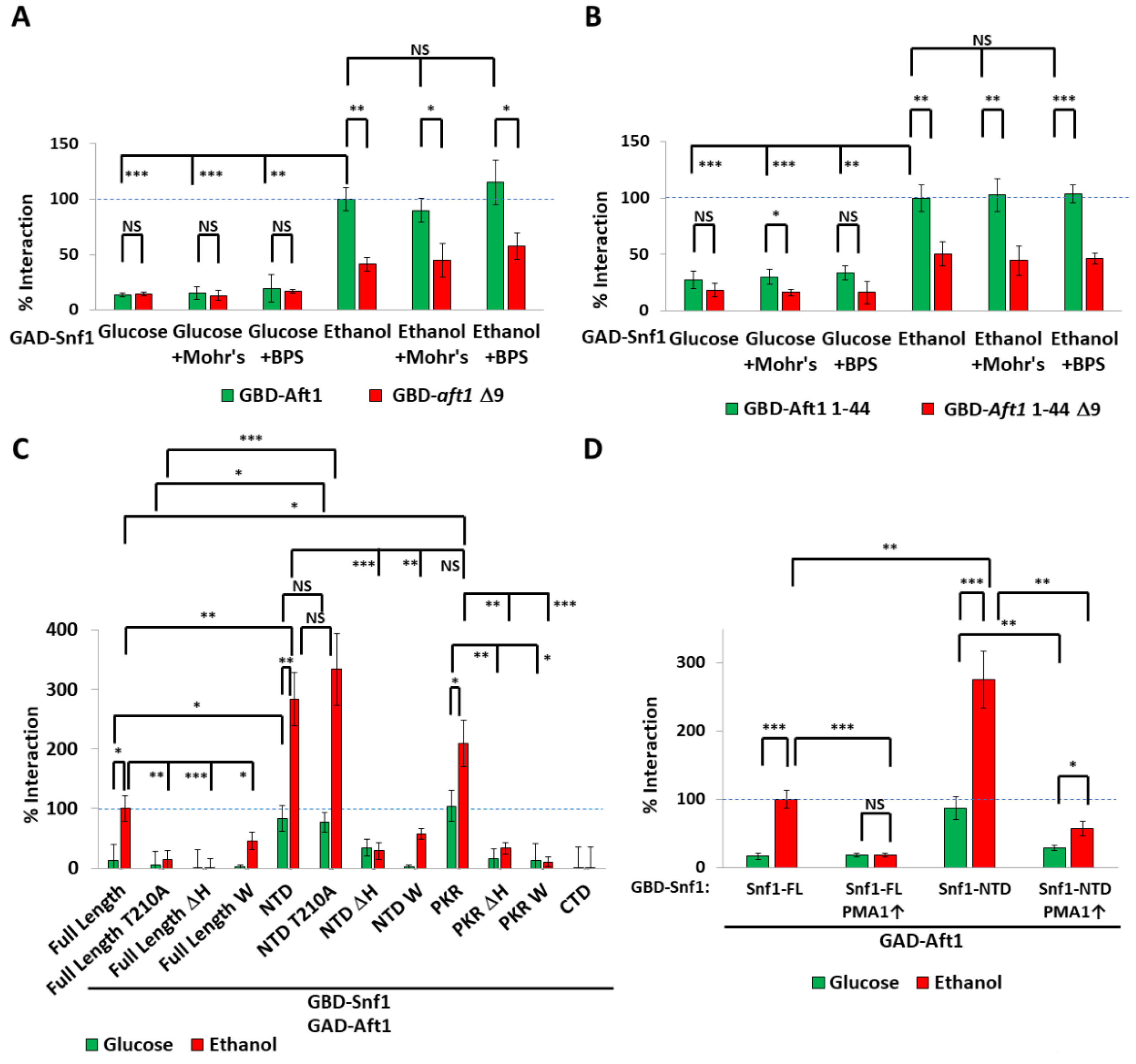
2.2. The Transcription Factor Aft1 Transmits a Low-Iron Signal to Snf1
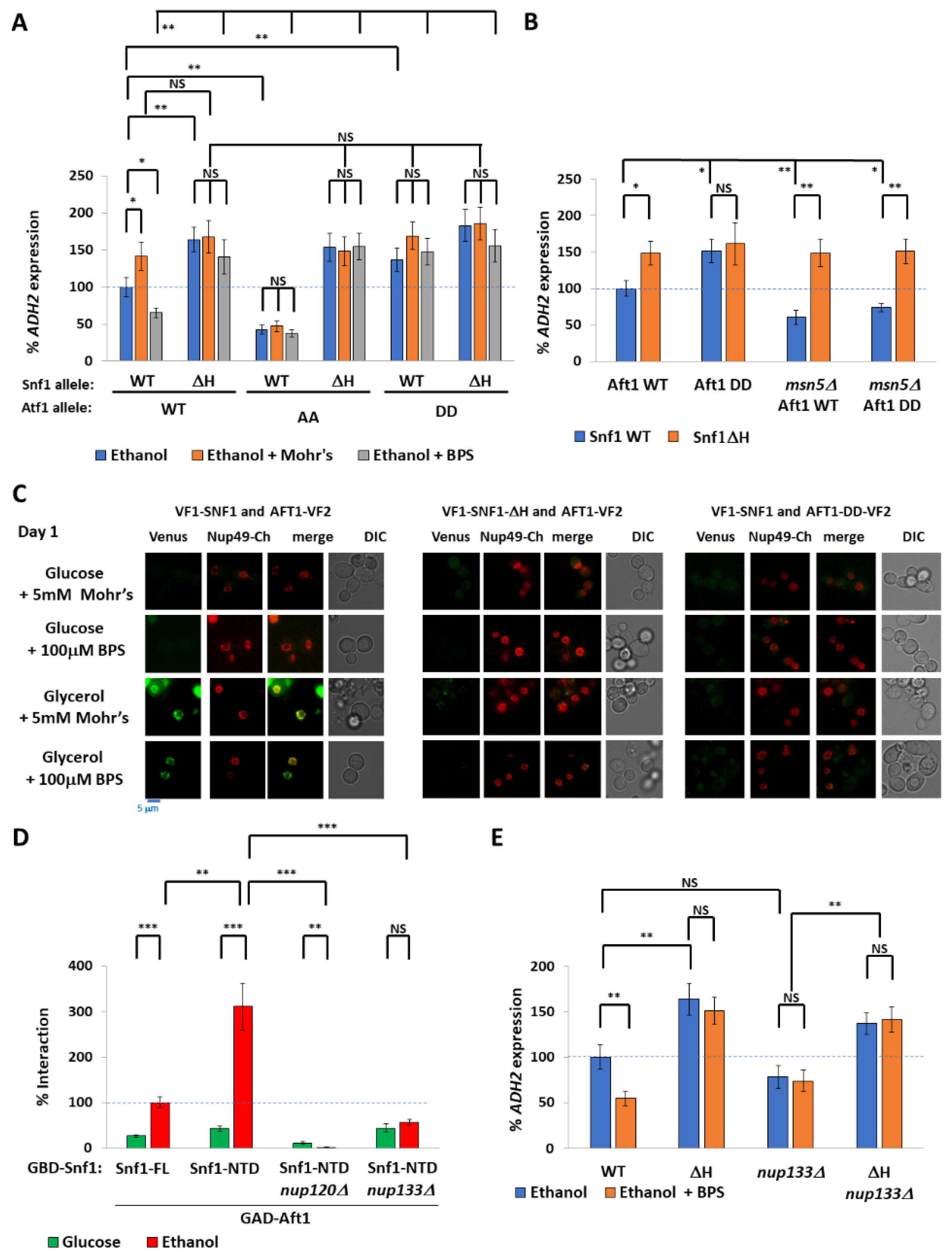
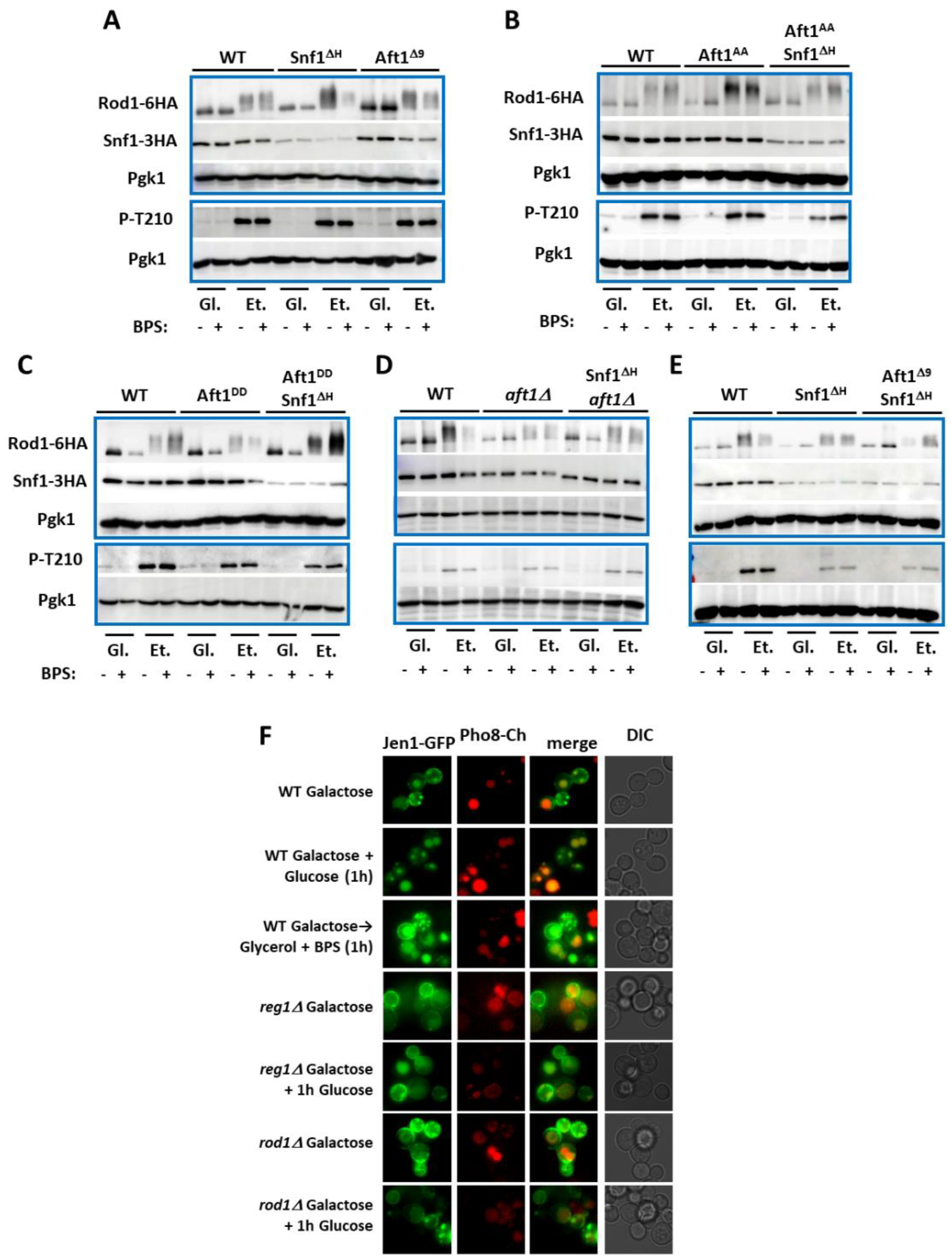
2.3. Nuclear Aft1 Inhibits Snf1
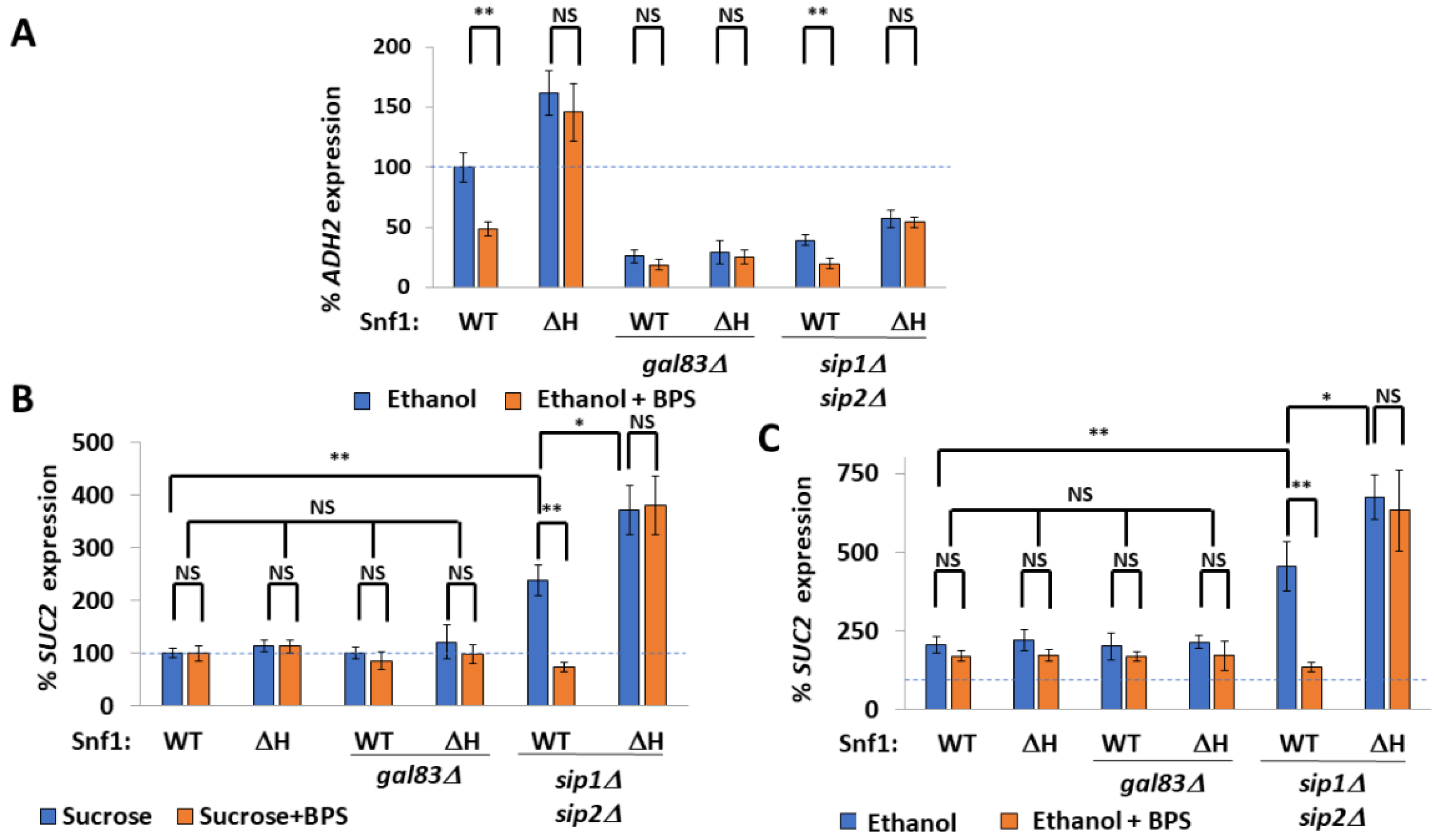
2.4. Spatiotemporal Regulation of Snf1 by Iron
2.5. Non-Nuclear Snf1 Substrates Are Not Regulated by Iron Deprivation
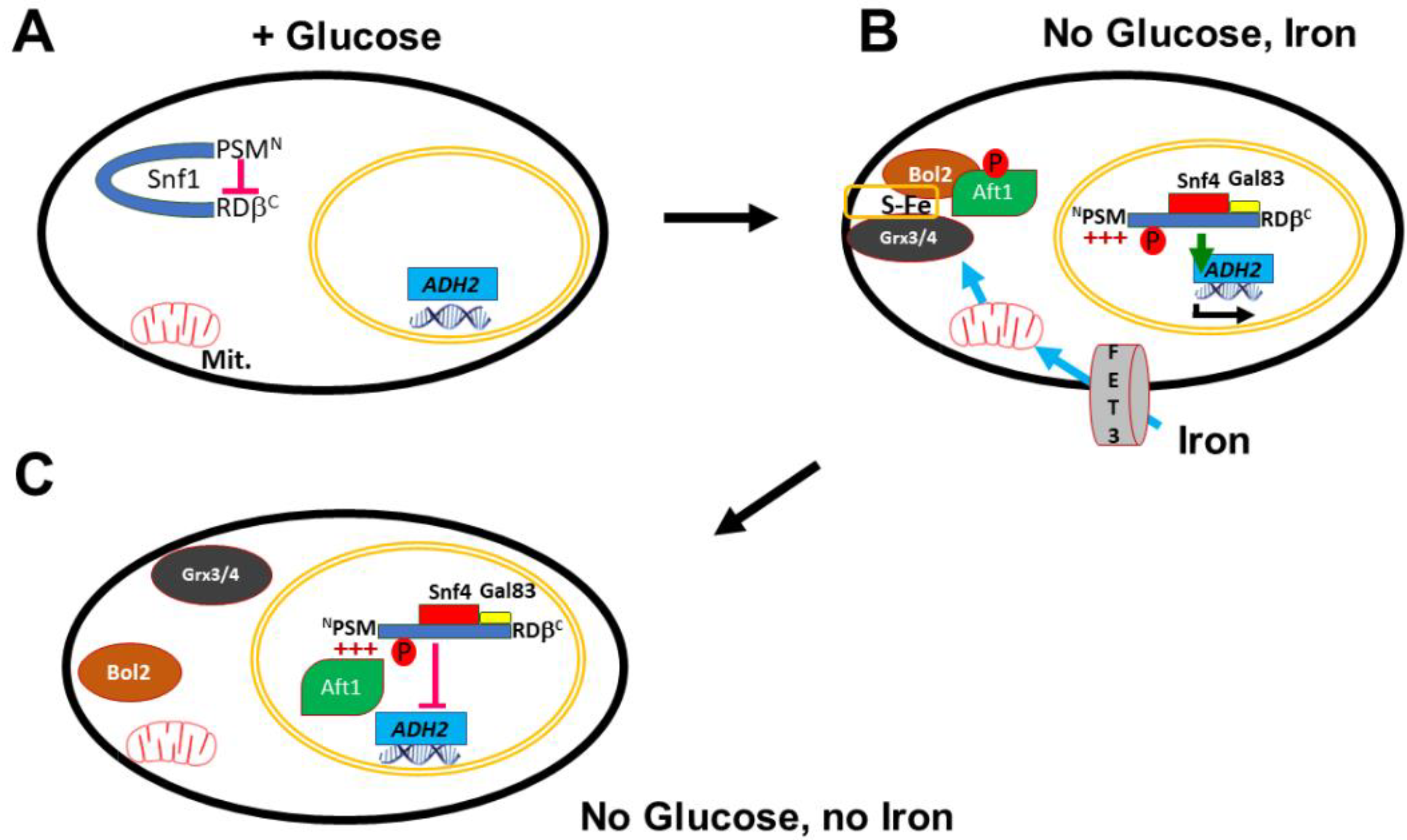
3. Discussion
3.1. The polyHIS Tract Has Different Interactions Depending upon Carbon Source and pH
3.2. The polyHIS Tract Links Mitochondrial Status to Nuclear Gene Expression
3.3. Restricted Localization of the Snf1-Aft1 Interaction
3.4. Spatial Regulation of Snf1 by Iron
3.5. Atf1 Regulates Snf1 Stability
3.6. Iron and the Diauxic Shift
3.7. Comparison with Other Species
4. Materials and Methods
4.1. Resource Availability
4.1.1. Lead Contact
4.1.2. Materials Availability
4.1.3. Data and Code Availability
4.2. Experimental Model and Subject Details
Yeasts, Plasmids, and Growth Conditions
4.3. Method Details
4.3.1. β-Galactosidase Assays for Gene Expression
4.3.2. Yeast 2-Hybrid Experiments
4.3.3. Western Blots
4.3.4. Microscopy
4.3.5. Jen1-GFP Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Askwith, C.C.; de Silva, D.; Kaplan, J. Molecular biology of iron acquisition in Saccharomyces cerevisiae. Mol. Microbiol. 1996, 20, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Kwast, K.E.; Burke, P.V.; Poyton, R.O. Oxygen sensing and the transcriptional regulation of oxygen-responsive genes in yeast. J. Exp. Biol. 1998, 201, 1177–1195. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.J.; Mallory, J.C.; Hand, R.A. Regulation of iron homeostasis mediated by the heme-binding protein Dap1 (damage resistance protein 1) via the P450 protein Erg11/Cyp51. J. Biol. Chem. 2007, 282, 36543–36551. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhong, F.; Alden, S.L.; Hoke, K.R.; Pletneva, E.V. The K79G Mutation Reshapes the Heme Crevice and Alters Redox Properties of Cytochrome c. Biochemistry 2018, 57, 5827–5840. [Google Scholar] [CrossRef]
- Hansberg, W.; Salas-Lizana, R.; Domínguez, L. Fungal catalases: Function, phylogenetic origin and structure. Arch. Biochem. Biophys. 2012, 525, 170–180. [Google Scholar] [CrossRef]
- Puig, S.; Ramos-Alonso, L.; Romero, A.M.; Martínez-Pastor, M.T. The elemental role of iron in DNA synthesis and repair. Metallomics 2017, 9, 1483–1500. [Google Scholar] [CrossRef]
- Martínez-Pastor, M.T.; de Llanos, R.; Romero, A.M.; Puig, S. Post-Transcriptional Regulation of Iron Homeostasis in Saccharomyces cerevisiae. Int. J. Mol. Sci. 2013, 14, 15785–15809. [Google Scholar] [CrossRef]
- Becker, T.; Franckenberg, S.; Wickles, S.; Shoemaker, C.J.; Anger, A.M.; Armache, J.-P.; Sieber, H.; Ungewickell, C.; Berninghausen, O.; Daberkow, I.; et al. Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature 2012, 482, 501–506. [Google Scholar] [CrossRef]
- Li, L.; Jia, X.; Ward, D.M.; Kaplan, J. Yap5 protein-regulated transcription of the TYW1 gene protects yeast from high iron toxicity. J. Biol. Chem. 2011, 286, 38488–38497. [Google Scholar] [CrossRef]
- Li, L.; Kaplan, J.; Ward, D.M. The glucose sensor Snf1 and the transcription factors Msn2 and Msn4 regulate transcription of the vacuolar iron importer gene CCC1 and iron resistance in yeast. J. Biol. Chem. 2017, 292, 15577–15586. [Google Scholar] [CrossRef]
- Raguzzi, F.; Lesuisse, E.; Crichton, R.R. Iron storage in Saccharomyces cerevisiae. FEBS Lett. 1988, 231, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, M.E.; Liu, X.F.; Culotta, V.C. Saccharomyces cerevisiae expresses three functionally distinct homologues of the nramp family of metal transporters. Mol. Cell. Biol. 2000, 20, 7893–7902. [Google Scholar] [CrossRef]
- Courel, M.; Lallet, S.; Camadro, J.-M.; Blaiseau, P.-L. Direct activation of genes involved in intracellular iron use by the yeast iron-responsive transcription factor Aft2 without its paralog Aft1. Mol. Cell. Biol. 2005, 25, 6760–6771. [Google Scholar] [CrossRef] [PubMed]
- Ros-Carrero, C.; Ramos-Alonso, L.; Romero, A.M.; Bañó, M.C.; Martínez-Pastor, M.T.; Puig, S. The yeast Aft1 transcription factor activates ribonucleotide reductase catalytic subunit RNR1 in response to iron deficiency. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194522. [Google Scholar] [CrossRef] [PubMed]
- Puig, S.; Askeland, E.; Thiele, D.J. Coordinated remodeling of cellular metabolism during iron deficiency through targeted mRNA degradation. Cell 2005, 120, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, L.; Keller, G.; Muhlenhoff, U.; Rutherford, J.C.; Lill, R.; Winge, D.R. Role of glutaredoxin-3 and glutaredoxin-4 in the iron regulation of the Aft1 transcriptional activator in Saccharomyces cerevisiae. J. Biol. Chem. 2006, 281, 17661–17669. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mapolelo, D.T.; Dingra, N.N.; Naik, S.G.; Lees, N.S.; Hoffman, B.M.; Riggs-Gelasco, P.J.; Huynh, B.H.; Johnson, M.K.; Outten, C.E. The yeast iron regulatory proteins Grx3/4 and Fra2 form heterodimeric complexes containing a 2Fe-2S cluster with cysteinyl and histidyl ligation. Biochemistry 2009, 48, 9569–9581. [Google Scholar] [CrossRef]
- Kumánovics, A.; Chen, O.S.; Li, L.; Bagley, D.; Adkins, E.M.; Lin, H.; Dingra, N.N.; Outten, C.E.; Keller, G.; Winge, D.; et al. Identification of FRA1 and FRA2 as genes involved in regulating the yeast iron regulon in response to decreased mitochondrial iron-sulfur cluster synthesis. J. Biol. Chem. 2008, 283, 10276–10286. [Google Scholar] [CrossRef]
- Martínez-Pastor, M.T.; Perea-García, A.; Puig, S. Mechanisms of iron sensing and regulation in the yeast Saccharomyces cerevisiae. World J. Microbiol. Biotechnol. 2017, 33, 75. [Google Scholar] [CrossRef]
- Poor, C.B.; Wegner, S.V.; Li, H.; Dlouhy, A.C.; Schuermann, J.P.; Sanishvili, R.; Hinshaw, J.R.; Riggs-Gelasco, P.J.; Outten, C.E.; He, C. Molecular mechanism and structure of the Saccharomyces cerevisiae iron regulator Aft2. Proc. Natl. Acad. Sci. USA 2014, 111, 4043–4048. [Google Scholar] [CrossRef]
- Hamza, A.; Baetz, K. Iron-responsive transcription factor Aft1 interacts with kinetochore protein Iml3 and promotes pericentromeric cohesin. J. Biol. Chem. 2012, 287, 4139–4147. [Google Scholar] [CrossRef] [PubMed]
- Kang, C.-M.; Kang, S.; Park, Y.-S.; Yun, C.-W. Physical interaction between Sit1 and Aft1 upregulates FOB uptake activity by inhibiting protein degradation of Sit1 in Saccharomyces cerevisiae. FEMS Yeast Res. 2015, 15, fov080. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Andreadis, C.; Nikolaou, C.; Fragiadakis, G.S.; Tsiliki, G.; Alexandraki, D. Rad9 interacts with Aft1 to facilitate genome surveillance in fragile genomic sites under non-DNA damage-inducing conditions in S. cerevisiae. Nucleic Acids Res. 2014, 42, 12650–12667. [Google Scholar] [CrossRef] [PubMed]
- Ghillebert, R.; Swinnen, E.; Wen, J.; Vandesteene, L.; Ramon, M.; Norga, K.; Rolland, F.; Winderickx, J. The AMPK/SNF1/SnRK1 fuel gauge and energy regulator: Structure, function and regulation. FEBS J. 2011, 278, 3978–3990. [Google Scholar] [CrossRef] [PubMed]
- Ratnakumar, S.; Kacherovsky, N.; Arms, E.; Young, E.T. Snf1 controls the activity of adr1 through dephosphorylation of Ser230. Genetics 2009, 182, 735–745. [Google Scholar] [CrossRef][Green Version]
- Charbon, G.; Breunig, K.D.; Wattiez, R.; Vandenhaute, J.; Noël-Georis, I. Key Role of Ser562/661 in Snf1-Dependent Regulation of Cat8p in Saccharomyces cerevisiae and Kluyveromyces lactis. Mol. Cell. Biol. 2004, 24, 4083–4091. [Google Scholar] [CrossRef]
- Zhou, H.; Winston, F. NRG1 is required for glucose repression of the SUC2 and GAL genes of Saccharomyces cerevisiae. BMC Genet. 2001, 2, 5. [Google Scholar] [CrossRef]
- Treitel, M.A.; Kuchin, S.; Carlson, M. Snf1 protein kinase regulates phosphorylation of the Mig1 repressor in Saccharomyces cerevisiae. Mol. Cell. Biol. 1998, 18, 6273–6280. [Google Scholar] [CrossRef]
- Woods, A.; Munday, M.R.; Scott, J.; Yang, X.; Carlson, M.; Carling, D. Yeast SNF1 is functionally related to mammalian AMP-activated protein kinase and regulates acetyl-CoA carboxylase in vivo. J. Biol. Chem. 1994, 269, 19509–19515. [Google Scholar] [CrossRef]
- DeMille, D.; Badal, B.D.; Evans, J.B.; Mathis, A.D.; Anderson, J.F.; Grose, J.H. PAS kinase is activated by direct SNF1-dependent phosphorylation and mediates inhibition of TORC1 through the phosphorylation and activation of Pbp1. Mol. Biol. Cell 2015, 26, 569–582. [Google Scholar] [CrossRef]
- Nicastro, R.; Tripodi, F.; Gaggini, M.; Castoldi, A.; Reghellin, V.; Nonnis, S.; Tedeschi, G.; Coccetti, P. Snf1 Phosphorylates Adenylate Cyclase and Negatively Regulates Protein Kinase A-dependent Transcription in Saccharomyces cerevisiae. J. Biol. Chem. 2015, 290, 24715–24726. [Google Scholar] [CrossRef] [PubMed]
- Llopis-Torregrosa, V.; Ferri-Blázquez, A.; Adam-Artigues, A.; Deffontaines, E.; van Heusden, G.P.H.; Yenush, L. Regulation of the Yeast Hxt6 Hexose Transporter by the Rod1 α-Arrestin, the Snf1 Protein Kinase, and the Bmh2 14-3-3 Protein. J. Biol. Chem. 2016, 291, 14973–14985. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-P.; Leiper, F.C.; Woods, A.; Carling, D.; Carlson, M. Activation of yeast Snf1 and mammalian AMP-activated protein kinase by upstream kinases. Proc. Natl. Acad. Sci. USA 2003, 100, 8839–8843. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Xu, X.; Carlson, M. Ptc1 protein phosphatase 2C contributes to glucose regulation of SNF1/AMP-activated protein kinase (AMPK) in Saccharomyces cerevisiae. J. Biol. Chem. 2013, 288, 31052–31058. [Google Scholar] [CrossRef]
- Elbing, K.; Rubenstein, E.M.; McCartney, R.R.; Schmidt, M.C. Subunits of the Snf1 kinase heterotrimer show interdependence for association and activity. J. Biol. Chem. 2006, 281, 26170–26180. [Google Scholar] [CrossRef]
- Vincent, O.; Townley, R.; Kuchin, S.; Carlson, M. Subcellular localization of the Snf1 kinase is regulated by specific beta subunits and a novel glucose signaling mechanism. Genes Dev. 2001, 15, 1104–1114. [Google Scholar] [CrossRef]
- Simpson-Lavy, K.J.; Bronstein, A.; Kupiec, M.; Johnston, M. Cross-Talk between Carbon Metabolism and the DNA Damage Response in S. cerevisiae. Cell Rep. 2015, 12, 1865–1875. [Google Scholar] [CrossRef]
- Jiang, R.; Carlson, M. Glucose regulates protein interactions within the yeast SNF1 protein kinase complex. Genes Dev. 1996, 10, 3105–3115. [Google Scholar] [CrossRef]
- Amodeo, G.A.; Rudolph, M.J.; Tong, L. Crystal structure of the heterotrimer core of Saccharomyces cerevisiae AMPK homologue SNF1. Nature 2007, 449, 492–495. [Google Scholar] [CrossRef]
- Jiang, R.; Carlson, M. The Snf1 protein kinase and its activating subunit, Snf4, interact with distinct domains of the Sip1/Sip2/Gal83 component in the kinase complex. Mol. Cell. Biol. 1997, 17, 2099–2106. [Google Scholar] [CrossRef]
- Celenza, J.L.; Carlson, M. Mutational analysis of the Saccharomyces cerevisiae SNF1 protein kinase and evidence for functional interaction with the SNF4 protein. Mol. Cell. Biol. 1989, 9, 5034–5044. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Lavy, K.J.; Kupiec, M. Regulation of yeast Snf1 (AMPK) by a polyhistidine containing pH sensing module. iScience 2022, 25, 105083. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Mapolelo, D.T.; Dingra, N.N.; Keller, G.; Riggs-Gelasco, P.J.; Winge, D.R.; Johnson, M.K.; Outten, C.E. Histidine 103 in Fra2 is an iron-sulfur cluster ligand in the 2Fe-2S Fra2-Grx3 complex and is required for in vivo iron signaling in yeast. J. Biol. Chem. 2011, 286, 867–876. [Google Scholar] [CrossRef] [PubMed]
- Ueta, R.; Fujiwara, N.; Iwai, K.; Yamaguchi-Iwai, Y. Iron-induced dissociation of the Aft1p transcriptional regulator from target gene promoters is an initial event in iron-dependent gene suppression. Mol. Cell. Biol. 2012, 32, 4998–5008. [Google Scholar] [CrossRef]
- Ueta, R.; Fujiwara, N.; Iwai, K.; Yamaguchi-Iwai, Y. Mechanism Underlying the Iron-dependent Nuclear Export of the Iron-responsive Transcription Factor Aft1p in Saccharomyces cerevisiae. Mol. Biol. Cell 2007, 18, 2980–2990. [Google Scholar] [CrossRef]
- Simpson-Lavy, K.; Kupiec, M. Carbon Catabolite Repression in Yeast is Not Limited to Glucose. Sci. Rep. 2019, 9, 6491. [Google Scholar] [CrossRef]
- Kim, M.S.; Cho, K.H.; Park, K.H.; Jang, J.; Hahn, J.-S. Activation of Haa1 and War1 transcription factors by differential binding of weak acid anions in Saccharomyces cerevisiae. Nucleic Acids Res. 2019, 47, 1211–1224. [Google Scholar] [CrossRef]
- Collins, M.E.; Black, J.J.; Liu, Z. Casein Kinase I Isoform Hrr25 Is a Negative Regulator of Haa1 in the Weak Acid Stress Response Pathway in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2017, 83, e00672-17. [Google Scholar] [CrossRef]
- Mormino, M.; Siewers, V.; Nygård, Y. Development of an Haa1-based biosensor for acetic acid sensing in Saccharomyces cerevisiae. FEMS Yeast Res. 2021, 21, foab049. [Google Scholar] [CrossRef]
- Martins, T.S.; Pereira, C.; Canadell, D.; Vilaça, R.; Teixeira, V.; Moradas-Ferreira, P.; de Nadal, E.; Posas, F.; Costa, V. The Hog1p kinase regulates Aft1p transcription factor to control iron accumulation. Biochim. Biophys. Acta (BBA)-Mol. Cell Biol. Lipids 2018, 1863, 61–70. [Google Scholar] [CrossRef]
- Malleshaiah, M.; Tchekanda, E.; Michnick, S.W. Real-Time Protein-Fragment Complementation Assays for Studying Temporal, Spatial, and Spatiotemporal Dynamics of Protein-Protein Interactions in Living Cells. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Sarma, N.J.; Buford, T.D.; Haley, T.; Barbara-Haley, K.; Santangelo, G.M.; Willis, K.A. The nuclear pore complex mediates binding of the Mig1 repressor to target promoters. PLoS ONE 2011, 6, e27117. [Google Scholar] [CrossRef]
- Sloan, J.S.; Dombek, K.M.; Young, E.T. Post-translational regulation of Adr1 activity is mediated by its DNA binding domain. J. Biol. Chem. 1999, 274, 37575–37582. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, G.W.; Welkenhuysen, N.; Ye, T.; Cvijovic, M.; Hohmann, S. Mig1 localization exhibits biphasic behavior which is controlled by both metabolic and regulatory roles of the sugar kinases. Mol. Genet. Genom. 2020, 295, 1489–1500. [Google Scholar] [CrossRef]
- Carlson, M. Glucose repression in yeast. Curr. Opin. Microbiol. 1999, 2, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Galdieri, L.; Vancura, A. The yeast AMPK homolog SNF1 regulates acetyl coenzyme A homeostasis and histone acetylation. Mol. Cell. Biol. 2013, 33, 4701–4717. [Google Scholar] [CrossRef] [PubMed]
- Becuwe, M.; Vieira, N.; Lara, D.; Gomes-Rezende, J.; Soares-Cunha, C.; Casal, M.; Haguenauer-Tsapis, R.; Vincent, O.; Paiva, S.; Léon, S. A molecular switch on an arrestin-like protein relays glucose signaling to transporter endocytosis. J. Cell Biol. 2012, 196, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Osiro, K.O.; Borgstrom, C.; Brink, D.P.; Fjolnisdottir, B.L.; Gorwa-Grauslund, M.F. Exploring the xylose paradox in Saccharomyces cerevisiae through in vivo sugar signalomics of targeted deletants. Microb. Cell Fact. 2019, 18, 88. [Google Scholar] [CrossRef]
- Sato, T.K.; Tremaine, M.; Parreiras, L.S.; Hebert, A.S.; Myers, K.S.; Higbee, A.J.; Sardi, M.; McIlwain, S.J.; Ong, I.M.; Breuer, R.J.; et al. Directed Evolution Reveals Unexpected Epistatic Interactions that Alter Metabolic Regulation and Enable Anaerobic Xylose Use by Saccharomyces cerevisiae. PLoS Genet. 2016, 12, e1006372. [Google Scholar] [CrossRef]
- Lee, S.-B.; Tremaine, M.; Place, M.; Liu, L.; Pier, A.; Krause, D.J.; Xie, D.; Zhang, Y.; Landick, R.; Gasch, A.P.; et al. Crabtree/Warburg-like aerobic xylose fermentation by engineered Saccharomyces cerevisiae. Metab. Eng. 2021, 68, 119–130. [Google Scholar] [CrossRef]
- Patnaik, P.K.; Beaupere, C.; Barlit, H.; Romero, A.M.; Tsuchiya, M.; Muir, M.; Martínez-Pastor, M.T.; Puig, S.; Kaeberlein, M.; Labunskyy, V.M. Deficiency of the RNA-binding protein Cth2 extends yeast replicative lifespan by alleviating its repressive effects on mitochondrial function. Cell Rep. 2022, 40, 111113. [Google Scholar] [CrossRef] [PubMed]
- Fontana, G.A.; Hess, D.; Reinert, J.K.; Mattarocci, S.; Falquet, B.; Klein, D.; Shore, D.; Thomä, N.H.; Rass, U. Rif1 S-acylation mediates DNA double-strand break repair at the inner nuclear membrane. Nat. Commun. 2019, 10, 2535. [Google Scholar] [CrossRef] [PubMed]
- Gaillard, H.; Santos-Pereira, J.M.; Aguilera, A. The Nup84 complex coordinates the DNA damage response to warrant genome integrity. Nucleic Acids Res. 2019, 47, 4054–4067. [Google Scholar] [CrossRef] [PubMed]
- Therizols, P.; Fairhead, C.; Cabal, G.G.; Genovesio, A.; Olivo-Marin, J.-C.; Dujon, B.; Fabre, E. Telomere tethering at the nuclear periphery is essential for efficient DNA double strand break repair in subtelomeric region. J. Cell Biol. 2006, 172, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Simpson-Lavy, K.J.; Johnston, M. SUMOylation regulates the SNF1 protein kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 17432–17437. [Google Scholar] [CrossRef]
- Ruiz, A.; Xu, X.; Carlson, M. Roles of two protein phosphatases, Reg1-Glc7 and Sit4, and glycogen synthesis in regulation of SNF1 protein kinase. Proc. Natl. Acad. Sci. USA 2011, 108, 6349–6354. [Google Scholar] [CrossRef]
- Haurie, V.; Boucherie, H.; Sagliocco, F. The Snf1 protein kinase controls the induction of genes of the iron uptake pathway at the diauxic shift in Saccharomyces cerevisiae. J. Biol. Chem. 2003, 278, 45391–45396. [Google Scholar] [CrossRef]
- Pujol-Carrion, N.; Pavón-Vergés, M.; Arroyo, J.; de la Torre-Ruiz, M.A. The MAPK Slt2/Mpk1 plays a role in iron homeostasis through direct regulation of the transcription factor Aft1. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118974. [Google Scholar] [CrossRef]
- Horie, T.; Kawamata, T.; Matsunami, M.; Ohsumi, Y. Recycling of iron via autophagy is critical for the transition from glycolytic to respiratory growth. J. Biol. Chem. 2017, 292, 8533–8543. [Google Scholar] [CrossRef]
- Gerwien, F.; Safyan, A.; Wisgott, S.; Hille, F.; Kaemmer, P.; Linde, J.; Brunke, S.; Kasper, L.; Hube, B. A Novel Hybrid Iron Regulation Network Combines Features from Pathogenic and Nonpathogenic Yeasts. mBio 2016, 7, e01782-16. [Google Scholar] [CrossRef]
- Massé, E.; Gottesman, S. A small RNA regulates the expression of genes involved in iron metabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 4620–4625. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.W.; Kim, D.; Latif, H.; O’Brien, E.J.; Szubin, R.; Palsson, B.O. Deciphering Fur transcriptional regulatory network highlights its complex role beyond iron metabolism in Escherichia coli. Nat. Commun. 2014, 5, 4910. [Google Scholar] [CrossRef] [PubMed]
- Serafini, A. Interplay between central carbon metabolism and metal homeostasis in mycobacteria and other human pathogens. Microbiology 2021, 167, 001060. [Google Scholar] [CrossRef]
- Rosas, H.D.; Chen, Y.I.; Doros, G.; Salat, D.H.; Chen, N.; Kwong, K.K.; Bush, A.; Fox, J.; Hersch, S.M. Alterations in brain transition metals in Huntington disease: An evolving and intricate story. Arch. Neurol. 2012, 69, 887–893. [Google Scholar] [CrossRef] [PubMed]
- Lan, A.P.; Chen, J.; Chai, Z.F.; Hu, Y. The neurotoxicity of iron, copper and cobalt in Parkinson’s disease through ROS-mediated mechanisms. Biometals 2016, 29, 665–678. [Google Scholar] [CrossRef]
- Sun, Y.; Pham, A.N.; Waite, T.D. Elucidation of the interplay between Fe(II), Fe(III), and dopamine with relevance to iron solubilization and reactive oxygen species generation by catecholamines. J. Neurochem. 2016, 137, 955–968. [Google Scholar] [CrossRef]
- Fernández-Real, J.M.; López-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef]
- Huang, J.; Simcox, J.; Mitchell, T.C.; Jones, D.; Cox, J.; Luo, B.; Cooksey, R.C.; Boros, L.G.; McClain, D.A. Iron regulates glucose homeostasis in liver and muscle via AMP-activated protein kinase in mice. FASEB J. 2013, 27, 2845–2854. [Google Scholar] [CrossRef]
- Zheng, Q.; Zhao, Y.; Guo, J.; Zhao, S.; Fei, C.; Xiao, C.; Wu, D.; Wu, L.; Li, X.; Chang, C. Iron overload promotes mitochondrial fragmentation in mesenchymal stromal cells from myelodysplastic syndrome patients through activation of the AMPK/MFF/Drp1 pathway. Cell Death Dis. 2018, 9, 515. [Google Scholar] [CrossRef]
- Higashida, K.; Takeuchi, N.; Inoue, S.; Hashimoto, T.; Nakai, N. Iron deficiency attenuates catecholamine-stimulated lipolysis via downregulation of lipolysis-related proteins and glucose utilization in 3T3-L1 adipocytes. Mol. Med. Rep. 2020, 21, 1383–1389. [Google Scholar] [CrossRef]
- Knop, M.; Siegers, K.; Pereira, G.; Zachariae, W.; Winsor, B.; Nasmyth, K.; Schiebel, E. Epitope tagging of yeast genes using a PCR-based strategy: More tags and improved practical routines. Yeast 1999, 15, 963–972. [Google Scholar] [CrossRef]
- Ma, H.; Kunes, S.; Schatz, P.J.; Botstein, D. Plasmid construction by homologous recombination in yeast. Gene 1987, 58, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Xu, X.; Carlson, M. Interaction of SNF1 protein kinase with its activating kinase Sak1. Eukaryot. Cell 2011, 10, 313–319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Germann, S.M.; Schramke, V.; Pedersen, R.T.; Gallina, I.; Eckert-Boulet, N.; Oestergaard, V.H.; Lisby, M. TopBP1/Dpb11 binds DNA anaphase bridges to prevent genome instability. J. Cell Biol. 2014, 204, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.; Orlova, M.; Maziarz, M.; Kuchin, S. Protein kinase A contributes to the negative control of Snf1 protein kinase in Saccharomyces cerevisiae. Eukaryot. Cell 2012, 11, 119–128. [Google Scholar] [CrossRef]
- Coonrod, E.M.; Graham, L.A.; Carpp, L.N.; Carr, T.M.; Stirrat, L.; Bowers, K.; Bryant, N.J.; Stevens, T.H. Homotypic vacuole fusion in yeast requires organelle acidification and not the V-ATPase membrane domain. Dev. Cell 2013, 27, 462–468. [Google Scholar] [CrossRef]
- Remy, I.; Michnick, S.W. Mapping Biochemical Networks with Protein-Fragment Complementation Assays. In Protein-Protein Interactions; Fu, H., Ed.; Humana Press: Totowa, NJ, USA, 2004; pp. 411–426. ISBN 1-59259-762-9. [Google Scholar]
- Orlova, M.; Barrett, L.; Kuchin, S. Detection of endogenous Snf1 and its activation state: Application to Saccharomyces and Candida species. Yeast 2008, 25, 745–754. [Google Scholar] [CrossRef]
- Wilson, W.A.; Hawley, S.A.; Hardie, D. Glucose repression/derepression in budding yeast: SNF1 protein kinase is activated by phosphorylation under derepressing conditions, and this correlates with a high AMP:ATP ratio. Curr. Biol. 1996, 6, 1426–1434. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Name | Genotype | From |
|---|---|---|---|
| 1 | WT | W303 MATa ade2-1 his3-11,15 trp1-1 leu2-3,112 ura3-1 rad5-535 bud4 | Lab collection |
| 2 | snf1 | W303a snf1::HYG | E. Young [25] |
| 3 | reg1 | W303a MATα reg1::NAT | E. Young [25] |
| 4 | snf1 reg1 | W303a snf1:: HYG reg1::NAT | E. Young [25] |
| 5 | snf1 hsp30 | W303a snf1:: HYG hsp30::KAN | [42] |
| 6 | PJ694 | trp1-901 leu2-3,112, ura3-52, his3Δ200, gal4Δ, gal80Δ, prGAL2:ADE2 lys2::prGAL1::HIS3 met2::prGAL7-LacZ | Lab collection |
| 7 | adr1 | W303a adr1::NAT | E. Young [25] |
| 11 | snf1 aft1 | W303a snf1::HYG aft1::NAT | This study |
| 12 | snf1 aft1 Rod1-6HA | W303a snf1::HYG Rod1-6HA::NAT aft1::KAN | This study |
| 13 | snf1 aft2 | W303a snf1::HYG aft2::NAT | This study |
| 14 | snf1 aft1 msn5 | W303a snf1::HYG aft1::KAN msn5::NAT | This study |
| 15 | snf1 msn5 | W303a snf1::HYG msn5::NAT | This study |
| 16 | snf1 sip1 sip2 | W303a snf1::HYG sip1::NAT sip2::KAN | This study |
| 17 | gal83 | W303a gal83::NAT | This study |
| 18 | snf1 gal83 | W303a snf1::HYG gal83::NAT | This study |
| 19 | rod1 | W303a rod1::NAT | This study |
| 20 | snf1 nup133 | W303a snf1::HYG nup133::NAT | This study |
| 21 | PJ694 hsp30 | PJ694 hsp30::NAT | [42] |
| 22 | PJ694 nup120 | PJ694nup120::NAT | This study |
| 23 | PJ694 nup133 | PJ694nup133::NAT | This study |
| Number | Genotype | Backbone | Source/Notes |
|---|---|---|---|
| 152 | Snf1-GFP | pPRS313 | M. Carlson [36] |
| 728 | Snf1-GFP | pRS315 | Marker switch of 152 |
| 847 | Snf1-ΔH-GFP | pRS315 | polyHIS of 728 deleted |
| 742 | Snf1-A-GFP | pRS315 | Alanine substituted for histidine in PKR of 728 |
| 745 | Snf1-W -GFP | pRS315 | Tryptophan substituted for histidine in PKR of 728 |
| 731 | Snf1-Y-GFP | pRS315 | Tyrosine substituted for histidine in PKR of 728 |
| 1244 | Snf1-4H -GFP | pRS315 | Four histidines inserted into 847 |
| 1248 | Snf1-8H-GFP | pRS315 | Eight histidines inserted into 847 |
| 816 | Snf1-3HA | pRS313 | M. Carlson [83] |
| 819 | Snf1-ΔH-3HA | pRS313 | polyHIS of 816 deleted |
| 1 or 919 | prADH2::LacZ | pRS316 | E. Young [25] |
| 63 | prADH2::LacZ | pRS313 | Marker switch of 1 |
| 164 | prADH2::LacZ | pRS315 | Marker switch of 1 |
| 526 | Nup49-Cherry | pRS314 | M. Lisby [84] |
| 589 | Nup49-Cherry | pRS316 | Marker switch of 526 |
| 590 | Nup49-Cherry | pRS315 | Marker switch of 526 |
| 1020 | Adr1-3HA | 2μ TRP1 KanMX | E. Young [25] |
| 1021 | Adr1S230A-3HA | 2μ TRP1 KanMX | E. Young [25] |
| 1081 | Aft1-1up | pRS416 | D. Winge [16] |
| 1160 | Aft1-1up | pRS413 | Marker switch of 1081 |
| 935 | prFET3::LacZ | Yep354 (2μ URA3) | V. Costa [50] |
| 931 | Aft1-HA | pRS416 | V. Costa [50] |
| 932 | Aft1-S210D S224D-HA | pRS416 | V. Costa [50] |
| 933 | GFP-Aft1 | pRS426 | V. Costa |
| 934 | 4t-2GST-Aft1-S210A S224A | pGEX | V. Costa [50] |
| 1208 | Aft1-S210A S224A-HA | pRS416 | Fragment from 934 replacing fragment in 931 |
| 1206 | Aft1-HA | pRSs415 | Marker switch of 931 |
| 1202 | Aft1-S210D S224D-HA | pRS415 | Marker switch of 932 |
| 1174 | GFP-Aft1 | pRS425 | Marker switch of 933 |
| 1212 | Aft1-S210A S224-HA | pRS415 | Marker switch of 1208 |
| 1172 | GFP-Aft1 Δ9 | pRS426 | Aa16-24 deleted in 933 |
| 1173 | GFP-Aft1 Δ9 | pRS425 | Marker switch of 1172 |
| 1195 | GFP-Aft1 Δ24 | pRS426 | Aa16-36 deleted in 933 |
| 1199 | GFP-Aft1 Δ24 | pRS425 | Marker switch of 1195 |
| 1204 | GFP-Aft1 S210A, S224A | pRS426 | S210A S224A mutations in 933 |
| 1205 | GFP-Aft1 S210D, S224D | pRS426 | S210D S224D mutations in 933 |
| 1234 | Pma1 | pRS314 | Pma1 inserted into prs314 |
| 1238 | Pma1-Δ901 | pRS314 | Final 18aa of Pma1 deleted |
| 216 | prSUC2::LacZ | Yep354 (2μ URA3) | S. Kuchin [85] |
| 722 | prYRO2::LacZ | pRS416 | Z. Liu [48] |
| 771 | prGAL1::Jen1-GFP | pRS316 | S. Leon [57] |
| 230 | prVph1::Cherry-Pho8 | pRS315 | T. Stevens [86] |
| Yeast 2-hybrid plasmids | |||
| 153 | prADH1::GAD | LEU2, 2μ | pACT2 yeast 2-hybrid empty vector |
| 154 | prADH1::GBD | TRP1, 2μ | pGBT9 yeast 2-hybrid empty vector |
| 155 | prADH1::GBD | URA3, 2μ | pGBU9 yeast 2-hybrid empty vector |
| 757 | GAD-Gal83 | LEU2, 2μ | Gal83 in 153 |
| 762 | GBD-Snf1 | URA3, 2μ | Snf1 in 155 |
| 764 | GBD-Snf1 T210A | URA3, 2μ | Snf1 T210A in 155 |
| 789 | GBD-Snf1 ΔH | URA3, 2μ | polyHIS deletion of 762 |
| 780 | GBD-Snf1 W | URA3, 2μ | Tryptophan substituted for polyHIS in 762 |
| 781 | GBD-Snf1 1-391 | URA3, 2μ | Snf1 1-391 in 155 |
| 849 | GBD-Snf1 1-391 T210A | URA3, 2μ | Snf1 1-391 T210A in 155 |
| 795 | GBD-Snf1 1-391 ΔH | URA3, 2μ | Snf1 1-391 ΔH in 155 |
| 785 | GBD-Snf1 1-391 W | URA3, 2μ | Snf1 1-391 W in 155 |
| 828 | GBD-Snf1 1-53 | URA3, 2μ | Snf1 1-53 in 155 |
| 836 | GBD-Snf1 1-53 ΔH | URA3, 2μ | Snf1 1-53 ΔH in 155 |
| 829 | GBD-Snf1 1-53 A | URA3, 2μ | Snf1 1-53 A in 155 |
| 831 | GBD-Snf1 1-53 R | URA3, 2μ | Snf1 1-53 R in 155 |
| 837 | GBD-Snf1 1-53 W | URA3, 2μ | Snf1 1-53 W in 155 |
| 786 | GBD-Snf1 392-633 | URA3, 2μ | Snf1 392-633 in 155 |
| 991 | GBD-Snf1 1-53 | TRP1, 2μ | Snf1 1-53 in 154 |
| 993 | GBD-Snf1 1-53 ΔH | TRP1, 2μ | Snf1 1-53 ΔH in 154 |
| 1013 | GBD-Snf1 1-53 A | TRP1, 2μ | Snf1 1-53 A in 154 |
| 1261 | GBD-Snf1 1-53 R | TRP1, 2μ | Snf1 1-53 R in 154 |
| 1012 | prADH1::Snf1 1-53 | TRP1, 2μ | Snf1 1-53 in 154 (no GBD) |
| 1004 | prADH1::Snf1 1-53 ΔH | TRP1, 2μ | Snf1 1-53 ΔH in 154 (no GBD) |
| 1019 | prADH1::Snf1 1-53 A | TRP1, 2μ | Snf1 1-53 A in 154 (no GBD) |
| 1267 | prADH1::Snf1 1-53 R | TRP1, 2μ | Snf1 1-53 R in 154 (no GBD) |
| 857 | GAD-Snf1 | LEU2, 2μ | Snf1 1-633 in 153 |
| 1210 | GBD-Aft1 | TRP1, 2μ | Aft1 in 154 |
| 1217 | GBD-Aft1 1-44 | TRP1, 2μ | Aft1 1-44 in 154 |
| 1211 | GBD-Aft1 Δ9 | TRP1, 2μ | Aft1 Δ9 in 154 |
| 1218 | GBD-Aft1 1-44 Δ9 | TRP1, 2μ | Aft1 1-44 Δ9 in 154 |
| 1229 | GAD-Aft1 | LEU2, 2μ | Aft1 in 153 |
| Venus Constructs | |||
| 4 | prADH1::VF1 | pRS413 | P. Chartrand [87] |
| 8 | prADH1::VF2 | pRS415 | P. Chartrand [87] |
| 1168 | prSNF1::VF1-Snf1-3HA | pRS413 | VF1 inserted at N-terminus of 816 |
| 1170 | prSNF1::VF1-Snf1 ΔH-3HA | pRS413 | VF1 inserted at N-terminus of 819 |
| 1149 | prADH1::Aft1-VF2 | pRS415 | Aft1 inserted into 8 |
| 1150 | prADH1::Aft1 S210D S224D-VF2 | pRS415 | Aft1 S210D S224D inserted into 8 |
| Name | Description | Sequence |
|---|---|---|
| Gap Repair Mutagenesis of polyHIS | ||
| C54 | Y-5 | CTACTATTATTACTATTATTACTACTATTACTATTATGGATATggcggaagcaactcgacg |
| C53 | Y-3 | cataatagtaatagtagtaataatagtaataatagtagtaGCTAGAATTTGCATTGGCAGGTG |
| C57 | A-5 | GCAGCTGCCGCAGCAGCCGCTGCCGCAGCAGCTGCAGCAGGAGCCggcggaagcaactcgacg |
| C58 | A-3 | ggctcctgctgcagctgctgcggcagcggctgctgcggcagctgcGCTAGAATTTGCATTGGCAGGTG |
| C74 | F-5 | CTTTTTCTTCTTCTTTTTCTTCTTCTTCTTCTTCTTTGGATTTggcggaagcaactcgacg |
| C73 | F-3 | caaagaagaagaagaagaagaaaaagaagaagaaaaagaaGCTAGAATTTGCATTGGCAGGTG |
| C107 | K-5 | aaaaagaaaaagaaaaagaagaaaaaaaagaaaaaaaaaggtaagGGCGGAAGCAACTCGACG |
| C106 | K-3 | cttacctttttttttcttttttttcttctttttctttttctttttGCTAGAATTTGCATTGGCAGGTG |
| C115 | R-5 | cgtagaaggagacgtcgtaggagaagacgtaggcgtaggggtaggGGCGGAAGCAACTCGACG |
| C114 | R-3 | cctacccctacgcctacgtcttctcctacgacgtctccttctacgGCTAGAATTTGCATTGGCAGGTG |
| C104 | W-5 | tggtggtggtggtggtggtggtggtggtggtggtggtggggttggGGCGGAAGCAACTCGACG |
| C103 | W-3 | ccaaccccaccaccaccaccaccaccaccaccaccaccaccaccaGCTAGAATTTGCATTGGCAGGTG |
| C117 | Delta-5 | GGCGGAAGCAACTCGACG |
| C116 | Delta-3 | caccatccgctaaggacgacttgggattgtttagcgtcgagttgcttccgccGCTAGAATTTGCATTGGCAG |
| Mutagenesis of Aft1 | ||
| C271 | I15-rev | AATCGGTGACGCATGTTCTATG |
| C272 | Δ9-fwd | GAAGGCTTCAATCCGGCTGACATAGAACATGCGTCACCGATTTTTGTATATGCTCTACCCAAAAGTGC |
| C275 | Δ21-fwd | GTAGTCAACCATAATGAGGGTCG |
| Mutagenesis of Snf1 | ||
| M69 * | 4H-3 | ATGGTGATGGTGGCTAGAATTTGCATTGGCAGGTGCTGTG |
| M70 * | 8H-3 | ATGGTGGTGATGGTGGTGGTGGTGGCTAGAATTTGCATTGGCAGGTGCTGTG |
| Pma1 Truncation | ||
| C292 | PMA1-5 | gaattgtaatacgactcactatagggcgaattggagctccgcttcctgaaacggagaaac |
| C293 | PMA1-3 | cctcactcattaggcaccccaggctttacactttatgcttccggctccgtaaaggtatttcgcggagg |
| C294 * | Pma1-trunc5 | TAAtcctgttgaagtagcatttaatc |
| C295 * | Pma1-trunc3 | GACACTTCTGGTAGACTTCTTTTC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Simpson-Lavy, K.J.; Kupiec, M. The polyHIS Tract of Yeast AMPK Coordinates Carbon Metabolism with Iron Availability. Int. J. Mol. Sci. 2023, 24, 1368. https://doi.org/10.3390/ijms24021368
Simpson-Lavy KJ, Kupiec M. The polyHIS Tract of Yeast AMPK Coordinates Carbon Metabolism with Iron Availability. International Journal of Molecular Sciences. 2023; 24(2):1368. https://doi.org/10.3390/ijms24021368
Chicago/Turabian StyleSimpson-Lavy, Kobi J., and Martin Kupiec. 2023. "The polyHIS Tract of Yeast AMPK Coordinates Carbon Metabolism with Iron Availability" International Journal of Molecular Sciences 24, no. 2: 1368. https://doi.org/10.3390/ijms24021368
APA StyleSimpson-Lavy, K. J., & Kupiec, M. (2023). The polyHIS Tract of Yeast AMPK Coordinates Carbon Metabolism with Iron Availability. International Journal of Molecular Sciences, 24(2), 1368. https://doi.org/10.3390/ijms24021368