

Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
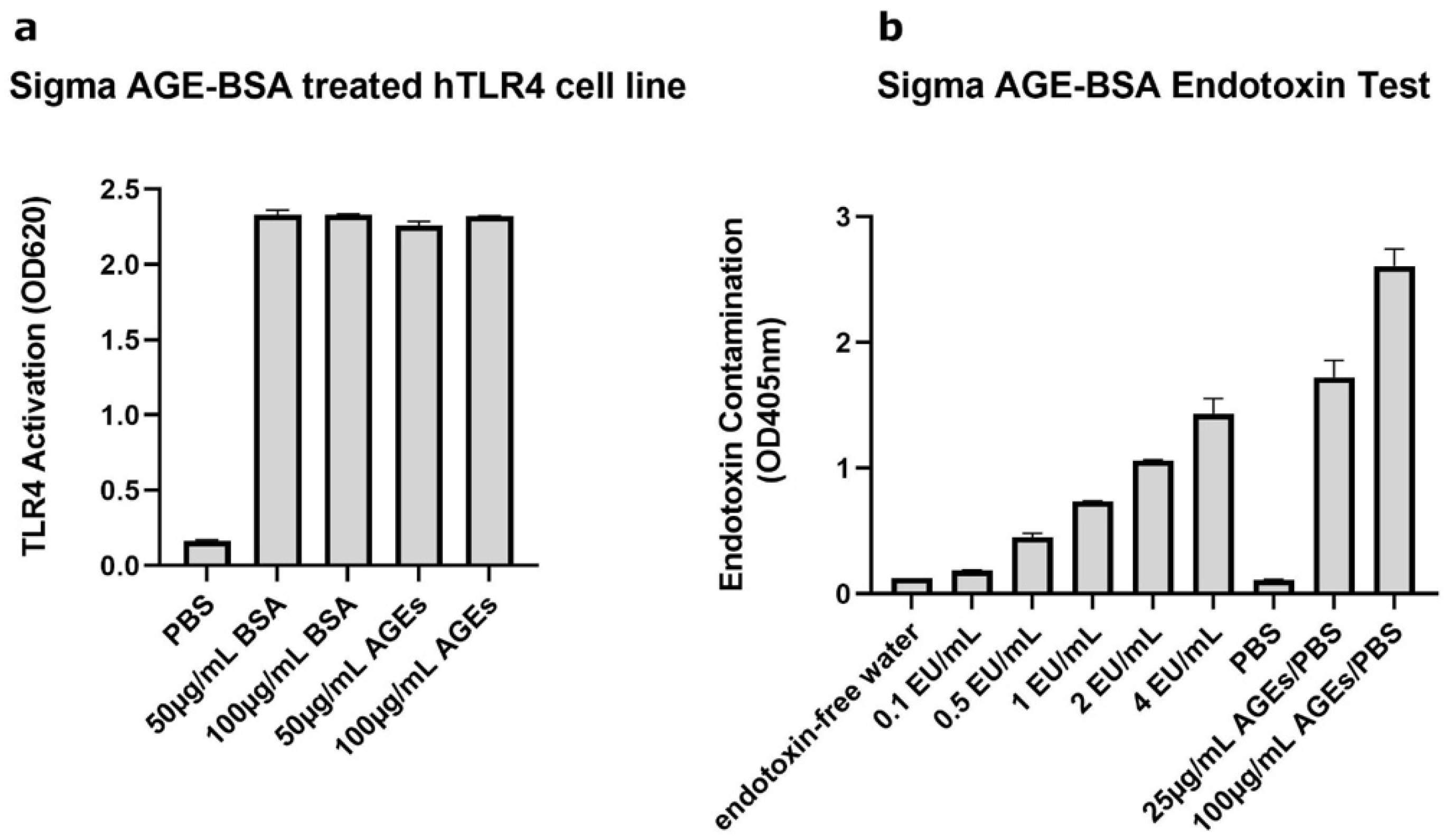
2.1. AGE-BSA Used Initially Was Contaminated with Endotoxin
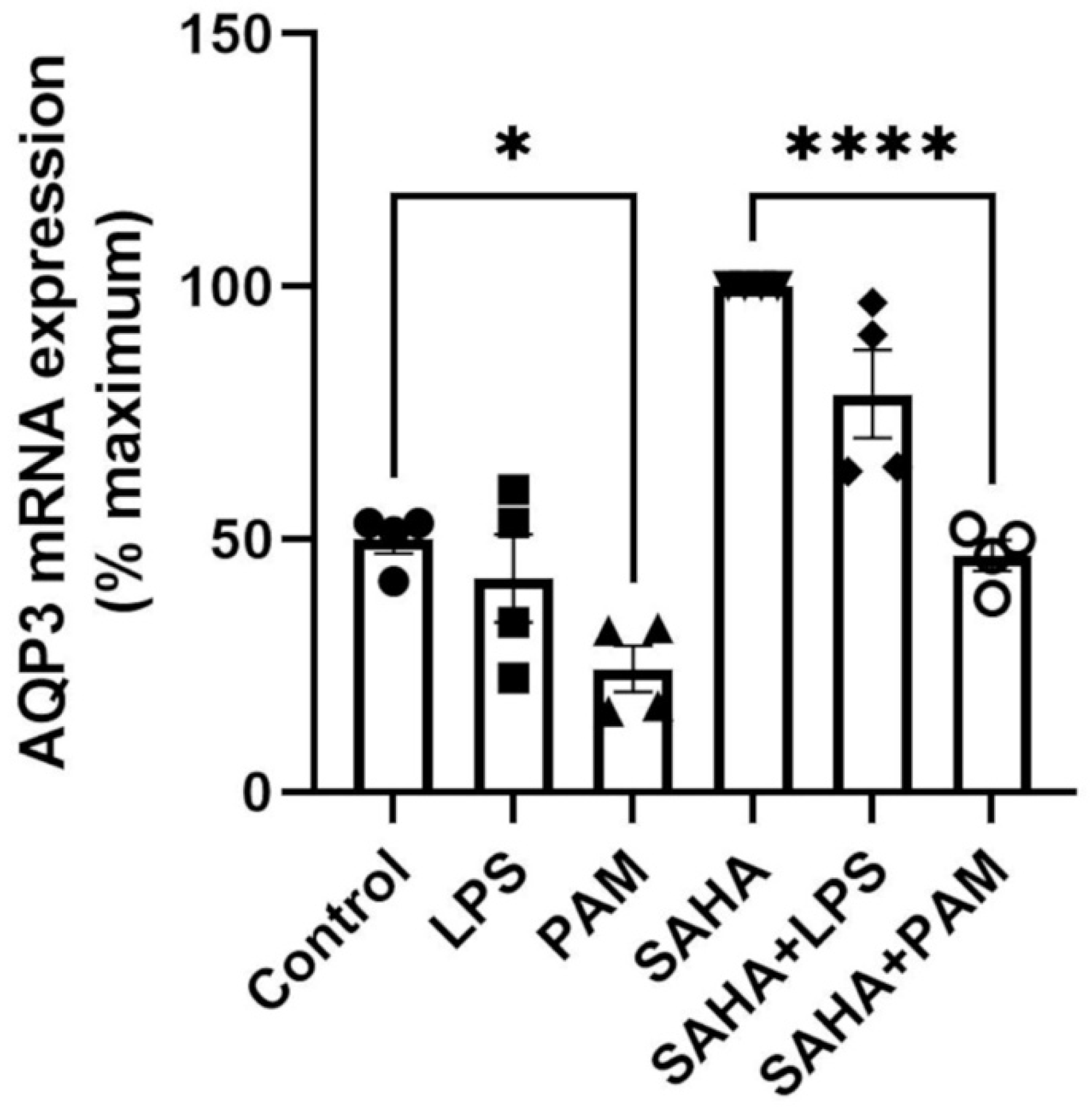
2.2. TLR2 Activation Downregulated AQP3 mRNA Expression; TLR2/TLR4 Activation Upregulated AQP3 Protein in the Absence of SAHA, and TLR2 Activation Downregulated AQP3 Protein in the Presence of SAHA in Primary Mouse Epidermal Keratinocytes
2.3. AGE-BSA from BioVision, Inc. Was Not Contaminated with TLR-Activating Endotoxin
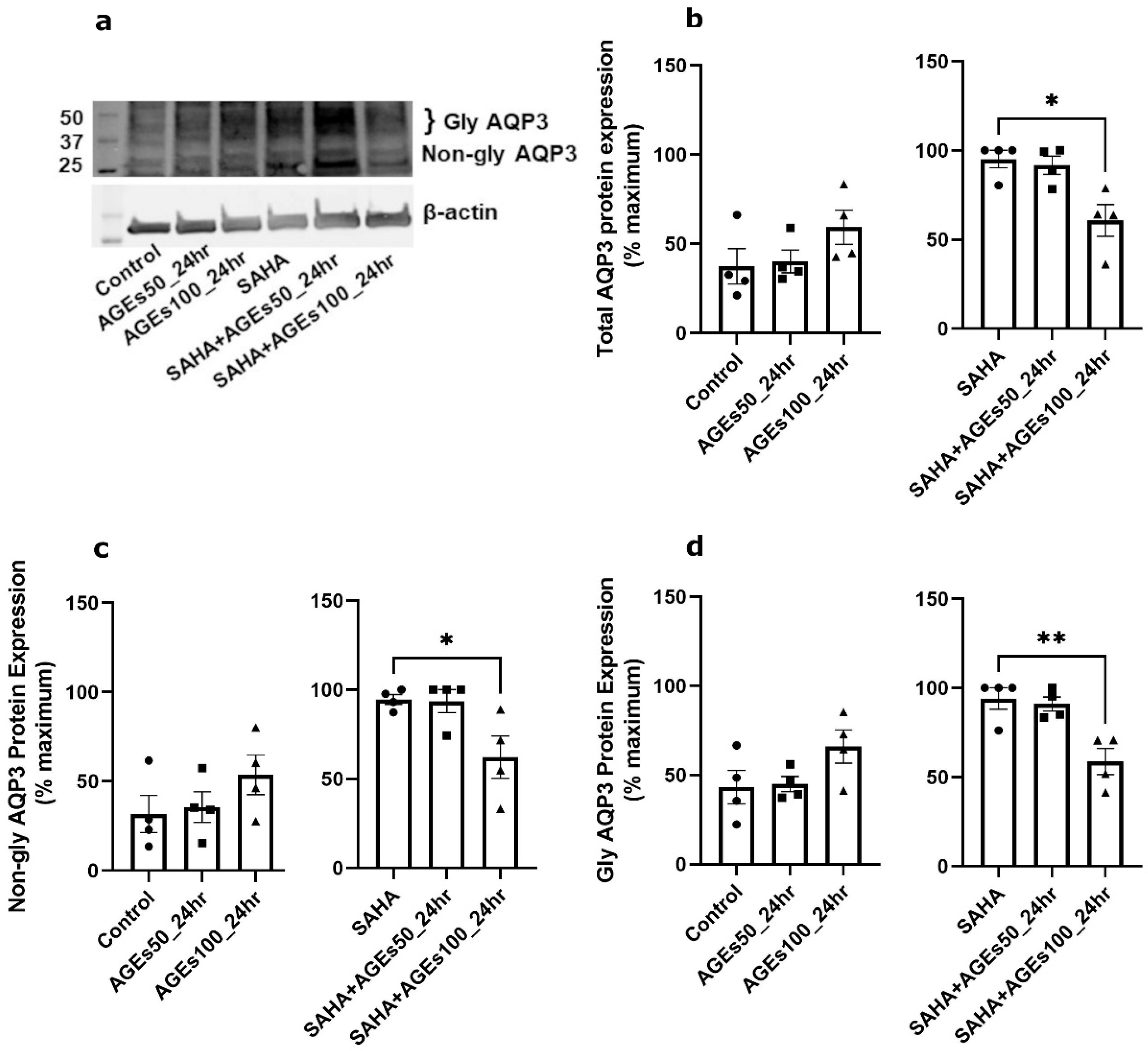
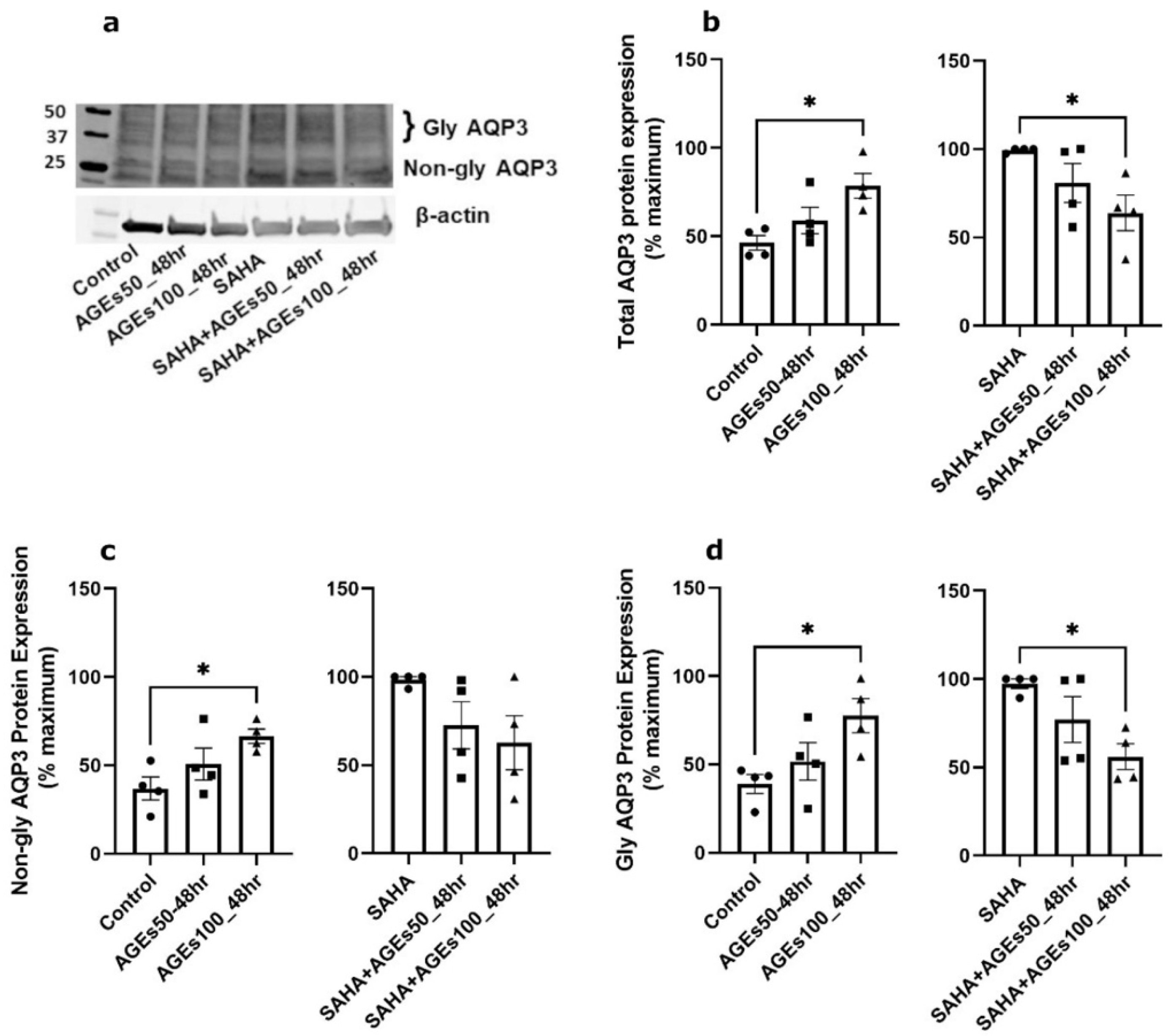
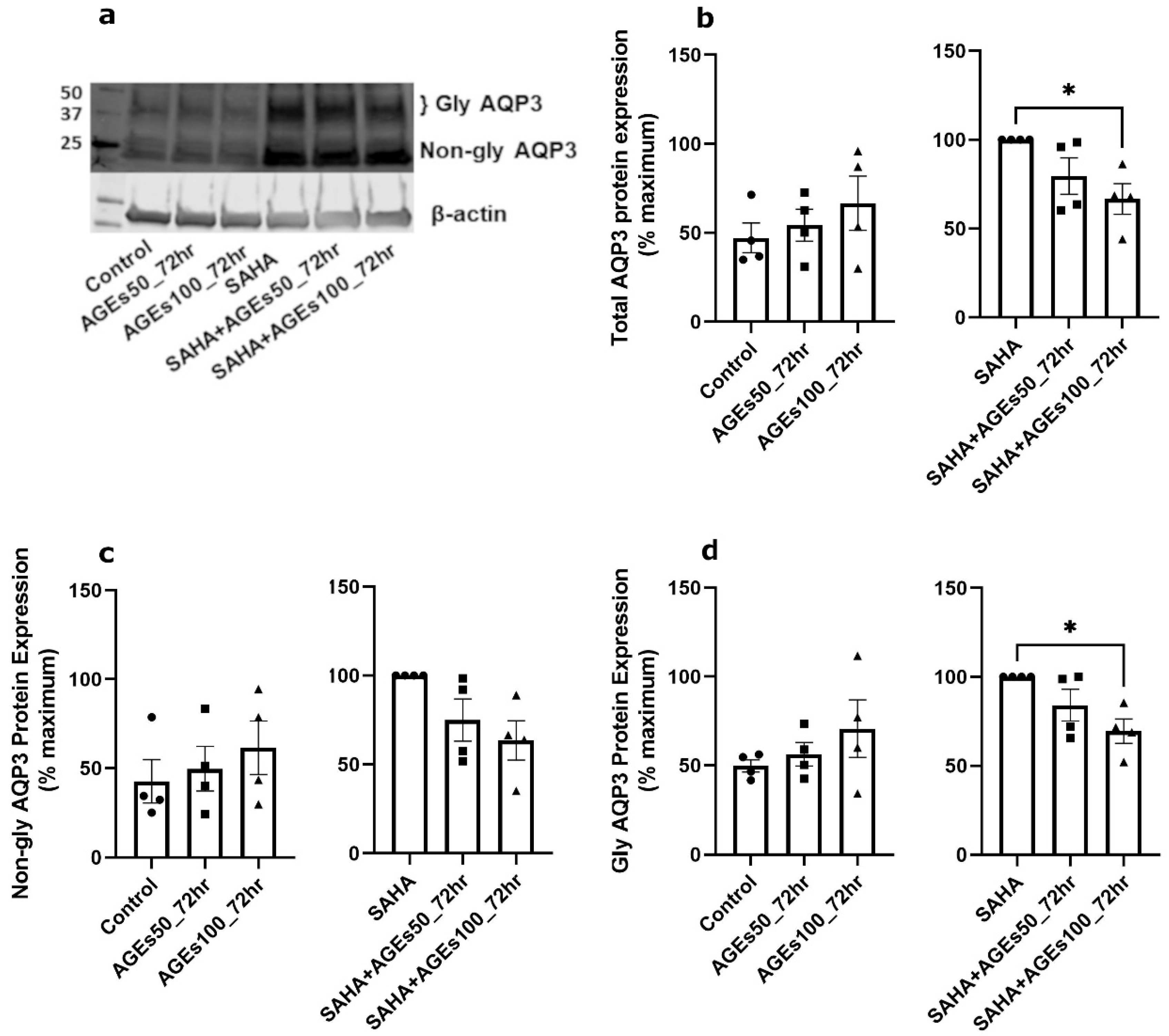
2.4. AGEs Increased AQP3 Protein in the Absence of SAHA but Reduced AQP3 Protein in Its Presence in Primary Mouse Epidermal Keratinocytes
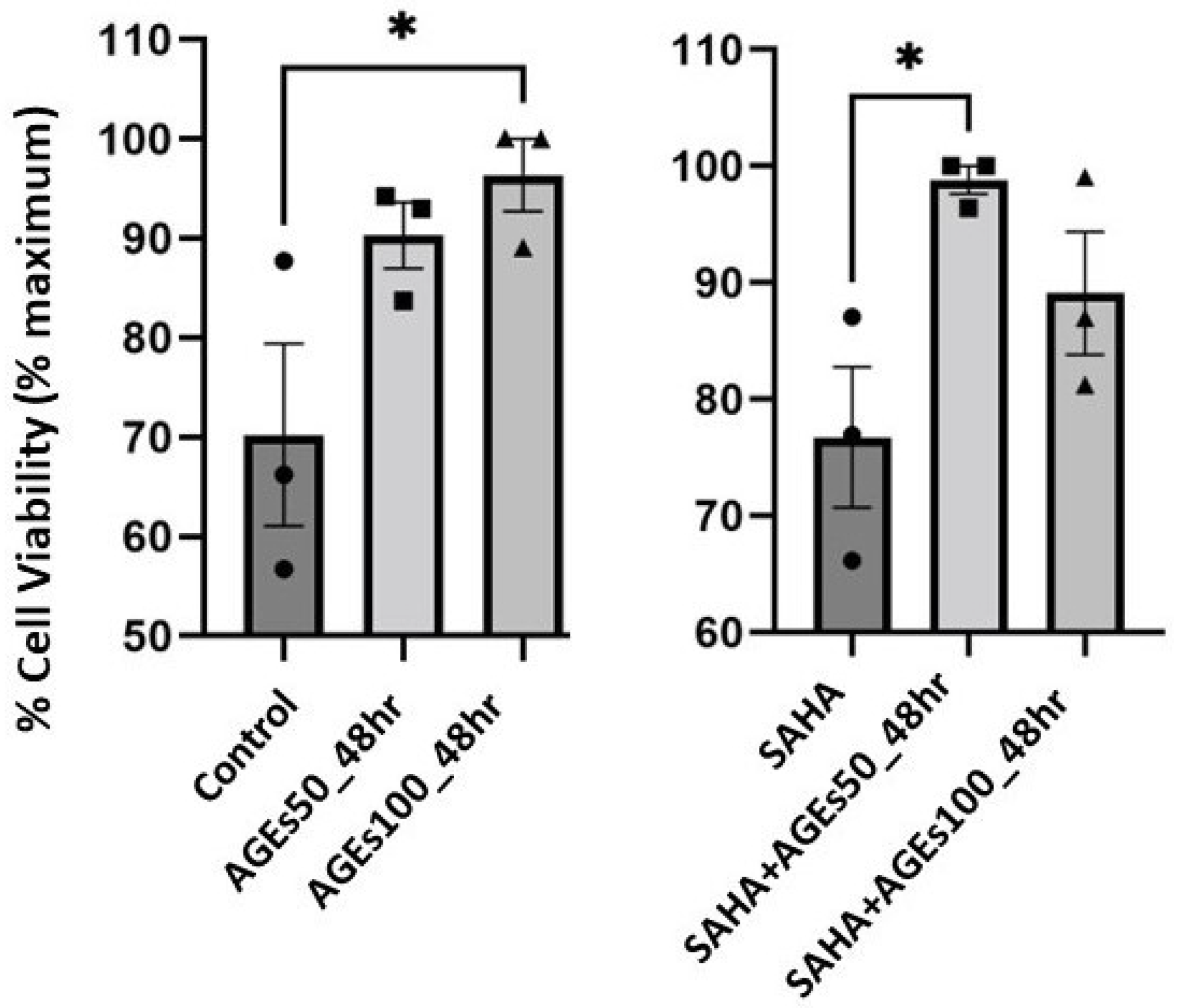
2.5. In Primary Mouse Epidermal Keratinocytes, AGEs, but Not SAHA, Increased Cell Number, a Measure of Proliferation
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. Quantitative RT-PCR
4.3. Western Blotting
4.4. BSA-AGEs Contamination Test
4.5. MTT Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; DiPietro, L.A. Toll-Like Receptor Function in Acute Wounds. Adv. Wound Care 2017, 6, 344–355. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, H.N.; Hardman, M.J. Wound healing: Cellular mechanisms and pathological outcomes. Open Biol. 2020, 10, 200223. [Google Scholar] [CrossRef] [PubMed]
- Seite, S.; Khemis, A.; Rougier, A.; Ortonne, J.P. Importance of treatment of skin xerosis in diabetes. J. Eur. Acad. Dermatol. Venereol. 2011, 25, 607–609. [Google Scholar] [CrossRef] [PubMed]
- Hara-Chikuma, M.; Satooka, H.; Watanabe, S.; Honda, T.; Miyachi, Y.; Watanabe, T.; Verkman, A.S. Aquaporin-3-mediated hydrogen peroxide transport is required for NF-kappaB signalling in keratinocytes and development of psoriasis. Nat. Commun. 2015, 6, 7454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara-Chikuma, M.; Chikuma, S.; Sugiyama, Y.; Kabashima, K.; Verkman, A.S.; Inoue, S.; Miyachi, Y. Chemokine-dependent T cell migration requires aquaporin-3-mediated hydrogen peroxide uptake. J. Exp. Med. 2012, 209, 1743–1752. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.W.; Dickinson, B.C.; Chang, C.J. Aquaporin-3 mediates hydrogen peroxide uptake to regulate downstream intracellular signaling. Proc. Natl. Acad. Sci. USA 2010, 107, 15681–15686. [Google Scholar] [CrossRef] [Green Version]
- Ecelbarger, C.A.; Terris, J.; Frindt, G.; Echevarria, M.; Marples, D.; Nielsen, S.; Knepper, M.A. Aquaporin-3 water channel localization and regulation in rat kidney. Am. J. Physiol. 1995, 269, F663–F672. [Google Scholar] [CrossRef] [Green Version]
- Echevarria, M.; Windhager, E.E.; Frindt, G. Selectivity of the renal collecting duct water channel aquaporin-3. J. Biol. Chem. 1996, 271, 25079–25082. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Verkman, A.S. Water and glycerol permeabilities of aquaporins 1-5 and MIP determined quantitatively by expression of epitope-tagged constructs in Xenopus oocytes. J. Biol. Chem. 1997, 272, 16140–16146. [Google Scholar] [CrossRef] [Green Version]
- Hara-Chikuma, M.; Verkman, A.S. Aquaporin-3 facilitates epidermal cell migration and proliferation during wound healing. J. Mol. Med. 2008, 86, 221–231. [Google Scholar] [CrossRef]
- Hara-Chikuma, M.; Takahashi, K.; Chikuma, S.; Verkman, A.S.; Miyachi, Y. The expression of differentiation markers in aquaporin-3 deficient epidermis. Arch. Dermatol. Res. 2009, 301, 245–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahigashi, K.; Kabashima, K.; Ikoma, A.; Verkman, A.S.; Miyachi, Y.; Hara-Chikuma, M. Upregulation of aquaporin-3 is involved in keratinocyte proliferation and epidermal hyperplasia. J. Investig. Dermatol. 2011, 131, 865–873. [Google Scholar] [CrossRef] [Green Version]
- Bollag, W.B.; Aitkens, L.; White, J.; Hyndman, K.A. Aquaporin-3 in the epidermis: More than skin deep. Am. J. Physiol. Cell Physiol. 2020, 318, C1144–C1153. [Google Scholar] [CrossRef] [PubMed]
- Bollag, W.B.; Xie, D.; Zheng, X.; Zhong, X. A potential role for the phospholipase D2-aquaporin-3 signaling module in early keratinocyte differentiation: Production of a phosphatidylglycerol signaling lipid. J. Investig. Dermatol. 2007, 127, 2823–2831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voss, K.E.; Bollag, R.J.; Fussell, N.; By, C.; Sheehan, D.J.; Bollag, W.B. Abnormal aquaporin-3 protein expression in hyperproliferative skin disorders. Arch. Dermatol. Res. 2011, 303, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, V.; Olala, L.O.; Qin, H.; Helwa, I.; Pan, Z.Q.; Tsai, Y.Y.; Frohman, M.A.; Kaddour-Djebbar, I.; Bollag, W.B. Aquaporin-3 re-expression induces differentiation in a phospholipase D2-dependent manner in aquaporin-3-knockout mouse keratinocytes. J. Investig. Dermatol. 2015, 135, 499–507. [Google Scholar] [CrossRef] [Green Version]
- Kim, N.H.; Lee, A.Y. Reduced aquaporin3 expression and survival of keratinocytes in the depigmented epidermis of vitiligo. J. Investig. Dermatol. 2010, 130, 2231–2239. [Google Scholar] [CrossRef] [Green Version]
- Hara-Chikuma, M.; Verkman, A.S. Prevention of skin tumorigenesis and impairment of epidermal cell proliferation by targeted aquaporin-3 gene disruption. Mol. Cell. Biol. 2008, 28, 326–332. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Chen, H.; Li, Y.; Zhou, Q.; Sui, Y. An aquaporin 3-notch1 axis in keratinocyte differentiation and inflammation. PLoS ONE 2013, 8, e80179. [Google Scholar] [CrossRef]
- Hara, M.; Ma, T.; Verkman, A.S. Selectively reduced glycerol in skin of aquaporin-3-deficient mice may account for impaired skin hydration, elasticity, and barrier recovery. J. Biol. Chem. 2002, 277, 46616–46621. [Google Scholar] [CrossRef]
- Ma, T.; Song, Y.; Yang, B.; Gillespie, A.; Carlson, E.J.; Epstein, C.J.; Verkman, A.S. Nephrogenic diabetes insipidus in mice lacking aquaporin-3 water channels. Proc. Natl. Acad. Sci. USA 2000, 97, 4386–4391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikarashi, N.; Mizukami, N.; Pei, C.; Uchino, R.; Fujisawa, I.; Fukuda, N.; Kon, R.; Sakai, H.; Kamei, J. Role of Cutaneous Aquaporins in the Development of Xeroderma in Type 2 Diabetes. Biomedicines 2021, 9, 104. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Huang, L.; Minematsu, T.; Yamamoto, Y.; Asada, M.; Nakagami, G.; Akase, T.; Nagase, T.; Oe, M.; Mori, T.; et al. Impaired aquaporin 3 expression in reepithelialization of cutaneous wound healing in the diabetic rat. Biol. Res. Nurs. 2013, 15, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ikarashi, N.; Mizukami, N.; Kon, R.; Kaneko, M.; Uchino, R.; Fujisawa, I.; Fukuda, N.; Sakai, H.; Kamei, J. Study of the Mechanism Underlying the Onset of Diabetic Xeroderma Focusing on an Aquaporin-3 in a Streptozotocin-Induced Diabetic Mouse Model. Int. J. Mol. Sci. 2019, 20, 3782. [Google Scholar] [CrossRef] [Green Version]
- Goh, S.Y.; Cooper, M.E. Clinical review: The role of advanced glycation end products in progression and complications of diabetes. J. Clin. Endocrinol. Metab. 2008, 93, 1143–1152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gkogkolou, P.; Bohm, M. Advanced glycation end products: Key players in skin aging? Dermatoendocrinol 2012, 4, 259–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luevano-Contreras, C.; Chapman-Novakofski, K. Dietary advanced glycation end products and aging. Nutrients 2010, 2, 1247–1265. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Zhu, G.; Cao, X.; Dong, J.; Song, F.; Niu, Y. Blocking AGE-RAGE Signaling Improved Functional Disorders of Macrophages in Diabetic Wound. J. Diabetes Res. 2017, 2017, 1428537. [Google Scholar] [CrossRef] [Green Version]
- Goova, M.T.; Li, J.; Kislinger, T.; Qu, W.; Lu, Y.; Bucciarelli, L.G.; Nowygrod, S.; Wolf, B.M.; Caliste, X.; Yan, S.F.; et al. Blockade of receptor for advanced glycation end-products restores effective wound healing in diabetic mice. Am. J. Pathol. 2001, 159, 513–525. [Google Scholar] [CrossRef] [Green Version]
- Kadiyala, C.S.; Zheng, L.; Du, Y.; Yohannes, E.; Kao, H.Y.; Miyagi, M.; Kern, T.S. Acetylation of retinal histones in diabetes increases inflammatory proteins: Effects of minocycline and manipulation of histone acetyltransferase (HAT) and histone deacetylase (HDAC). J. Biol. Chem. 2012, 287, 25869–25880. [Google Scholar] [CrossRef]
- Mosley, A.L.; Ozcan, S. Glucose regulates insulin gene transcription by hyperacetylation of histone h4. J. Biol. Chem. 2003, 278, 19660–19666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosley, A.L.; Corbett, J.A.; Ozcan, S. Glucose regulation of insulin gene expression requires the recruitment of p300 by the beta-cell-specific transcription factor Pdx-1. Mol. Endocrinol. 2004, 18, 2279–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampley, M.L.; Ozcan, S. Regulation of insulin gene transcription by multiple histone acetyltransferases. DNA Cell Biol. 2012, 31, 8–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhong, J.; Zhang, X.; Liu, Z.; Yang, Y.; Gong, Q.; Ren, B. The Role of HMGB1 in the Pathogenesis of Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 2543268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasu, M.R.; Isseroff, R.R. Toll-like receptors in wound healing: Location, accessibility, and timing. J. Investig. Dermatol. 2012, 132, 1955–1958. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Guo, S.; Ranzer, M.J.; DiPietro, L.A. Toll-like receptor 4 has an essential role in early skin wound healing. J. Investig. Dermatol. 2013, 133, 258–267. [Google Scholar] [CrossRef] [Green Version]
- Suga, H.; Sugaya, M.; Fujita, H.; Asano, Y.; Tada, Y.; Kadono, T.; Sato, S. TLR4, rather than TLR2, regulates wound healing through TGF-beta and CCL5 expression. J. Dermatol. Sci. 2014, 73, 117–124. [Google Scholar] [CrossRef]
- Dasu, M.R.; Thangappan, R.K.; Bourgette, A.; DiPietro, L.A.; Isseroff, R.; Jialal, I. TLR2 expression and signaling-dependent inflammation impair wound healing in diabetic mice. Lab. Investig. 2010, 90, 1628–1636. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Jialal, I. Amelioration in wound healing in diabetic toll-like receptor-4 knockout mice. J. Diabetes Complicat. 2013, 27, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, V.; Olala, L.O.; Kagha, K.; Pan, Z.Q.; Chen, X.; Yang, R.; Cline, A.; Helwa, I.; Marshall, L.; Kaddour-Djebbar, I.; et al. Regulation of the Glycerol Transporter, Aquaporin-3, by Histone Deacetylase-3 and p53 in Keratinocytes. J. Investig. Dermatol. 2017, 137, 1935–1944. [Google Scholar] [CrossRef]
- Hodgkinson, C.P.; Laxton, R.C.; Patel, K.; Ye, S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2275–2281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.J.; Sheu, M.L.; Tsai, K.S.; Yang, R.S.; Liu, S.H. Advanced glycation end products induce peroxisome proliferator-activated receptor gamma down-regulation-related inflammatory signals in human chondrocytes via Toll-like receptor-4 and receptor for advanced glycation end products. PLoS ONE 2013, 8, e66611. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, V.; Uaratanawong, R.; Patel, R.R.; Patel, H.; Bao, W.; Hartney, B.; Cohen, E.; Chen, X.; Zhong, Q.; Isales, C.M.; et al. Phosphatidylglycerol Inhibits Toll-Like Receptor-Mediated Inflammation by Danger-Associated Molecular Patterns. J. Investig. Dermatol. 2019, 139, 868–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, V.; Griffith, S.; Chen, X.; Bollag, W.B. Pathogen-Associated Molecular Pattern-Induced TLR2 and TLR4 Activation Increases Keratinocyte Production of Inflammatory Mediators and is Inhibited by Phosphatidylglycerol. Mol. Pharmacol. 2020, 97, 324–335. [Google Scholar] [CrossRef]
- Baum, M.A.; Ruddy, M.K.; Hosselet, C.A.; Harris, H.W. The perinatal expression of aquaporin-2 and aquaporin-3 in developing kidney. Pediatr. Res. 1998, 43, 783–790. [Google Scholar] [CrossRef] [Green Version]
- Kamiloglu, S.; Sari, G.; Ozdal, T.; Capanoglu, E. Guidelines for cell viability assays. Food Front. 2020, 1, 332–349. [Google Scholar] [CrossRef]
- Sorci, G.; Riuzzi, F.; Giambanco, I.; Donato, R. RAGE in tissue homeostasis, repair and regeneration. Biochim. Biophys. Acta 2013, 1833, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erridge, C. Endogenous ligands of TLR2 and TLR4: Agonists or assistants? J. Leukoc. Biol. 2010, 87, 989–999. [Google Scholar] [CrossRef]
- Xing, Y.; Pan, S.; Zhu, L.; Cui, Q.; Tang, Z.; Liu, Z.; Liu, F. Advanced Glycation End Products Induce Atherosclerosis via RAGE/TLR4 Signaling Mediated-M1 Macrophage Polarization-Dependent Vascular Smooth Muscle Cell Phenotypic Conversion. Oxid. Med. Cell. Longev. 2022, 2022, 9763377. [Google Scholar] [CrossRef]
- Da Silva, I.V.; Cardoso, C.; Martinez-Banaclocha, H.; Casini, A.; Pelegrin, P.; Soveral, G. Aquaporin-3 is involved in NLRP3-inflammasome activation contributing to the setting of inflammatory response. Cell. Mol. Life Sci. 2021, 78, 3073–3085. [Google Scholar] [CrossRef]
- Ito, H.; Kanbe, A.; Sakai, H.; Seishima, M. Activation of NLRP3 signalling accelerates skin wound healing. Exp. Dermatol. 2018, 27, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Vinaik, R.; Abdullahi, A.; Barayan, D.; Jeschke, M.G. NLRP3 inflammasome activity is required for wound healing after burns. Transl. Res. 2020, 217, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Qin, H.; Zheng, X.; Zhong, X.; Shetty, A.K.; Elias, P.M.; Bollag, W.B. Aquaporin-3 in keratinocytes and skin: Its role and interaction with phospholipase D2. Arch. Biochem. Biophys. 2011, 508, 138–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, C.; Belville, C.; Lavergne, M.; Choltus, H.; Jabaudon, M.; Blondonnet, R.; Constantin, J.M.; Chiambaretta, F.; Blanchon, L.; Sapin, V. Advanced Glycation End Products and Receptor (RAGE) Promote Wound Healing of Human Corneal Epithelial Cells. Investig. Ophthalmol. Vis. Sci. 2020, 61, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nass, N.; Trau, S.; Paulsen, F.; Kaiser, D.; Kalinski, T.; Sel, S. The receptor for advanced glycation end products RAGE is involved in corneal healing. Ann. Anat. 2017, 211, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Christensen, D.P.; Dahllof, M.; Lundh, M.; Rasmussen, D.N.; Nielsen, M.D.; Billestrup, N.; Grunnet, L.G.; Mandrup-Poulsen, T. Histone deacetylase (HDAC) inhibition as a novel treatment for diabetes mellitus. Mol. Med. 2011, 17, 378–390. [Google Scholar] [CrossRef]
- Makkar, R.; Behl, T.; Arora, S. Role of HDAC inhibitors in diabetes mellitus. Curr. Res. Transl. Med. 2020, 68, 45–50. [Google Scholar] [CrossRef]
- Chen, Y.; Du, J.; Zhao, Y.T.; Zhang, L.; Lv, G.; Zhuang, S.; Qin, G.; Zhao, T.C. Histone deacetylase (HDAC) inhibition improves myocardial function and prevents cardiac remodeling in diabetic mice. Cardiovasc. Diabetol. 2015, 14, 99. [Google Scholar] [CrossRef] [Green Version]
- Dokmanovic, M.; Clarke, C.; Marks, P.A. Histone deacetylase inhibitors: Overview and perspectives. Mol. Cancer Res. 2007, 5, 981–989. [Google Scholar] [CrossRef] [Green Version]
- Hancock, W.W.; Akimova, T.; Beier, U.H.; Liu, Y.; Wang, L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann. Rheum. Dis. 2012, 71 (Suppl. 2), i46–i54. [Google Scholar] [CrossRef]
- Van Putte, L.; De Schrijver, S.; Moortgat, P. The effects of advanced glycation end products (AGEs) on dermal wound healing and scar formation: A systematic review. Scars Burn Heal. 2016, 2, 2059513116676828. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, G.; Koudijs, M.; van Balkom, B.W.; Oorschot, V.; Klumperman, J.; Deen, P.M.; van der Sluijs, P. Glycosylation is important for cell surface expression of the water channel aquaporin-2 but is not essential for tetramerization in the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 2975–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, L.J.; Choudhary, V.; Merai, P.; Bollag, W.B. Preparation of primary cultures of mouse epidermal keratinocytes and the measurement of phospholipase D activity. Methods Mol. Biol. 2014, 1195, 111–131. [Google Scholar] [CrossRef]
- Tian, M.; Lu, S.; Niu, Y.; Xie, T.; Dong, J.; Cao, X.; Song, F.; Jin, S.; Qing, C. Effects of advanced glycation end-products (AGEs) on skin keratinocytes by nuclear factor-kappa B (NF-κB) activation. Afr. J. Biotechnol. 2012, 11, 11132–11142. [Google Scholar]
- Zhu, P.; Yang, C.; Chen, L.-H.; Ren, M.; Lao, G.-j.; Yan, L. Impairment of human keratinocyte mobility and proliferation by advanced glycation end products-modified BSA. Arch. Dermatol. Res. 2011, 303, 339–350. [Google Scholar] [CrossRef]
- Zhu, P.; Chen, C.; Wu, D.; Chen, G.; Tan, R.; Ran, J. AGEs-induced MMP-9 activation mediated by Notch1 signaling is involved in impaired wound healing in diabetic rats. Diabetes Res. Clin. Pract. 2022, 186, 109831. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, Y.; Uaratanawong, R.; Choudhary, V.; Hardin, M.; Zhang, C.; Melnyk, S.; Chen, X.; Bollag, W.B. Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes. Int. J. Mol. Sci. 2023, 24, 1376. https://doi.org/10.3390/ijms24021376
Luo Y, Uaratanawong R, Choudhary V, Hardin M, Zhang C, Melnyk S, Chen X, Bollag WB. Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes. International Journal of Molecular Sciences. 2023; 24(2):1376. https://doi.org/10.3390/ijms24021376
Chicago/Turabian StyleLuo, Yonghong, Rawipan Uaratanawong, Vivek Choudhary, Mary Hardin, Catherine Zhang, Samuel Melnyk, Xunsheng Chen, and Wendy B. Bollag. 2023. "Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes" International Journal of Molecular Sciences 24, no. 2: 1376. https://doi.org/10.3390/ijms24021376
APA StyleLuo, Y., Uaratanawong, R., Choudhary, V., Hardin, M., Zhang, C., Melnyk, S., Chen, X., & Bollag, W. B. (2023). Advanced Glycation End Products and Activation of Toll-like Receptor-2 and -4 Induced Changes in Aquaporin-3 Expression in Mouse Keratinocytes. International Journal of Molecular Sciences, 24(2), 1376. https://doi.org/10.3390/ijms24021376